

Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Metal–Tf Sample Preparation
2.3. Biolayer Interferometry Measurements
2.4. Protein Aggregation Measurements, Gel Electrophoresis, and Crystal Structure Analysis
2.5. Cell Culture and Treatment
2.6. Measurements of Cellular Ru and Fe Uptake
2.7. Determination of Ru–Protein Binding in Cell Culture Medium
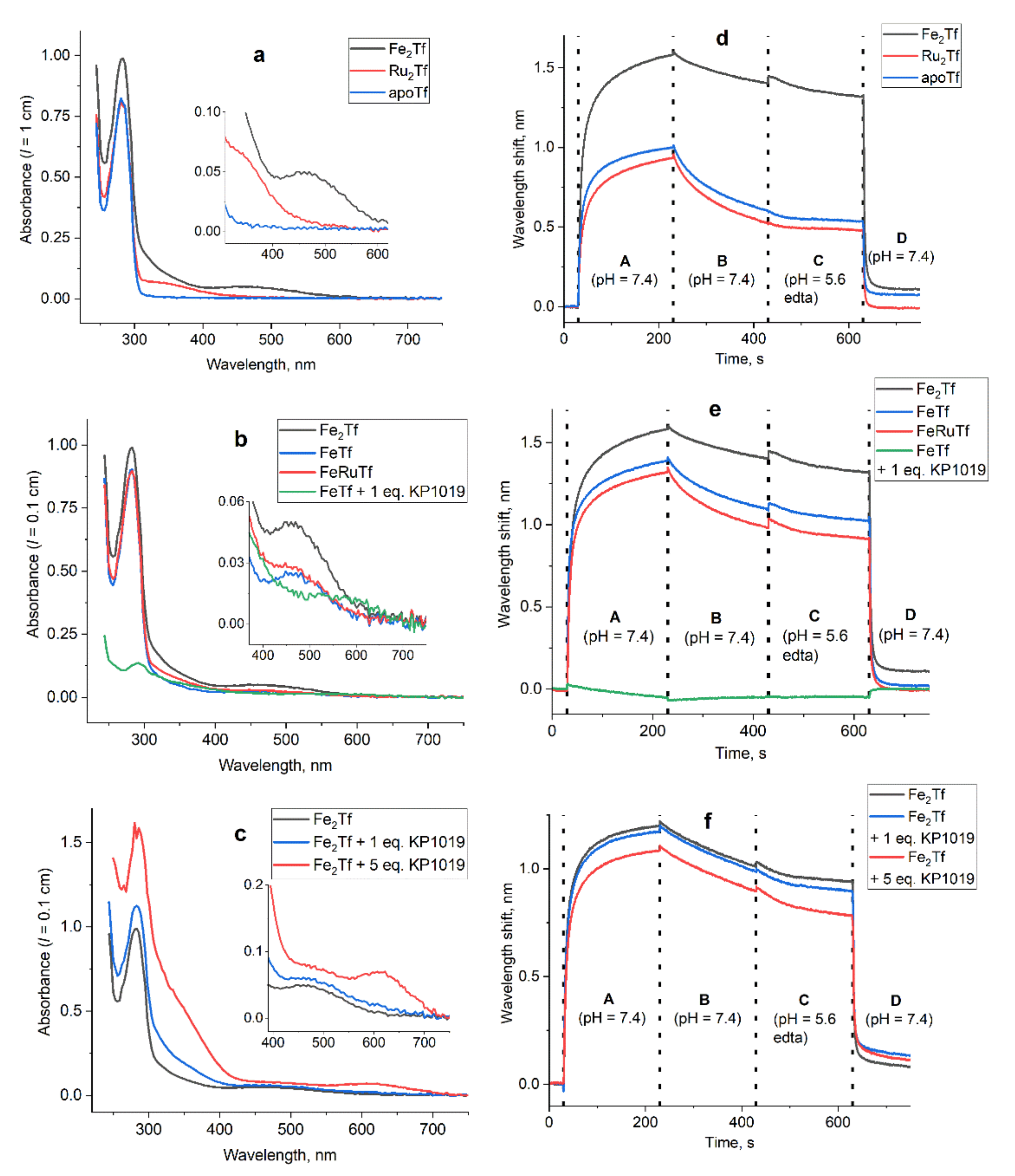
3. Results
3.1. Effect of Ru Complexes on Tf–TfR1 Binding in Cell-Free Systems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample b | λmax, nm | εmax, M−1 cm−1 | KD, nM c | Mean Particle Size, nm |
|---|---|---|---|---|
| apoTf | 280 | 8.2 × 104 d | 43 | 7 ± 1 |
| Ru2Tf (A1) | 380 e | 7.0 × 102 | 162 | 7 ± 1 |
| Fe2Tf (A2) | 470 | 4.9 × 102 d | 4.6 f | 7 ± 2 |
| monoFeTf (A3) | 460 | 2.5 × 102 | 19 | 7 ± 1 |
| RuFeTf (A4) | 460 | 2.8 × 102 | 31 | 6.5 ± 1 |
| monoFeTf + 1.0 eq. KP1019 (A5) | 290 | 1.4 × 104 | no binding | 175, 805 |
| apoTf + 2.0 eq. KP1019 (A6) | 290 | 2.0 × 104 | no binding | 450, 970 |
| apoTf + 4.0 eq. KP1019 (A7) | 290 | 2.4 × 104 | no binding | >1000 |
| Fe2Tf + 1.0 eq. KP1019 (A8) | 460 | 6.2 × 102 | 5 | 7 ± 2 |
| Fe2Tf + 5.0 eq. KP1019 (A9) | 620 | 7.5 × 102 | 10 | 9 ± 2 |
3.2. Effect of Tf on the Uptake of Ru Complexes by HepG2 Cells
| Conditions b | Ru Uptake c | Fe Content c | % Ru–Protein d |
|---|---|---|---|
| no Ru added | 0 | 1.9 ± 0.7 | 0 |
| KP1019 fresh (B1) | 6.6 ± 2.8 e | 11.2 ± 6.5 f | 46 ± 4 |
| KP1019 decomposed (B2) | 0.23 ± 0.09 e | 1.2 ± 0.8 | 49 ± 1 |
| KP1019 + HSA (B3) | 0.062 ± 0.005 | 1.6 ± 0.5 | 73 ± 3 g |
| KP1019 + apoTf (B4) | 0.13 ± 0.04 e | 1.3 ± 0.6 | 64 ± 1 g |
| KP1019 + Fe0.6Tf (B5) | 0.13 ± 0.03 e | 3.4 ± 3.5 | 64 ± 1 g |
| KP1019 + Fe2Tf (B6) | 0.26 ± 0.03 e | 8.9 ± 1.7 f | 53 ± 4 |
| [Ru(nta)2]3− (B7) | 0.09 ± 0.03 | 1.4 ± 0.4 | 2 ± 1 g |
| [Ru(nta)2]3− + Fe2Tf (B8) | 2.9 ± 0.3 e | 5.8 ± 1.7 f | 3 ± 1 g |
| Ru2Tf (B9) | 0.07 ± 0.06 | 1.9 ± 0.7 | 74 ± 3 g |

4. Discussion
4.1. Changing Paradigm in Anticancer Ru(III) Complexes
4.2. Interactions of Ru(III) Complexes with Tf in Aqueous Buffers
4.3. Interactions of Ru(III) Complexes with Tf in Cell Culture Medium and in Blood Serum
4.4. Roles of Tf in Cellular Ru Uptake
4.5. Potential Application of KP1019 for Intratumoral Injections
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poetsch, I.; Baier, D.; Keppler, B.K.; Berger, W. Challenges and chances in the preclinical to clinical translation of anticancer metallodrugs. RSC Metallobiol. 2019, 14, 308–347. [Google Scholar]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Future Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure-activity relationships for ruthenium and osmium anticancer agents-towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.S.; Dyson, P.J. Recent progress in the development of organometallics for the treatment of cancer. Curr. Opin. Chem. Biol. 2020, 56, 28–34. [Google Scholar] [CrossRef]
- Pragti; Kundu, B.K.; Mukhopadhyay, S. Target based chemotherapeutic advancement of ruthenium complexes. Coord. Chem. Rev. 2021, 448, 214169. [Google Scholar]
- Paprocka, R.; Wiese-Szadkowska, M.; Janciauskiene, S.; Kosmalski, T.; Kulik, M.; Helmin-Basa, A. Latest developments in metal complexes as anticancer agents. Coord. Chem. Rev. 2022, 452, 214307. [Google Scholar]
- Conti, L.; Macedi, E.; Giorgi, C.; Valtancoli, B.; Fusi, V. Combination of light and Ru(II) polypyridyl complexes: Recent advances in the development of new anticancer drugs. Coord. Chem. Rev. 2022, 469, 214656. [Google Scholar]
- Sen, S.; Won, M.; Levine, M.S.; Noh, Y.; Sedgwick, A.C.; Kim, J.S.; Sessler, J.L.; Arambula, J.F. Metal-based anticancer agents as immunogenic cell death inducers: The past, present, and future. Chem. Soc. Rev. 2022, 51, 1212–1233. [Google Scholar] [CrossRef]
- Toupin, N.; Herroon, M.K.; Thummel, R.P.; Turro, C.; Podgorski, I.; Gibson, H.; Kodanko, J.J. Metalloimmunotherapy with rhodium and ruthenium complexes: Targeting tumor-associated macrophages. Chem. Eur. J. 2022, 28, e202104430. [Google Scholar]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Levina, A.; Crans, D.C.; Lay, P.A. Speciation of metal drugs, supplements and toxins in media and bodily fluids controls in vitro activities. Coord. Chem. Rev. 2017, 352, 473–498. [Google Scholar] [CrossRef]
- Pal, M.; Nandi, U.; Mukherjee, D. Detailed account on activation mechanisms of ruthenium coordination complexes and their role as antineoplastic agents. Eur. J. Med. Chem. 2018, 150, 419–445. [Google Scholar] [CrossRef] [PubMed]
- Rilak Simovic, A.; Masnikosa, R.; Bratsos, I.; Alessio, E. Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure. Coord. Chem. Rev. 2019, 398, 113011. [Google Scholar] [CrossRef]
- Levina, A.; Crans, D.C.; Lay, P.A. Advantageous reactivity of unstable metal complexes: Potential applications of metal-based anticancer drugs for intratumoral injections. Pharmaceutics 2022, 14, 790. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E. Thirty years of the drug candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: A personal perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Online Resource of the U.S. National Library of Medicine. Available online: www.ClinicalTrials.gov (accessed on 7 September 2022).
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef]
- Baier, D.; Schoenhacker-Alte, B.; Rusz, M.; Pirker, C.; Mohr, T.; Mendrina, T.; Kirchhofer, D.; Meier-Menches, S.M.; Hohenwallner, K.; Schaier, M.; et al. The anticancer ruthenium compound BOLD-100 targets glycolysis and generates a metabolic vulnerability towards glucose deprivation. Pharmaceutics 2022, 14, 238. [Google Scholar] [CrossRef]
- Silva, A.M.N.; Moniz, T.; de Castro, B.; Rangel, M. Human transferrin: An inorganic biochemistry perspective. Coord. Chem. Rev. 2021, 449, 214186. [Google Scholar] [CrossRef]
- Kratz, F.; Hartmann, M.; Keppler, B.; Messori, L. The binding properties of two antitumor ruthenium(III) complexes to apotransferrin. J. Biol. Chem. 1994, 269, 2581–2588. [Google Scholar] [CrossRef]
- Piccioli, F.; Sabatini, S.; Messori, L.; Orioli, P.; Hartinger, C.G.; Keppler, B.K. A comparative study of adduct formation between the anticancer ruthenium(III) compound HInd trans-[RuCl4(Ind)2] and serum proteins. J. Inorg. Biochem. 2004, 98, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Lay, P.A. Influence of an anti-metastatic ruthenium(III) prodrug on extracellular protein-protein interactions: Studies by bio-layer interferometry. Inorg. Chem. Front. 2014, 1, 44–48. [Google Scholar] [CrossRef]
- Guo, W.; Zheng, W.; Luo, Q.; Li, X.; Zhao, Y.; Xiong, S.; Wang, F. Transferrin serves as a mediator to deliver organometallic ruthenium(II) anticancer complexes into cells. Inorg. Chem. 2013, 52, 5328–5338. [Google Scholar] [CrossRef]
- Wang, M.; Wang, H.; Xu, X.; Lai, T.-P.; Zhou, Y.; Hao, Q.; Li, H.; Sun, H. Binding of ruthenium and osmium at non-iron sites of transferrin accounts for their iron-independent cellular uptake. J. Inorg. Biochem. 2022, 234, 111885. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Transferrin cycle and clinical roles of citrate and ascorbate in improved iron metabolism. ACS Chem. Biol. 2019, 14, 893–900. [Google Scholar] [CrossRef]
- Byrne, S.L.; Mason, A.B. Human serum transferrin: A tale of two lobes. Urea gel and steady state fluorescence analysis of recombinant transferrins as a function of pH, time, and the soluble portion of the transferrin receptor. J. Biol. Inorg. Chem. 2009, 14, 771–781. [Google Scholar] [CrossRef]
- Levina, A.; Wang, B.; Lay, P.A. Urea gel electrophoresis in studies of conformational changes of transferrin on binding and transport of non-ferric metal ions. Gels 2022, 8, 19. [Google Scholar] [CrossRef]
- Levina, A.; Pham, T.H.N.; Lay, P.A. Binding of chromium(III) to transferrin could be involved in detoxification of dietary chromium(III) rather than transport of an essential trace element. Angew. Chem. Int. Ed. 2016, 55, 8104–8107. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Vanadium(V/IV)-transferrin binding disrupts the transferrin cycle and reduces vanadium uptake and antiproliferative activity in human lung cancer cells. Inorg. Chem. 2020, 59, 16143–16153. [Google Scholar] [CrossRef]
- Spiewak, K.; Swiatek, S.; Jachimska, B.; Brindell, M. Induction of transferrin aggregation by indazolium [tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019) and its biological function. New J. Chem. 2019, 43, 11296–11306. [Google Scholar] [CrossRef]
- Alessio, E.; Balducci, G.; Calligaris, M.; Costa, G.; Attia, W.M.; Mestroni, G. Synthesis, molecular structure, and chemical behavior of hydrogen trans-bis(dimethyl sulfoxide)tetrachlororuthenate(III) and mer-trichlorotris(dimethyl sulfoxide)ruthenium(III): The first fully characterized chloride-dimethyl sulfoxide-ruthenium(III) complexes. Inorg. Chem. 1991, 30, 609–618. [Google Scholar]
- Lipponer, K.-G.; Vogel, E.; Keppler, B.K. Synthesis, Characterization and solution chemistry of trans-indazoliumtetrachlorobis(indazole)ruthenate(III), a new anticancer ruthenium complex. IR, UV, NMR, HPLC investigations and antitumor activity. Crystal structures of trans-1-methyl-indazoliumtetrachlorobis-(1-methylindazole)ruthenate(III) and its hydrolysis product trans-monoaquatrichlorobis-(1-methylindazole)-ruthenate(III). Metal-Based Drugs 1996, 3, 841465. [Google Scholar]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2pta)] (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 2001, 1396–1397. [Google Scholar] [CrossRef]
- Levina, A.; Aitken, J.B.; Gwee, Y.Y.; Lim, Z.J.; Liu, M.; Singharay, A.M.; Wong, P.F.; Lay, P.A. Biotransformations of anticancer ruthenium(III) complexes: An X-ray absorption spectroscopic study. Chem. Eur. J. 2013, 19, 3609–3619. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.-J. Ruthenium Arene Complexes as Anticancer Agents: An XAS Study. Ph.D. Thesis, The University of Sydney, Sydney, NSW, Australia, 2014. [Google Scholar]
- Patra, M.; Joshi, T.; Pierroz, V.; Ingram, K.; Kaiser, M.; Ferrari, S.; Spingler, B.; Keiser, J.; Gasser, G. DMSO-mediated ligand dissociation: Renaissance for biological activity of N-heterocyclic-[Ru(η6-arene)Cl2] drug candidates. Chem. Eur. J. 2013, 19, 14768–14772. [Google Scholar] [CrossRef]
- Aisen, P.; Aasa, R.; Malmstrom, B.G.; Vanngard, T. Bicarbonate and the binding of iron to transferrin. J. Biol. Chem. 1967, 242, 2484–2490. [Google Scholar] [CrossRef]
- Kratz, F.; Messori, L. Spectral characterization of ruthenium(III) transferrin. J. Inorg. Biochem. 1993, 49, 79–82. [Google Scholar] [CrossRef]
- James, N.G.; Mason, A.B. Protocol to determine accurate absorption coefficients for iron-containing transferrins. Anal. Biochem. 2008, 378, 202–207. [Google Scholar] [CrossRef]
- Ritchie, R.F.; Palomaki, G.E.; Neveux, L.M.; Navolotskaia, O.; Ledue, T.B.; Craig, W.Y. Reference distributions for serum iron and transferrin saturation: A comparison of a large cohort to the world’s literature. J. Clin. Lab. Anal. 2002, 16, 246–252. [Google Scholar] [CrossRef]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef]
- Lebron, J.A.; Bennett, M.J.; Vaughn, D.E.; Chirino, A.J.; Snow, P.M.; Mintier, G.A.; Feder, J.N.; Bjorkman, P.J. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell 1998, 93, 111–123. [Google Scholar] [CrossRef]
- Giannetti, A.M.; Snow, P.M.; Zak, O.; Bjoerkman, P.J. Mechanism for multiple ligand recognition by the human transferrin receptor. PLoS Biol. 2003, 1, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Lorber, B.; Fischer, F.; Bailly, M.; Roy, H.; Kern, D. Protein analysis by dynamic light scattering: Methods and techniques for students. Biochem. Mol. Biol. Educ. 2012, 40, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.P.; Gorman-Lewis, D.; Aryal, B.; Paunesku, T.; Vogt, S.; Rickert, P.G.; Seifert, S.; Lai, B.; Woloschak, G.E.; Soderholm, L. An iron-dependent and transferrin-mediated cellular uptake pathway for plutonium. Nat. Chem. Biol. 2011, 7, 560–565. [Google Scholar] [CrossRef]
- Bijelic, A.; Theiner, S.; Keppler, B.K.; Rompel, A. X-ray Structure analysis of indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019) bound to human serum albumin reveals two ruthenium binding sites and provides insights into the drug binding mechanism. J. Med. Chem. 2016, 59, 5894–5903. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Hann, S.; Koellensperger, G.; Sulyok, M.; Groessl, M.; Timerbaev, A.R.; Rudnev, A.V.; Stingeder, G.; Keppler, B.K. Interactions of a novel ruthenium-based anticancer drug (KP1019 or FFC14a) with serum proteins-significance for the patient. Int. J. Clin. Pharmacol. Ther. 2005, 43, 583–585. [Google Scholar] [CrossRef]
- Cetinbas, N.; Webb, M.I.; Dubland, J.A.; Walsby, C.J. Serum-protein interactions with anticancer Ru(III) complexes KP1019 and KP418 characterized by EPR. J. Biol. Inorg. Chem. 2010, 15, 131–145. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Tomaz, I. Transport of therapeutic vanadium and ruthenium complexes by blood plasma components. Curr. Med. Chem. 2010, 17, 3701–3738. [Google Scholar] [CrossRef]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef]
- Li, H.Y.; Sadler, P.J.; Sun, H.Z. Rationalization of the strength of metal binding to human serum transferrin. Eur. J. Biochem. 1996, 242, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent-Preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Reisner, E.; Arion, V.B.; Keppler, B.K.; Pombeiro, A.J.L. Electron-transfer activated metal-based anticancer drugs. Inorg. Chim. Acta 2008, 361, 1569–1583. [Google Scholar] [CrossRef]
- Candelaria, P.V.; Leoh, L.S.; Penichet, M.L.; Daniels-Wells, T.R. Antibodies targeting the transferrin receptor 1 (TfR1) as direct anti-cancer agents. Front. Immunol. 2021, 12, 607692. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.-F. Targeting hypoxia: Hypoxia-activated prodrugs in cancer therapy. Front. Oncol. 2021, 11, 700407. [Google Scholar] [CrossRef]
- Henke, M.M.; Richly, H.; Drescher, A.; Grubert, M.; Alex, D.; Thyssen, D.; Jaehde, U.; Scheulen, M.E.; Hilger, R.A. Pharmacokinetic study of sodium trans-[tetrachlorobis(1H-indazole)-ruthenate (III)]/-indazole hydrochloride (1:1:1) (FFC14A) in patients with solid tumors. Int. J. Clin. Pharmacol. Ther. 2009, 47, 58–60. [Google Scholar] [CrossRef]
- Timerbaev, A.R.; Foteeva, L.S.; Rudnev, A.V.; Abramski, J.K.; Polec-Pawlak, K.; Hartinger, C.G.; Jarosz, M.; Keppler, B.K. Probing the stability of serum protein-ruthenium(III) drug adducts in the presence of extracellular reductants using CE. Electrophoresis 2007, 28, 2235–2240. [Google Scholar] [CrossRef]
- Hummer, A.A.; Heffeter, P.; Berger, W.; Filipits, M.; Batchelor, D.; Buechel, G.E.; Jakupec, M.A.; Keppler, B.K.; Rompel, A. X-ray absorption near edge structure spectroscopy to resolve the in vivo chemistry of the redox-active indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019). J. Med. Chem. 2013, 56, 1182–1196. [Google Scholar] [CrossRef]
- Gransbury, G.K.; Kappen, P.; Glover, C.J.; Hughes, J.N.; Levina, A.; Lay, P.A.; Musgrave, I.F.; Harris, H.H. Comparison of KP1019 and NAMI-A in tumour-mimetic environments. Metallomics 2016, 8, 762–773. [Google Scholar] [CrossRef]
- El Hage Chahine, J.-M.; Hemadi, M.; Ha-Duong, N.-T. Uptake and release of metal ions by transferrin and interaction with receptor 1. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 334–347. [Google Scholar] [CrossRef]
- Pongratz, M.; Schluga, P.; Jakupec, M.A.; Arion, V.B.; Hartinger, C.G.; Allmaier, G.; Keppler, B.K. Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy. J. Anal. At. Spectrom. 2004, 19, 46–51. [Google Scholar] [CrossRef]
- Levina, A.; Armstrong, R.S.; Lay, P.A. Three-dimensional structure determination using multiple-scattering analysis of XAFS: Applications to metalloproteins and coordination chemistry. Coord. Chem. Rev. 2005, 249, 141–160. [Google Scholar] [CrossRef]
- Yang, N.; Zhang, H.; Wang, M.; Hao, Q.; Sun, H. Iron and bismuth bound human serum transferrin reveals a partially-opened conformation in the N-lobe. Sci. Rep. 2012, 2, srep00999. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Saxena, M.; Sharma, S.; Noinaj, N.; Delgado, Y.; Quinones Gonzalez, E.P.; Conklin, S.E.; Zambrana, N.; Loza-Rosas, S.A.; Parks, T.B. Unusual synergism of transferrin and citrate in the regulation of Ti(IV) speciation, transport, and toxicity. J. Am. Chem. Soc. 2016, 138, 5659–5665. [Google Scholar] [CrossRef] [PubMed]
- Spiewak, K.; Brindell, M. Impact of low- and high-molecular-mass components of human serum on NAMI-A binding to transferrin. J. Biol. Inorg. Chem. 2015, 20, 695–703. [Google Scholar] [CrossRef]
- Steere, A.N.; Byrne, S.L.; Chasteen, N.D.; Smith, V.C.; MacGillivray, R.T.A.; Mason, A.B. Evidence that His349 acts as a pH-inducible switch to accelerate receptor-mediated iron release from the C-lobe of human transferrin. J. Biol. Inorg. Chem. 2010, 15, 1341–1352. [Google Scholar] [CrossRef][Green Version]
- Eckenroth, B.E.; Steere, A.N.; Chasteen, N.D.; Everse, S.J.; Mason, A.B. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc. Natl. Acad. Sci. USA 2011, 108, 13089–13094. [Google Scholar] [CrossRef]
- Dayon, L.; Kussmann, M. Proteomics of human plasma: A critical comparison of analytical workflows in terms of effort, throughput and outcome. EuPa Open Proteom. 2013, 1, 8–16. [Google Scholar] [CrossRef]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anti-Cancer Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef]
- Sanna, D.; Micera, G.; Garribba, E. Interaction of insulin-enhancing vanadium compounds with human serum holo-transferrin. Inorg. Chem. 2013, 52, 11975–11985. [Google Scholar] [CrossRef]
- Bryce, N.S.; Pham, B.T.T.; Fong, N.W.S.; Jain, N.; Pan, E.H.; Whan, R.M.; Hambley, T.W.; Hawkett, B.S. The composition and end-group functionality of sterically stabilized nanoparticles enhances the effectiveness of co-administered cytotoxins. Biomater. Sci. 2013, 1, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Pan, Y.; Cai, R.; Guo, S.; Zhang, X.; Xue, Y.; Wang, J.; Huang, J.; Wang, J.; Gu, Y.; et al. Salvianolic acid A increases the accumulation of doxorubicin in brain tumors through caveolae endocytosis. Neuropharmacology 2020, 167, 107980. [Google Scholar] [CrossRef] [PubMed]
- Naves, M.A.; Graminha, A.E.; Vegas, L.C.; Luna-Dulcey, L.; Honorato, J.; Menezes, A.C.S.; Batista, A.A.; Cominetti, M.R. Transport of the ruthenium complex [Ru(GA)(dppe)2]PF6 into triple-negative breast cancer cells is facilitated by transferrin receptors. Mol. Pharm. 2019, 16, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Pires Vieira, A.; Wijetunga, A.; Kaur, R.; Koehn, J.T.; Crans, D.C.; Lay, P.A. A Short-lived but highly cytotoxic vanadium(V) complex as a potential drug lead for brain cancer treatment by intratumoral injections. Angew. Chem. Int. Ed. 2020, 59, 15834–15838. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lim, Z.J.; Gwee, Y.Y.; Levina, A.; Lay, P.A. Characterization of a ruthenium(III)/NAMI-A adduct with bovine serum albumin that exhibits a high anti-metastatic activity. Angew. Chem. Int. Ed. 2010, 49, 1661–1664. [Google Scholar] [CrossRef]
- Munteanu, A.-C.; Uivarosi, V. Ruthenium complexes in the fight against pathogenic microorganisms. an extensive review. Pharmaceutics 2021, 13, 874. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Paduano, L.; Montesarchio, D. Anticancer ruthenium(III) complexes and Ru(III)-containing nanoformulations: An update on the mechanism of action and biological activity. Pharmaceuticals 2019, 12, 146. [Google Scholar] [CrossRef]





| Designation a | Description b |
|---|---|
| A1 | apoTf (100 µM) + 2.0 eq. [RuIII(nta)2]3−→Ru2Tf |
| A2 | apoTf (100 µM) + 2.0 eq. [FeIII(nta)2]3−→Fe2Tf |
| A3 | apoTf (100 µM) + 1.0 eq. [FeIII(nta)2]3−→monoFeTf |
| A4 | apoTf (100 µM) + 1.0 eq. [FeIII(nta)2]3−, then 1.0 eq. [RuIII(nta)2]3−→RuFeTf |
| A5 | apoTf (100 µM) + 1.0 eq. [FeIII(nta)2]3−, then 1.0 eq. KP1019 |
| A6 | apoTf (100 µM) + 2.0 eq. KP1019 |
| A7 | apoTf (100 µM) + 4.0 eq. KP1019 |
| A8 | Fe2Tf (84 µM) + 1.0 eq. KP1019 |
| A9 | Fe2Tf (84 µM) + 5.0 eq. KP1019 |
| B1 | KP1019 (60 µM), freshly added to the base medium |
| B2 | KP1019 (60 µM) |
| B3 | KP1019 (60 µM) + HSA (30 µM) |
| B4 | KP1019 (60 µM) + apoTf (30 µM) |
| B5 | KP1019 (60 µM) + Fe0.6Tf (30 µM) |
| B6 | KP1019 (60 µM) + Fe2Tf (30 µM) |
| B7 | [RuIII(nta)2]3− (60 µM) |
| B8 | [RuIII(nta)2]3− (60 µM) + Fe2Tf (30 µM) |
| B9 | Ru2Tf (30 µM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levina, A.; Chetcuti, A.R.M.; Lay, P.A. Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs. Biomolecules 2022, 12, 1319. https://doi.org/10.3390/biom12091319
Levina A, Chetcuti ARM, Lay PA. Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs. Biomolecules. 2022; 12(9):1319. https://doi.org/10.3390/biom12091319
Chicago/Turabian StyleLevina, Aviva, Anthony R. M. Chetcuti, and Peter A. Lay. 2022. "Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs" Biomolecules 12, no. 9: 1319. https://doi.org/10.3390/biom12091319
APA StyleLevina, A., Chetcuti, A. R. M., & Lay, P. A. (2022). Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs. Biomolecules, 12(9), 1319. https://doi.org/10.3390/biom12091319