

New Insights into the Alveolar Epithelium as a Driver of Acute Respiratory Distress Syndrome

 , , ,
, , , 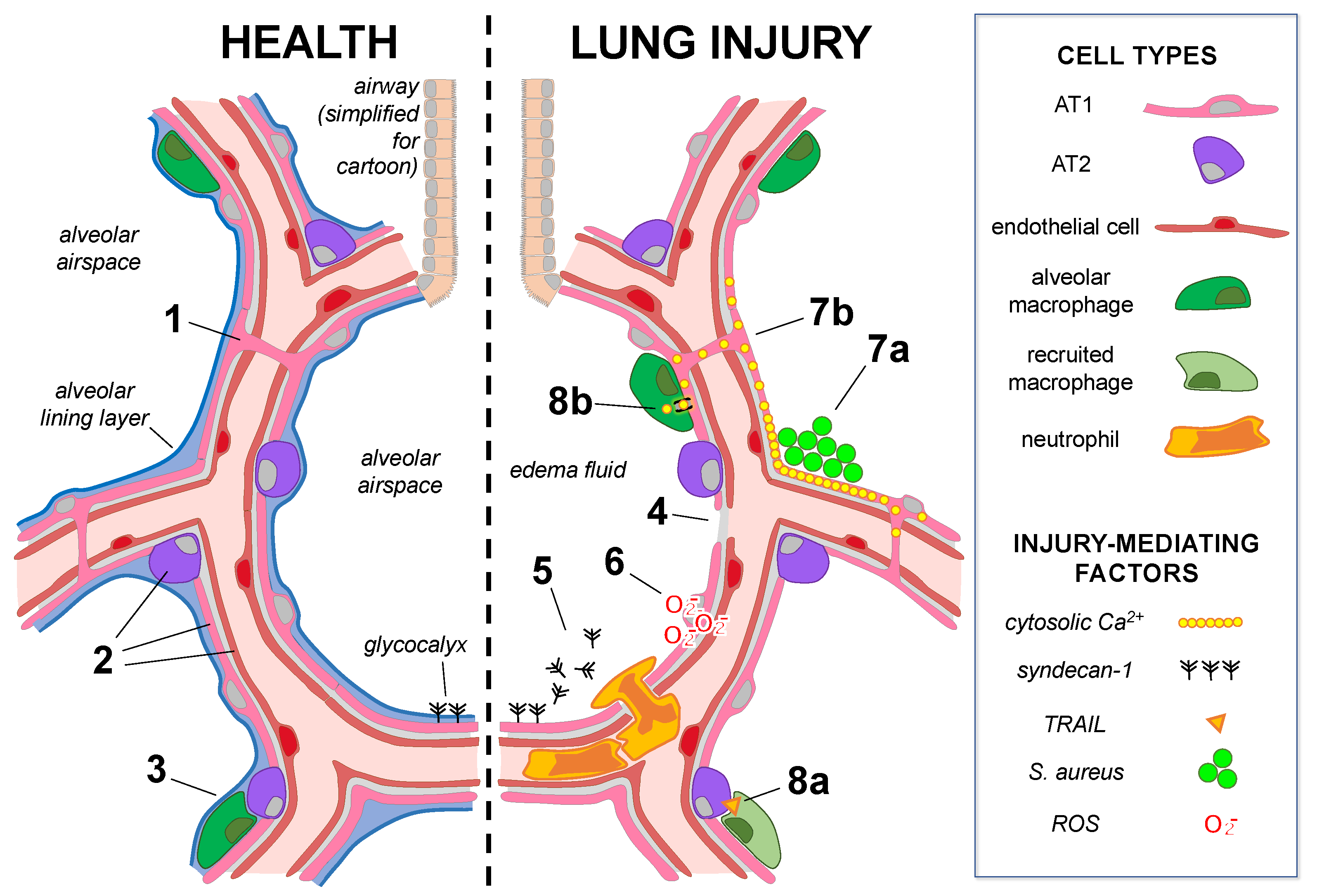{kind=link}
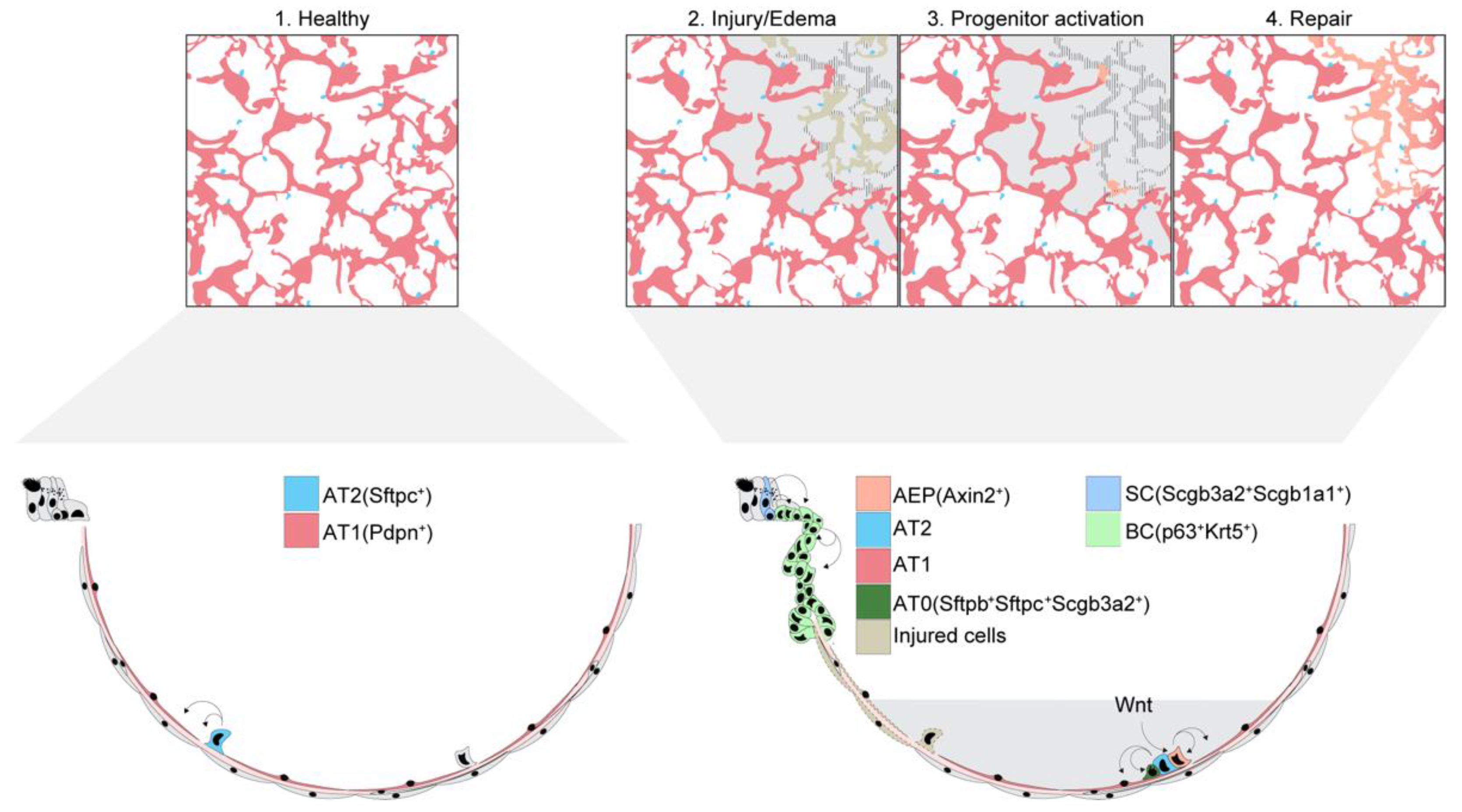{kind=link}
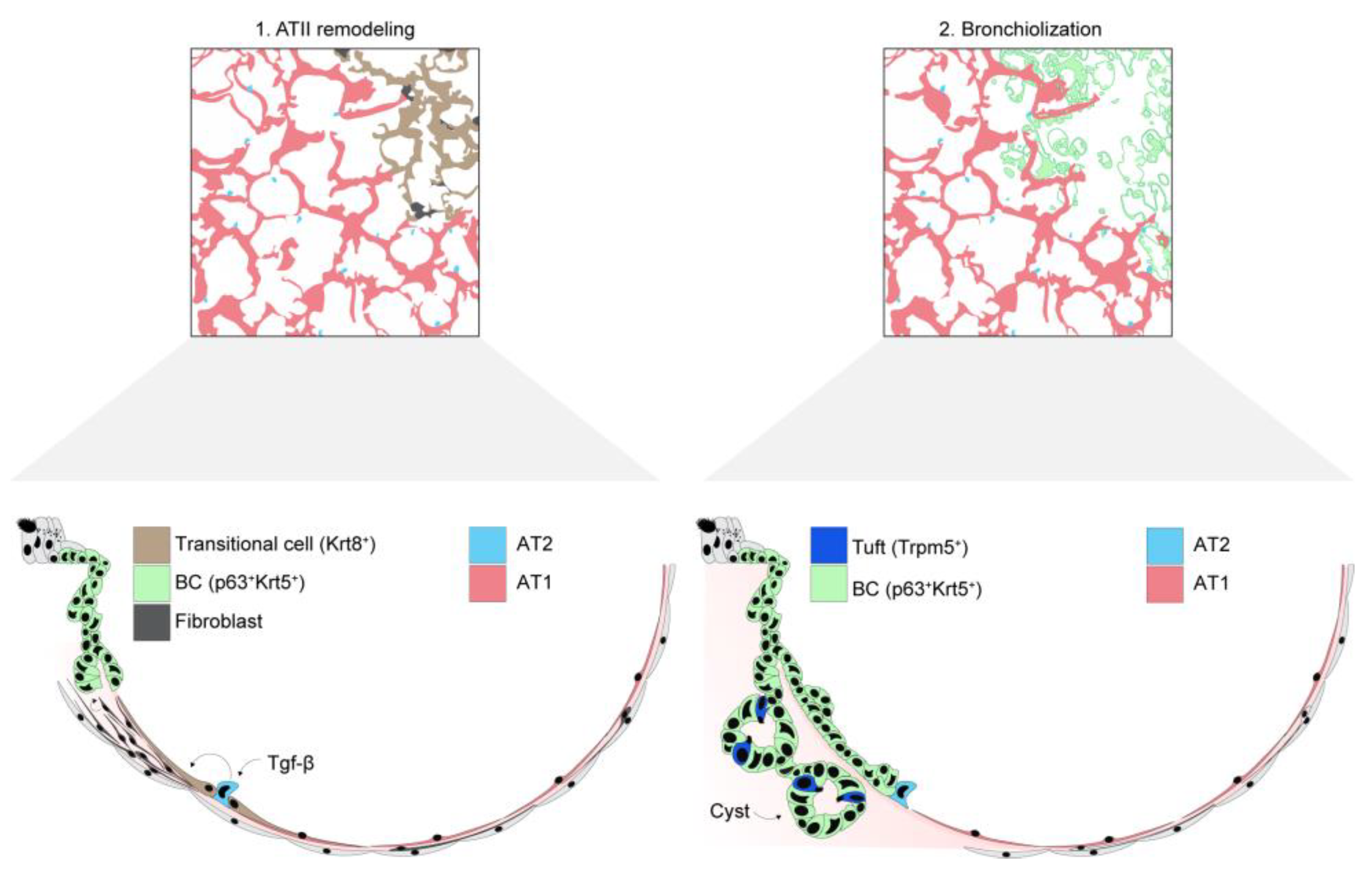{kind=link}
Abstract
1. Introduction
2. The Alveolar Epithelium at Homeostasis
2.1. Alveolar Epithelial Structure
2.2. Alveolar Epithelial Homeostatic Function
3. Alveolar Epithelial Responses That Lead to Lung Injury
3.1. New Insights into ARDS Pathogenesis from Human Studies during the COVID-19 Pandemic
3.2. Direct Mechanisms of Alveolar Epithelial Damage
3.3. Alveolar Epithelial Cell–Cell Communication Amplifies Injury Responses
3.4. Alveolar Epithelial Cells Transmit Injury Signals to Microvessels
3.5. Alveolar Epithelial Cells Organize Signaling Networks with Innate Immunity
4. Alveolar Epithelial Repair and Regeneration
4.1. Alveolar Epithelial Survival after Lung Injury
4.2. Alveolar Epithelial Cell Contributions to Alveolar Epithelial Repair and Regeneration
4.3. Distal Airway Cell Contributions to Alveolar Epithelial Repair and Regeneration
5. Maladaptive Alveolar Epithelial Responses That Cause Lung Remodeling
5.1. Evidence of Maladaptive Lung Remodeling in ARDS
5.2. Mechanisms of Post-ARDS Maladaptive Remodeling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weibel, E.R. The Pathway for Oxygen: Structure and Function in the Mammalian Respiratory System; Harvard University Press: Cambridge, MA, USA, 1984; p. 425. [Google Scholar]
- Gehr, P.; Bachofen, M.; Weibel, E.R. The normal human lung: Ultrastructure and morphometric estimation of diffusion capacity. Respir. Physiol. 1978, 32, 121–140. [Google Scholar] [CrossRef]
- Crapo, J.D.; Barry, B.E.; Gehr, P.; Bachofen, M.; Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Wrede, C.; Hegermann, J.; Weibel, E.R.; Mühlfeld, C.; Ochs, M. On the Topological Complexity of Human Alveolar Epithelial Type 1 Cells. Am. J. Respir. Crit. Care Med. 2019, 199, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.C.; Mercer, R.R.; Gehr, P.; Stockstill, B.; Crapo, J.D. Allometric Relationships of Cell Numbers and Size in the Mammalian Lung. Am. J. Respir. Cell Mol. Biol. 1992, 6, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, H.; Ng, C. Estimating alveolar surface area during life. Respir. Physiol. 1992, 88, 163–170. [Google Scholar] [CrossRef]
- Bastacky, J.; Lee, C.Y.; Goerke, J.; Koushafar, H.; Yager, D.; Kenaga, L.; Speed, T.; Chen, Y.; Clements, J.A. Alveolar lining layer is thin and continuous: Low-temperature scanning electron microscopy of rat lung. J. Appl. Physiol. 1995, 79, 1615–1628. [Google Scholar] [CrossRef]
- Weibel, E.R.; Gil, J. Electron microscopic demonstration of an extracellular duplex lining layer of alveoli. Respir. Physiol. 1968, 4, 42–57. [Google Scholar] [CrossRef]
- Gil, J.; Weibel, E.R. Improvements in demonstration of lining layer of lung alveoli by electron microscopy. Respir. Physiol. 1969, 8, 13–36. [Google Scholar] [CrossRef]
- Ochs, M.; Hegermann, J.; Lopez-Rodriguez, E.; Timm, S.; Nouailles, G.; Matuszak, J.; Simmons, S.; Witzenrath, M.; Kuebler, W.M. On Top of the Alveolar Epithelium: Surfactant and the Glycocalyx. Int. J. Mol. Sci. 2020, 21, 3075. [Google Scholar] [CrossRef]
- Goodale, R.; Goetzman, B.; Visscher, M. Hypoxia and iodoacetic acid and alveolocapillary barrier permeability to albumin. Am. J. Physiol. Content 1970, 219, 1226–1230. [Google Scholar] [CrossRef][Green Version]
- Schneeberger, E.E. Ultrastructural basis for alveolar-capillary permeability to protein. In Ciba Foundation Symposium 38-Lung Liquids; Wiley: Hoboken, NJ, USA, 1976; pp. 3–28. [Google Scholar] [CrossRef]
- Gorin, A.B.; Stewart, P.A. Differential permeability of endothelial and epithelial barriers to albumin flux. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1979, 47, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Song, Y.; Bai, C.; Koller, B.H.; Matthay, M.A.; Verkman, A.S. Pleural surface fluorescence measurement of Na+ and Cl− transport across the air space-capillary barrier. J. Appl. Physiol. 2003, 94, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.; Rahner, C.; Anderson, J.M. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J. Clin. Investig. 2001, 107, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, M.D.; Jeansonne, B.G.; Renegar, R.H.; Tatum, R.; Chen, Y.-H. The first extracellular domain of claudin-7 affects paracellular Cl− permeability. Biochem. Biophys. Res. Commun. 2007, 357, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Flodby, P.; Luo, J.; Kage, H.; Sipos, A.; Gao, D.; Ji, Y.; Beard, L.L.; Marconett, C.N.; DeMaio, L.; et al. Knockout Mice Reveal Key Roles for Claudin 18 in Alveolar Barrier Properties and Fluid Homeostasis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 210–222. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J.; Bander, N.H.; Soler, A.P.; Rajasekaran, A.K. Na,K-ATPase β-Subunit Is Required for Epithelial Polarization, Suppression of Invasion, and Cell Motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The Role of the β1 Subunit of the Na,K-ATPase and Its Glycosylation in Cell-Cell Adhesion. J. Biol. Chem. 2006, 281, 39573–39587. [Google Scholar] [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Shoshani, L.; Vagin, O. Epithelial Junctions Depend on Intercellular Trans-interactions between the Na,K-ATPase β1 Subunits. J. Biol. Chem. 2011, 286, 25801–25812. [Google Scholar] [CrossRef]
- Wang, Y.; Minshall, R.D.; Schwartz, D.E.; Hu, G. Cyclic stretch induces alveolar epithelial barrier dysfunction via calpain-mediated degradation of p120-catenin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L197–L206. [Google Scholar] [CrossRef]
- Vagin, O.; Dada, L.A.; Tokhtaeva, E.; Sachs, G. The Na-K-ATPase α1β1heterodimer as a cell adhesion molecule in epithelia. Am. J. Physiol. Cell. Physiol. 2012, 302, C1271–C1281. [Google Scholar] [CrossRef]
- Vadász, I.; Raviv, S.; Sznajder, J.I. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensiv. Care Med. 2007, 33, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Sigrid, A.R.; A Rajasekaran, S.; Rajasekaran, A.K. Na,K-ATPase and epithelial tight junctions. Front. Biosci. 2009, 14, 2130–2148. [Google Scholar] [CrossRef]
- Factor, P.; Saldias, F.; Ridge, K.; Dumasius, V.; Zabner, J.; A Jaffe, H.; Blanco, G.; Barnard, M.; Mercer, R.; Perrin, R.; et al. Augmentation of lung liquid clearance via adenovirus-mediated transfer of a Na,K-ATPase beta1 subunit gene. J. Clin. Investig. 1998, 102, 1421–1430. [Google Scholar] [CrossRef]
- Ridge, K.; Olivera, W.; Saldias, F.; Azzam, Z.; Horowitz, S.; Rutschman, D.; Dumasius, V.; Factor, P.; Sznajder, J. Alveolar Type 1 Cells Express the α2 Na,K-ATPase, Which Contributes to Lung Liquid Clearance. Circ. Res. 2003, 92, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Cheek, J.M.; Kim, K.J.; Crandall, E.D. Tight monolayers of rat alveolar epithelial cells: Bioelectric properties and active sodium transport. Am. J. Physiol. Physiol. 1989, 256, C688–C693. [Google Scholar] [CrossRef] [PubMed]
- Hummler, E.; Barker, P.; Gatzy, J.; Beermann, F.; Verdumo, C.; Schmidt, A.; Boucher, R.; Rossier, B.C. Early death due to defective neonatal lung liquid clearance in αENaC-deficient mice. Nat. Genet. 1996, 12, 325–328. [Google Scholar] [CrossRef]
- Borok, Z.; Liebler, J.M.; Lubman, R.L.; Foster, M.J.; Zhou, B.; Li, X.; Zabski, S.M.; Kim, K.-J.; Crandall, E.D. Na transport proteins are expressed by rat alveolar epithelial type I cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L599–L608. [Google Scholar] [CrossRef]
- Li, T.; Folkesson, H.G. RNA interference for α-ENaC inhibits rat lung fluid absorption in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L649–L660. [Google Scholar] [CrossRef]
- Threefoot, S.A. Gross and microscopic anatomy of the lymphatic vessels and lymphaticovenous communications. Cancer Chemother. Rep. 1968, 52, 1–20. [Google Scholar]
- Bhattacharya, J.; Gropper, M.A.; Staub, N.C. Interstitial fluid pressure gradient measured by micropuncture in excised dog lung. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 271–277. [Google Scholar] [CrossRef]
- Clements, J.A. Surface Tension of Lung Extracts. Proc. Soc. Exp. Biol. Med. 1957, 95, 170–172. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Clements, J. Surface active materials from dog lung. II. Composition and physiological correlations. Am. J. Physiol. Content 1972, 223, 715–726. [Google Scholar] [CrossRef]
- Schürch, S.; Goerke, J.; A Clements, J. Direct determination of volume- and time-dependence of alveolar surface tension in excised lungs. Proc. Natl. Acad. Sci. USA 1978, 75, 3417–3421. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, L.G. Pulmonary Surfactant. Annu. Rev. Med. 1989, 40, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.; Ochs, M. The micromechanics of lung alveoli: Structure and function of surfactant and tissue components. Histochem. Cell Biol. 2018, 150, 661–676. [Google Scholar] [CrossRef]
- Islam, M.N.; Gusarova, G.A.; Monma, E.; Das, S.R.; Bhattacharya, J. F-actin scaffold stabilizes lamellar bodies during surfactant secretion. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L50–L57. [Google Scholar] [CrossRef]
- Andreeva, A.V.; Kutuzov, M.; Voyno-Yasenetskaya, T.A. Regulation of surfactant secretion in alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L259–L271. [Google Scholar] [CrossRef]
- Lindert, J.; Perlman, C.E.; Parthasarathi, K.; Bhattacharya, J. Chloride-Dependent Secretion of Alveolar Wall Liquid Determined by Optical-Sectioning Microscopy. Am. J. Respir. Cell Mol. Biol. 2007, 36, 688–696. [Google Scholar] [CrossRef]
- Bove, P.F.; Grubb, B.R.; Okada, S.F.; Ribeiro, C.M.; Rogers, T.D.; Randell, S.H.; O’Neal, W.K.; Boucher, R.C. Human Alveolar Type II Cells Secrete and Absorb Liquid in Response to Local Nucleotide Signaling. J. Biol. Chem. 2010, 285, 34939–34949. [Google Scholar] [CrossRef]
- Li, X.; Comellas, A.; Karp, P.H.; Ernst, S.E.; Moninger, T.O.; Gansemer, N.D.; Taft, P.J.; Pezzulo, A.; Rector, M.V.; Rossen, N.; et al. CFTR is required for maximal transepithelial liquid transport in pig alveolar epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L152–L160. [Google Scholar] [CrossRef]
- Gschwend, J.; Sherman, S.P.; Ridder, F.; Feng, X.; Liang, H.-E.; Locksley, R.M.; Becher, B.; Schneider, C. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kuzmenko, A.; Wan, S.; Schaffer, L.; Weiss, A.; Fisher, J.H.; Kim, K.S.; McCormack, F.X. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Investig. 2003, 111, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- McCormack, F.X.; Gibbons, R.; Ward, S.R.; Kuzmenko, A.; Wu, H.; Deepe, G.S., Jr. Macrophage-independent Fungicidal Action of the Pulmonary Collectins. J. Biol. Chem. 2003, 278, 36250–36256. [Google Scholar] [CrossRef] [PubMed]
- Schürch, S.; Gehr, P.; Hof, V.I.; Geiser, M.; Green, F. Surfactant displaces particles toward the epithelium in airways and alveoli. Respir. Physiol. 1990, 80, 17–32. [Google Scholar] [CrossRef]
- Nielson, D.W.; Goerke, J.; Clements, J.A. Alveolar subphase pH in the lungs of anesthetized rabbits. Proc. Natl. Acad. Sci. USA 1981, 78, 7119–7123. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef]
- Jansing, N.L.; McClendon, J.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Zemans, R.L. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 519–526. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Chen, Q.; Kumar, V.S.; Finn, J.; Jiang, D.; Liang, J.; Zhao, Y.-Y.; Liu, Y. CD44high alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L41–L51. [Google Scholar] [CrossRef]
- Gillich, A.; Zhang, F.; Farmer, C.G.; Travaglini, K.J.; Tan, S.Y.; Gu, M.; Zhou, B.; Feinstein, J.A.; Krasnow, M.A.; Metzger, R.J. Capillary cell-type specialization in the alveolus. Nature 2020, 586, 785–789. [Google Scholar] [CrossRef]
- Konopka, K.E.; Nguyen, T.; Jentzen, J.M.; Rayes, O.; Schmidt, C.J.; Wilson, A.M.; Farver, C.F.; Myers, J.L. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 Infection is Morphologically Indistinguishable from Other Causes of DAD. Histopathology 2020, 77, 570–578. [Google Scholar] [CrossRef] [PubMed]
- D’Agnillo, F.; Walters, K.-A.; Xiao, Y.; Sheng, Z.-M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790. [Google Scholar] [CrossRef]
- Cardinal-Fernández, P.; Lorente, J.A.; Ballén-Barragán, A.; Matute-Bello, G. Acute Respiratory Distress Syndrome and Diffuse Alveolar Damage. New Insights on a Complex Relationship. Ann. Am. Thorac. Soc. 2017, 14, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 2021, 593, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y.; Li, G.; Zhang, Y.; Li, J.; Haosheng, C. Fluorescent reconstitution on deposition of PM2.5 in lung and extrapulmonary organs. Proc. Natl. Acad. Sci. USA 2019, 116, 2488–2493. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Schuler, B.A.; Habermann, A.C.; Plosa, E.J.; Taylor, C.J.; Jetter, C.; Negretti, N.M.; Kapp, M.E.; Benjamin, J.T.; Gulleman, P.; Nichols, D.S.; et al. Age-determined expression of priming protease TMPRSS2 and localization of SARS-CoV-2 in lung epithelium. J. Clin. Investig. 2021, 131, e140766. [Google Scholar] [CrossRef]
- Ganna, A.; COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Kousathanas, A.; Pairo-Castineira, E.; Rawlik, K.; Stuckey, A.; Odhams, C.A.; Walker, S.; Russell, C.D.; Malinauskas, T.; Wu, Y.; Millar, J.; et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 2022, 607, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Bora, S.A.; Parimon, T.; Zaman, T.; Friedman, O.A.; Palatinus, J.A.; Surapaneni, N.S.; Matusov, Y.P.; Chiang, G.C.; Kassar, A.G.; et al. Cell-Type-Specific Immune Dysregulation in Severely Ill COVID-19 Patients. Cell Rep. 2021, 34, 108590. [Google Scholar] [CrossRef]
- Herold, S.; Gabrielli, N.M.; Vadász, I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L665–L681. [Google Scholar] [CrossRef]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef]
- Biasin, V.; Wygrecka, M.; Bärnthaler, T.; Jandl, K.; Jain, P.P.; Bálint, Z.; Kovacs, G.; Leitinger, G.; Kolb-Lenz, D.; Kornmueller, K.; et al. Docking of Meprin α to Heparan Sulphate Protects the Endothelium from Inflammatory Cell Extravasation. Thromb. Haemost. 2018, 118, 1790–1802. [Google Scholar] [CrossRef]
- Haeger, S.M.; Liu, X.; Han, X.; McNeil, J.B.; Oshima, K.; McMurtry, S.A.; Yang, Y.; Ouyang, Y.; Zhang, F.; Nozik-Grayck, E.; et al. Epithelial Heparan Sulfate Contributes to Alveolar Barrier Function and Is Shed during Lung Injury. Am. J. Respir. Cell Mol. Biol. 2018, 59, 363–374. [Google Scholar] [CrossRef]
- Rizzo, A.N.; Haeger, S.M.; Oshima, K.; Yang, Y.; Wallbank, A.M.; Jin, Y.; Lettau, M.; McCaig, L.A.; Wickersham, N.E.; McNeil, J.B.; et al. Alveolar epithelial glycocalyx degradation mediates surfactant dysfunction and contributes to acute respiratory distress syndrome. JCI Insight 2022, 7, e154573. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.E.; Nadler, S.T.; Li, Q.; Frevert, C.W.; Park, P.W.; Chen, P.; Parks, W.C. Shedding of Syndecan-1/CXCL1 Complexes by Matrix Metalloproteinase 7 Functions as an Epithelial Checkpoint of Neutrophil Activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 243–251. [Google Scholar] [CrossRef]
- Li, Q.; Park, P.W.; Wilson, C.L.; Parks, W.C. Matrilysin Shedding of Syndecan-1 Regulates Chemokine Mobilization and Transepithelial Efflux of Neutrophils in Acute Lung Injury. Cell 2002, 111, 635–646. [Google Scholar] [CrossRef]
- Brauer, R.; Ge, L.; Schlesinger, S.Y.; Birkland, T.P.; Huang, Y.; Parimon, T.; Lee, V.; McKinney, B.L.; McGuire, J.K.; Parks, W.C.; et al. Syndecan-1 Attenuates Lung Injury during Influenza Infection by Potentiating c-Met Signaling to Suppress Epithelial Apoptosis. Am. J. Respir. Crit. Care Med. 2016, 194, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, D.; Bigelow, D.B.; Petty, T.; Levine, B. ACUTE RESPIRATORY DISTRESS IN ADULTS. Lancet 1967, 290, 319–323. [Google Scholar] [CrossRef]
- Shaver, C.M.; Upchurch, C.P.; Janz, D.R.; Grove, B.S.; Putz, N.D.; Wickersham, N.E.; Dikalov, S.I.; Ware, L.B.; Bastarache, J.A. Cell-free hemoglobin: A novel mediator of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L532–L541. [Google Scholar] [CrossRef] [PubMed]
- Chintagari, N.R.; Jana, S.; Alayash, A.I. Oxidized Ferric and Ferryl Forms of Hemoglobin Trigger Mitochondrial Dysfunction and Injury in Alveolar Type I Cells. Am. J. Respir. Cell Mol. Biol. 2016, 55, 288–298. [Google Scholar] [CrossRef]
- Shaver, C.M.; Wickersham, N.; McNeil, J.B.; Nagata, H.; Miller, A.; Landstreet, S.R.; Kuck, J.L.; Diamond, J.M.; Lederer, D.J.; Kawut, S.M.; et al. Cell-free hemoglobin promotes primary graft dysfunction through oxidative lung endothelial injury. JCI Insight 2018, 3, e98546. [Google Scholar] [CrossRef]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef]
- Ruan, T.; Sun, J.; Liu, W.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. H1N1 Influenza Virus Cross-Activates Gli1 to Disrupt the Intercellular Junctions of Alveolar Epithelial Cells. Cell Rep. 2020, 31, 107801. [Google Scholar] [CrossRef]
- Peteranderl, C.; Morales-Nebreda, L.; Selvakumar, B.; Lecuona, E.; Vadász, I.; Morty, R.E.; Schmoldt, C.; Bespalowa, J.; Wolff, T.; Pleschka, S.; et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J. Clin. Investig. 2016, 126, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.D.; Lazrak, A.; Trombley, J.E.; Shei, R.-J.; Adewale, A.T.; Tipper, J.L.; Yu, Z.; Ashtekar, A.R.; Rowe, S.M.; Matalon, S.; et al. Influenza-mediated reduction of lung epithelial ion channel activity leads to dysregulated pulmonary fluid homeostasis. JCI Insight 2018, 3, e123467. [Google Scholar] [CrossRef] [PubMed]
- Kryvenko, V.; Wessendorf, M.; Tello, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadász, I. Hypercapnia Induces Inositol-Requiring Enzyme 1α–Driven Endoplasmic Reticulum–associated Degradation of the Na,K-ATPase β-Subunit. Am. J. Respir. Cell Mol. Biol. 2021, 65, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Woods, P.S.; Doolittle, L.M.; Rosas, L.E.; Joseph, L.M.; Calomeni, E.P.; Davis, I.C. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1160–L1169. [Google Scholar] [CrossRef]
- Cui, H.; Xie, N.; Banerjee, S.; Ge, J.; Guo, S.; Liu, G. Impairment of Fatty Acid Oxidation in Alveolar Epithelial Cells Mediates Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2019, 60, 167–178. [Google Scholar] [CrossRef]
- Lin, W.-C.; Fessler, M.B. Regulatory mechanisms of neutrophil migration from the circulation to the airspace. Experientia 2021, 78, 4095–4124. [Google Scholar] [CrossRef]
- Brazee, P.L.; Morales-Nebreda, L.; Magnani, N.D.; Garcia, J.G.; Misharin, A.V.; Ridge, K.M.; Budinger, G.S.; Iwai, K.; Dada, L.A.; Sznajder, J.I. Linear ubiquitin assembly complex regulates lung epithelial–driven responses during influenza infection. J. Clin. Investig. 2020, 130, 1301–1314. [Google Scholar] [CrossRef]
- Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin. Toxins 2015, 7, 4817–4831. [Google Scholar] [CrossRef]
- Massa, C.B.; Scott, P.; Abramova, E.; Gardner, C.; Laskin, D.L.; Gow, A.J. Acute chlorine gas exposure produces transient inflammation and a progressive alteration in surfactant composition with accompanying mechanical dysfunction. Toxicol. Appl. Pharmacol. 2014, 278, 53–64. [Google Scholar] [CrossRef]
- Jurkuvenaite, A.; Benavides, G.A.; Komarova, S.; Doran, S.F.; Johnson, M.; Aggarwal, S.; Zhang, J.; Darley-Usmar, V.M.; Matalon, S. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic. Biol. Med. 2015, 85, 83–94. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, Y.; Su, C.; Sun, H.; Zhang, H.; Zhu, B.; Zhang, H.; Xiao, H.; Wang, J.; Zhang, J. Epithelial sodium channel is involved in H2S-induced acute pulmonary edema. Inhal. Toxicol. 2015, 27, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Beitler, J.R.; Malhotra, A.; Thompson, B.T. Ventilator-induced Lung Injury. Clin. Chest Med. 2016, 37, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Cressoni, M.; Cadringher, P.; Chiurazzi, C.; Amini, M.; Gallazzi, E.; Marino, A.; Brioni, M.; Carlesso, E.; Chiumello, D.; Quintel, M.; et al. Lung Inhomogeneity in Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2014, 189, 149–158. [Google Scholar] [CrossRef]
- López-Alonso, I.; Blázquez-Prieto, J.; Amado-Rodríguez, L.; González-López, A.; Astudillo, A.; Sánchez, M.; Huidobro, C.; López-Martínez, C.; dos Santos, C.C.; Albaiceta, G.M. Preventing loss of mechanosensation by the nuclear membranes of alveolar cells reduces lung injury in mice during mechanical ventilation. Sci. Transl. Med. 2018, 10, eaam7598. [Google Scholar] [CrossRef]
- Lee, H.; Fei, Q.; Streicher, A.; Zhang, W.; Isabelle, C.; Patel, P.; Lam, H.C.; Arciniegas-Rubio, A.; Pinilla-Vera, M.; Amador-Munoz, D.P.; et al. mTORC1 is a mechanosensor that regulates surfactant function and lung compliance during ventilator-induced lung injury. JCI Insight 2021, 6, e137708. [Google Scholar] [CrossRef]
- Sinclair, S.E.; Molthen, R.C.; Haworth, S.T.; Dawson, C.A.; Waters, C.M. Airway Strain during Mechanical Ventilation in an Intact Animal Model. Am. J. Respir. Crit. Care Med. 2007, 176, 786–794. [Google Scholar] [CrossRef]
- Pires-Neto, R.C.; Morales, M.M.B.; Lancas, T.; Inforsato, N.; Duarte, M.I.S.; Amato, M.B.P.; de Carvalho, C.R.R.; da Silva, L.F.F.; Mauad, T.; Dolhnikoff, M. Expression of acute-phase cytokines, surfactant proteins, and epithelial apoptosis in small airways of human acute respiratory distress syndrome. J. Crit. Care 2013, 28. [Google Scholar] [CrossRef][Green Version]
- Muñoz-Moreno, R.; Martínez-Romero, C.; García-Sastre, A. Induction and Evasion of Type-I Interferon Responses during Influenza A Virus Infection. Cold Spring Harb. Perspect. Med. 2020, 11, a038414. [Google Scholar] [CrossRef]
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482. [Google Scholar] [CrossRef]
- Ramos, I.; Smith, G.; Ruf-Zamojski, F.; Martínez-Romero, C.; Fribourg, M.; Carbajal, E.A.; Hartmann, B.; Nair, V.D.; Marjanovic, N.; Monteagudo, P.L.; et al. Innate Immune Response to Influenza Virus at Single-Cell Resolution in Human Epithelial Cells Revealed Paracrine Induction of Interferon Lambda 1. J. Virol. 2019, 93, e00559-19. [Google Scholar] [CrossRef] [PubMed]
- Stifter, S.A.; Bhattacharyya, N.; Sawyer, A.J.; Cootes, T.A.; Stambas, J.; Doyle, S.E.; Feigenbaum, L.; Paul, W.E.; Britton, W.J.; Sher, A.; et al. Visualizing the Selectivity and Dynamics of Interferon Signaling In Vivo. Cell Rep. 2019, 29, 3539–3550.e4. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.B.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef]
- Rannels, D.E. Gap junction communication in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1083–L1084. [Google Scholar] [CrossRef] [PubMed]
- Hook, J.L.; Islam, M.N.; Parker, D.; Prince, A.S.; Bhattacharya, S.; Bhattacharya, J. Disruption of staphylococcal aggregation protects against lethal lung injury. J. Clin. Investig. 2018, 128, 1074–1086. [Google Scholar] [CrossRef]
- Kuebler, W.M.; Parthasarathi, K.; Wang, P.M.; Bhattacharya, J. A novel signaling mechanism between gas and blood compartments of the lung. J. Clin. Investig. 2000, 106, 607. [Google Scholar] [CrossRef]
- Westphalen, K.; Monma, E.; Islam, M.N.; Bhattacharya, J. Acid contact in the rodent pulmonary alveolus causes proinflammatory signaling by membrane pore formation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L107–L116. [Google Scholar] [CrossRef]
- Hough, R.F.; Islam, M.N.; Gusarova, G.A.; Jin, G.; Das, S.; Bhattacharya, J. Endothelial mitochondria determine rapid barrier failure in chemical lung injury. JCI Insight 2019, 4, e124329. [Google Scholar] [CrossRef]
- Rhodin, J.A. Ultrastructure of mammalian venous capillaries, venules, and small collecting veins. J. Ultrastruct. Res. 1968, 25, 452–500. [Google Scholar] [CrossRef]
- Kato, K.; Diéguez-Hurtado, R.; Park, D.Y.; Hong, S.P.; Kato-Azuma, S.; Adams, S.; Stehling, M.; Trappmann, B.; Wrana, J.L.; Koh, G.Y.; et al. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 2018, 9, 2448. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.E.; Westfall, J.A. Analysis of relationships between pericytes and gas exchange capillaries in neonatal and mature bovine lungs. Microvasc. Res. 1983, 25, 333–342. [Google Scholar] [CrossRef]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef]
- Hung, C.; Linn, G.; Chow, Y.-H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of Lung Pericytes and Resident Fibroblasts in the Pathogenesis of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.F.; Mittelsteadt, K.L.; Brauer, R.; McKinney, B.L.; Hallstrand, T.; Parks, W.C.; Chen, P.; Schnapp, L.M.; Liles, W.C.; Duffield, J.S.; et al. Lung pericyte-like cells are functional interstitial immune sentinel cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L556–L567. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.F.; Holton, S.; Chow, Y.; Liles, W.C.; Gharib, S.A.; Altemeier, W.A. Pericyte-like cells undergo transcriptional reprogramming and distinct functional adaptations in acute lung injury. FASEB J. 2021, 35, e21323. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.J.; Cain, M.P.; Lynch, A.M.; Flores, J.R.; Tuvim, M.J.; Dickey, B.F.; Chen, J. Intermediary Role of Lung Alveolar Type 1 Cells in Epithelial Repair Upon Sendai Virus Infection. Am. J. Respir. Cell Mol. Biol. 2022, 67, 389–401. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ferrari, J.D.; Cao, Y.; Ramirez, M.I.; Jones, M.; Quinton, L.; Mizgerd, J.P. Type I Alveolar Epithelial Cells Mount Innate Immune Responses during Pneumococcal Pneumonia. J. Immunol. 2012, 189, 2450–2459. [Google Scholar] [CrossRef]
- Dobbs, L.G.; Johnson, M.D. Alveolar epithelial transport in the adult lung. Respir. Physiol. Neurobiol. 2007, 159, 283–300. [Google Scholar] [CrossRef]
- Lin, W.-C.; Gowdy, K.M.; Madenspacher, J.H.; Zemans, R.L.; Yamamoto, K.; Lyons-Cohen, M.R.; Nakano, H.; Janardhan, K.; Williams, C.J.; Cook, D.N.; et al. Epithelial membrane protein 2 governs transepithelial migration of neutrophils into the airspace. J. Clin. Investig. 2020, 130, 157–170. [Google Scholar] [CrossRef]
- E Weiland, J.; Davis, W.B.; Holter, J.F.; Mohammed, J.R.; Dorinsky, P.M.; E Gadek, J. Lung neutrophils in the adult respiratory distress syndrome. Clinical and pathophysiologic significance. Am. Rev. Respir. Dis. 1986, 133, 218–225. [Google Scholar] [CrossRef]
- Eddy, W.E.; Gong, K.-Q.; Bell, B.; Parks, W.C.; Ziegler, S.F.; Manicone, A.M. Stat5 Is Required for CD103+ Dendritic Cell and Alveolar Macrophage Development and Protection from Lung Injury. J. Immunol. 2017, 198, 4813–4822. [Google Scholar] [CrossRef] [PubMed]
- Cardani, A.; Boulton, A.; Kim, T.S.; Braciale, T.J. Alveolar Macrophages Prevent Lethal Influenza Pneumonia By Inhibiting Infection Of Type-1 Alveolar Epithelial Cells. PLOS Pathog. 2017, 13, e1006140. [Google Scholar] [CrossRef]
- Westphalen, K.; Gusarova, G.A.; Islam, M.N.; Subramanian, M.; Cohen, T.; Prince, A.S.; Bhattacharya, J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 2014, 506, 503–506. [Google Scholar] [CrossRef]
- McQuattie-Pimentel, A.C.; Ren, Z.; Joshi, N.; Watanabe, S.; Stoeger, T.; Chi, M.; Lu, Z.; Sichizya, L.; Aillon, R.P.; Chen, C.-I.; et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J. Clin. Investig. 2021, 131, e140299. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.R.; Hagimoto, N.; Nakamura, M.; Matute-Bello, G. Apoptosis and Epithelial Injury in the Lungs. Proc. Am. Thorac. Soc. 2005, 2, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Patel, B.V.; Wilson, M.R.; O’Dea, K.P.; Takata, M. TNF-Induced Death Signaling Triggers Alveolar Epithelial Dysfunction in Acute Lung Injury. J. Immunol. 2013, 190, 4274–4282. [Google Scholar] [CrossRef]
- Fiege, J.K.; Stone, I.A.; Dumm, R.E.; Waring, B.M.; Fife, B.T.; Agudo, J.; Brown, B.D.; Heaton, N.S.; Langlois, R.A. Long-term surviving influenza infected cells evade CD8+ T cell mediated clearance. PLOS Pathog. 2019, 15, e1008077. [Google Scholar] [CrossRef]
- Suliman, H.B.; Kraft, B.; Bartz, R.; Chen, L.; Welty-Wolf, K.E.; Piantadosi, C.A. Mitochondrial quality control in alveolar epithelial cells damaged by S. aureus pneumonia in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L699–L709. [Google Scholar] [CrossRef]
- Traber, K.E.; Symer, E.M.; Allen, E.; Kim, Y.; Hilliard, K.L.; Wasserman, G.; Stewart, C.L.; Jones, M.R.; Mizgerd, J.; Quinton, L.J. Myeloid-epithelial cross talk coordinates synthesis of the tissue-protective cytokine leukemia inhibitory factor during pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L548–L558. [Google Scholar] [CrossRef] [PubMed]
- Na, E.; Allen, E.; Baird, L.A.; Odom, C.V.; Korkmaz, F.T.; Shenoy, A.T.; Matschulat, A.M.; Jones, M.R.; Kotton, D.N.; Mizgerd, J.P.; et al. Epithelial LIF signaling limits apoptosis and lung injury during bacterial pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L550–L563. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Renewal of alveolar epithelium in the rat following exposure to NO2. Am. J. Pathol. 1973, 70, 175–198. [Google Scholar] [PubMed]
- Desai, T.J.; Brownfield, D.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.B.; Peng, T.; Zepp, J.A.; Snitow, M.; Vincent, T.L.; Penkala, I.J.; Cui, Z.; Herriges, M.J.; Morley, M.P.; Zhou, S.; et al. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep. 2016, 17, 2312–2325. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.-E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.-K.; Han, N.; Lee, J.-H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7. [Google Scholar] [CrossRef]
- McClendon, J.; Jansing, N.L.; Redente, E.F.; Gandjeva, A.; Ito, Y.; Colgan, S.P.; Ahmad, A.; Riches, D.W.; Chapman, H.A.; Mason, R.J.; et al. Hypoxia-Inducible Factor 1α Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am. J. Pathol. 2017, 187, 1772–1786. [Google Scholar] [CrossRef]
- Finn, J.; Sottoriva, K.; Pajcini, K.V.; Kitajewski, J.K.; Chen, C.; Zhang, W.; Malik, A.B.; Liu, Y. Dlk1-Mediated Temporal Regulation of Notch Signaling Is Required for Differentiation of Alveolar Type II to Type I Cells during Repair. Cell Rep. 2019, 26, 2942–2954.e5. [Google Scholar] [CrossRef]
- Zacharias, W.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Liberti, D.C.; Kremp, M.M.; Liberti, W.A.; Penkala, I.J.; Li, S.; Zhou, S.; Morrisey, E.E. Alveolar epithelial cell fate is maintained in a spatially restricted manner to promote lung regeneration after acute injury. Cell Rep. 2021, 35, 109092. [Google Scholar] [CrossRef]
- Ahmadvand, N.; Khosravi, F.; Lingampally, A.; Wasnick, R.; Vazquez-Armendariz, A.I.; Carraro, G.; Heiner, M.; Rivetti, S.; Lv, Y.; Wilhelm, J.; et al. Identification of a novel subset of alveolar type 2 cells enriched in PD-L1 and expanded following pneumonectomy. Eur. Respir. J. 2021, 58, 2004168. [Google Scholar] [CrossRef] [PubMed]
- Murthy, P.K.L.; Sontake, V.; Tata, A.; Kobayashi, Y.; Macadlo, L.; Okuda, K.; Conchola, A.S.; Nakano, S.; Gregory, S.; Miller, L.A.; et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 2022, 604, 111–119. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nature 2020, 22, 934–946. [Google Scholar] [CrossRef]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx+ type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Zuo, W.; Zhang, T.; Wu, D.Z.; Guan, S.P.; Liew, A.-A.; Yamamoto, Y.; Wang, X.; Lim, S.J.; Vincent, M.; Lessard, M.; et al. p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nature 2015, 517, 616–620. [Google Scholar] [CrossRef]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal Airway Stem Cells Yield Alveoli In Vitro and during Lung Regeneration following H1N1 Influenza Infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 2020, 26, 346–358. [Google Scholar] [CrossRef]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Tang, M.; Jin, H.; Liu, Q.; He, L.; Zhu, H.; Liu, X.; Han, X.; Li, Y.; Zhang, L.; et al. Triple-cell lineage tracing by a dual reporter on a single allele. J. Biol. Chem. 2020, 295, 690–700. [Google Scholar] [CrossRef]
- Weibel, E.R. Morphometry of the Human Lung; Springer: Berlin/Heidelberg, Germany, 1963. [Google Scholar]
- Hsieh, M.-J.; Lee, W.-C.; Cho, H.-Y.; Wu, M.-F.; Hu, H.-C.; Kao, K.-C.; Chen, N.-H.; Tsai, Y.-H.; Huang, C.-C. Recovery of pulmonary functions, exercise capacity, and quality of life after pulmonary rehabilitation in survivors of ARDS due to severe influenza A (H1N1) pneumonitis. Influ. Other Respir. Viruses 2018, 12, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Herridge, M.S.; Tansey, C.M.; Matté, A.; Tomlinson, G.; Diaz-Granados, N.; Cooper, A.; Guest, C.B.; Mazer, C.D.; Mehta, S.; Stewart, T.E.; et al. Functional Disability 5 Years after Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2011, 364, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Zapol, W.M.; Trelstad, R.L.; Coffey, J.W.; Tsai, I.; A Salvador, R. Pulmonary fibrosis in severe acute respiratory failure. Am. Rev. Respir. Dis. 1979, 119, 547–554. [Google Scholar] [CrossRef]
- Raghu, G.; Striker, L.J.; Hudson, L.D.; E Striker, G. Extracellular matrix in normal and fibrotic human lungs. Am. Rev. Respir. Dis. 1985, 131, 281–289. [Google Scholar] [CrossRef]
- Patel, S.R.; Karmpaliotis, D.; Ayas, N.T.; Mark, E.J.; Wain, J.; Thompson, B.T.; Malhotra, A. The Role of Open-Lung Biopsy in ARDS. Chest 2004, 125, 197–202. [Google Scholar] [CrossRef]
- Taylor, M.S.; Chivukula, R.R.; Myers, L.; Jeck, W.R.; Tata, P.R.; O’Donnell, W.J.; Farver, C.F.; Thompson, B.T.; Rajagopal, J.; Kradin, R.L. Delayed Alveolar Epithelialization: A Distinct Pathology in Diffuse Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2018, 197, 522–524. [Google Scholar] [CrossRef]
- Ting, C.; Aspal, M.; Vaishampayan, N.; Huang, S.K.; Riemondy, K.A.; Wang, F.; Farver, C.; Zemans, R.L. Fatal COVID-19 and Non–COVID-19 Acute Respiratory Distress Syndrome Is Associated with Incomplete Alveolar Type 1 Epithelial Cell Differentiation from the Transitional State without Fibrosis. Am. J. Pathol. 2021, 192, 454–467. [Google Scholar] [CrossRef]
- Bharat, A.; Querrey, M.; Markov, N.S.; Kim, S.; Kurihara, C.; Garza-Castillon, R.; Manerikar, A.; Shilatifard, A.; Tomic, R.; Politanska, Y.; et al. Lung transplantation for patients with severe COVID-19. Sci. Transl. Med. 2020, 12, eabe4282. [Google Scholar] [CrossRef]
- Bharat, A.; Machuca, T.N.; Querrey, M.; Kurihara, C.; Garza-Castillon, R.; Kim, S.; Manerikar, A.; Pelaez, A.; Pipkin, M.; Shahmohammadi, A.; et al. Early outcomes after lung transplantation for severe COVID-19: A series of the first consecutive cases from four countries. Lancet Respir. Med. 2021, 9, 487–497. [Google Scholar] [CrossRef]
- Gao, J.; Chu, W.; Duan, J.; Li, J.; Ma, W.; Hu, C.; Yao, M.; Xing, L.; Yang, Y. Six-Month Outcomes of Post-ARDS Pulmonary Fibrosis in Patients With H1N1 Pneumonia. Front. Mol. Biosci. 2021, 8, 640763. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef] [PubMed]
- Nouno, T.; Okamoto, M.; Ohnishi, K.; Kaieda, S.; Tominaga, M.; Zaizen, Y.; Ichiki, M.; Momosaki, S.; Nakamura, M.; Fujimoto, K.; et al. Elevation of pulmonary CD163+ and CD204+ macrophages is associated with the clinical course of idiopathic pulmonary fibrosis patients. J. Thorac. Dis. 2019, 11, 4005–4017. [Google Scholar] [CrossRef]
- Riemondy, K.A.; Jansing, N.L.; Jiang, P.; Redente, E.F.; Gillen, A.E.; Fu, R.; Miller, A.J.; Spence, J.R.; Gerber, A.N.; Hesselberth, J.R.; et al. Single-cell RNA sequencing identifies TGF-β as a key regenerative cue following LPS-induced lung injury. JCI Insight 2019, 4, e123637. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual Type 2–to–Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8hi Transitional State. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.-K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Kanegai, C.M.; Xi, Y.; Donne, M.L.; Gotts, J.E.; Driver, I.H.; Amidzic, G.; Lechner, A.J.; Jones, K.D.; Vaughan, A.E.; Chapman, H.A.; et al. Persistent Pathology in Influenza-Infected Mouse Lungs. Am. J. Respir. Cell Mol. Biol. 2016, 55, 613–615. [Google Scholar] [CrossRef]
- Taylor, M.S.; Chivukula, R.R.; Myers, L.C.; Jeck, W.R.; Waghray, A.; Tata, P.R.; Selig, M.K.; O’Donnell, W.J.; Farver, C.F.; Thompson, B.T.; et al. A Conserved Distal Lung Regenerative Pathway in Acute Lung Injury. Am. J. Pathol. 2018, 188, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Rane, C.K.; Jackson, S.R.; Pastore, C.F.; Zhao, G.; Weiner, A.I.; Patel, N.N.; Herbert, D.R.; Cohen, N.A.; Vaughan, A.E. Development of solitary chemosensory cells in the distal lung after severe influenza injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1141–L1149. [Google Scholar] [CrossRef] [PubMed]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Ray, S.; Chiba, N.; Yao, C.; Guan, X.; McConnell, A.M.; Brockway, B.; Que, L.; McQualter, J.L.; Stripp, B.R. Rare SOX2 + Airway Progenitor Cells Generate KRT5 + Cells that Repopulate Damaged Alveolar Parenchyma following Influenza Virus Infection. Stem Cell Rep. 2016, 7, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Riccio, P.; Schotsaert, M.; Mori, M.; Lu, J.; Lee, D.-K.; García-Sastre, A.; Xu, J.; Cardoso, W.V. Spatial-Temporal Lineage Restrictions of Embryonic p63+ Progenitors Establish Distinct Stem Cell Pools in Adult Airways. Dev. Cell 2018, 44, 752–761.e4. [Google Scholar] [CrossRef] [PubMed]
- Suchyta, M.R.; Clemmer, T.P.; Elliott, C.G.; Orme, J.F.; Morris, A.H.; Jacobson, J.; Menlove, R. Increased Mortality of Older Patients With Acute Respiratory Distress Syndrome. Chest 1997, 111, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Morales-Nebreda, L.; Helmin, K.A.; Acosta, M.A.T.; Markov, N.S.; Hu, J.Y.-S.; Joudi, A.M.; Piseaux-Aillon, R.; Abdala-Valencia, H.; Politanska, Y.; Singer, B.D. Aging imparts cell-autonomous dysfunction to regulatory T cells during recovery from influenza pneumonia. JCI Insight 2021, 6, e141690. [Google Scholar] [CrossRef]
- Yazicioglu, T.; Mühlfeld, C.; Autilio, C.; Huang, C.-K.; Bär, C.; Dittrich-Breiholz, O.; Thum, T.; Pérez-Gil, J.; Schmiedl, A.; Brandenberger, C. Aging impairs alveolar epithelial type II cell function in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L755–L769. [Google Scholar] [CrossRef]
- Liang, J.; Huang, G.; Liu, X.; Taghavifar, F.; Liu, N.; Wang, Y.; Deng, N.; Yao, C.; Xie, T.; Kulur, V.; et al. The ZIP8/SIRT1 axis regulates alveolar progenitor cell renewal in aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2022, 132, e157338. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Rana, T.; Jiang, C.; Liu, G.; Miyata, T.; Antony, V.; Thannickal, V.J.; Liu, R.-M. PAI-1 Regulation of TGF-β1–induced Alveolar Type II Cell Senescence, SASP Secretion, and SASP-mediated Activation of Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2020, 62, 319–330. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanches Santos Rizzo Zuttion, M.; Moore, S.K.L.; Chen, P.; Beppu, A.K.; Hook, J.L. New Insights into the Alveolar Epithelium as a Driver of Acute Respiratory Distress Syndrome. Biomolecules 2022, 12, 1273. https://doi.org/10.3390/biom12091273
Sanches Santos Rizzo Zuttion M, Moore SKL, Chen P, Beppu AK, Hook JL. New Insights into the Alveolar Epithelium as a Driver of Acute Respiratory Distress Syndrome. Biomolecules. 2022; 12(9):1273. https://doi.org/10.3390/biom12091273
Chicago/Turabian StyleSanches Santos Rizzo Zuttion, Marilia, Sarah Kathryn Littlehale Moore, Peter Chen, Andrew Kota Beppu, and Jaime Lynn Hook. 2022. "New Insights into the Alveolar Epithelium as a Driver of Acute Respiratory Distress Syndrome" Biomolecules 12, no. 9: 1273. https://doi.org/10.3390/biom12091273
APA StyleSanches Santos Rizzo Zuttion, M., Moore, S. K. L., Chen, P., Beppu, A. K., & Hook, J. L. (2022). New Insights into the Alveolar Epithelium as a Driver of Acute Respiratory Distress Syndrome. Biomolecules, 12(9), 1273. https://doi.org/10.3390/biom12091273