
Prediction of Potential Commercially Available Inhibitors against SARS-CoV-2 by Multi-Task Deep Learning Model

Abstract
:1. Introduction
2. Results and Discussion
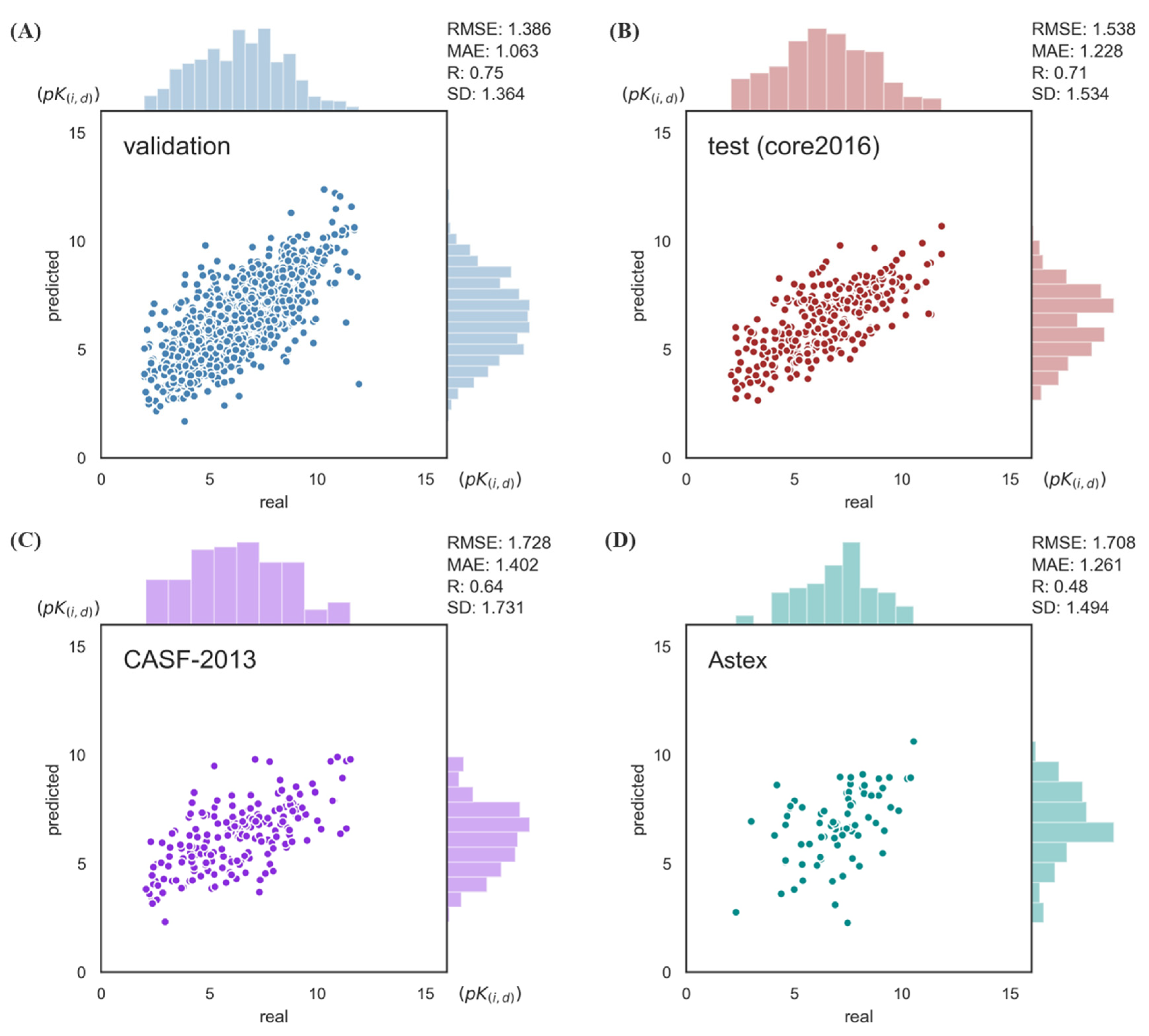
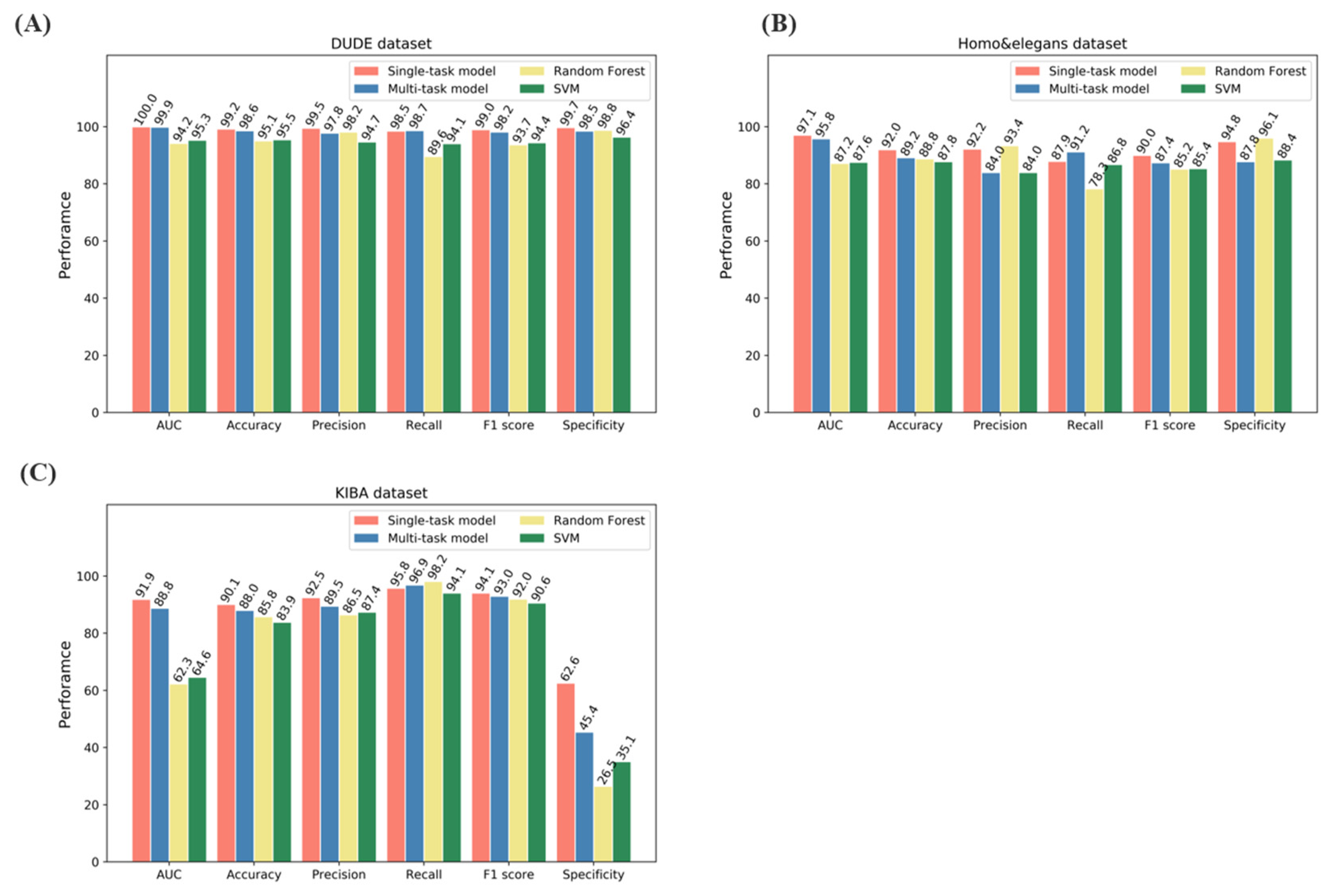
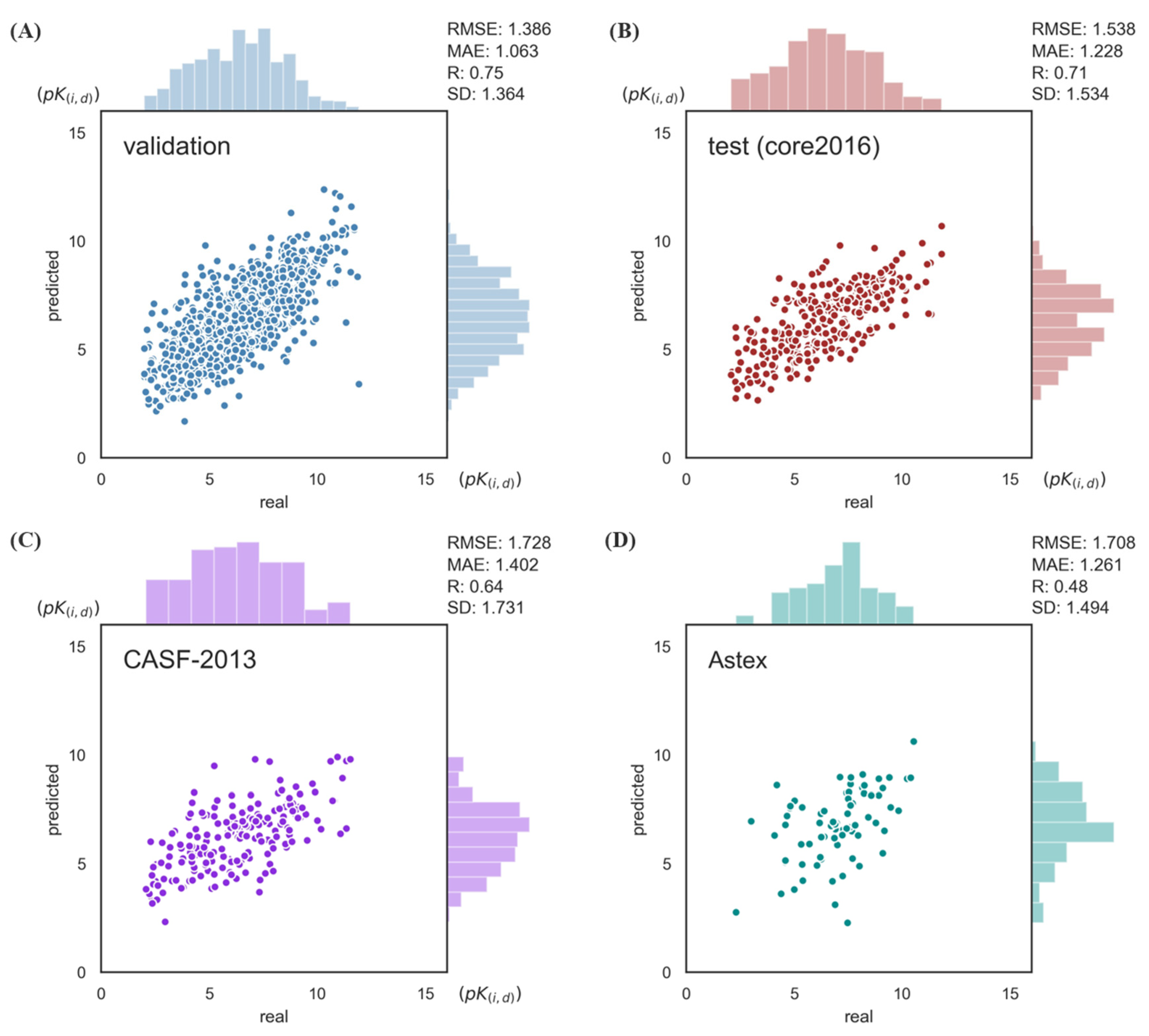
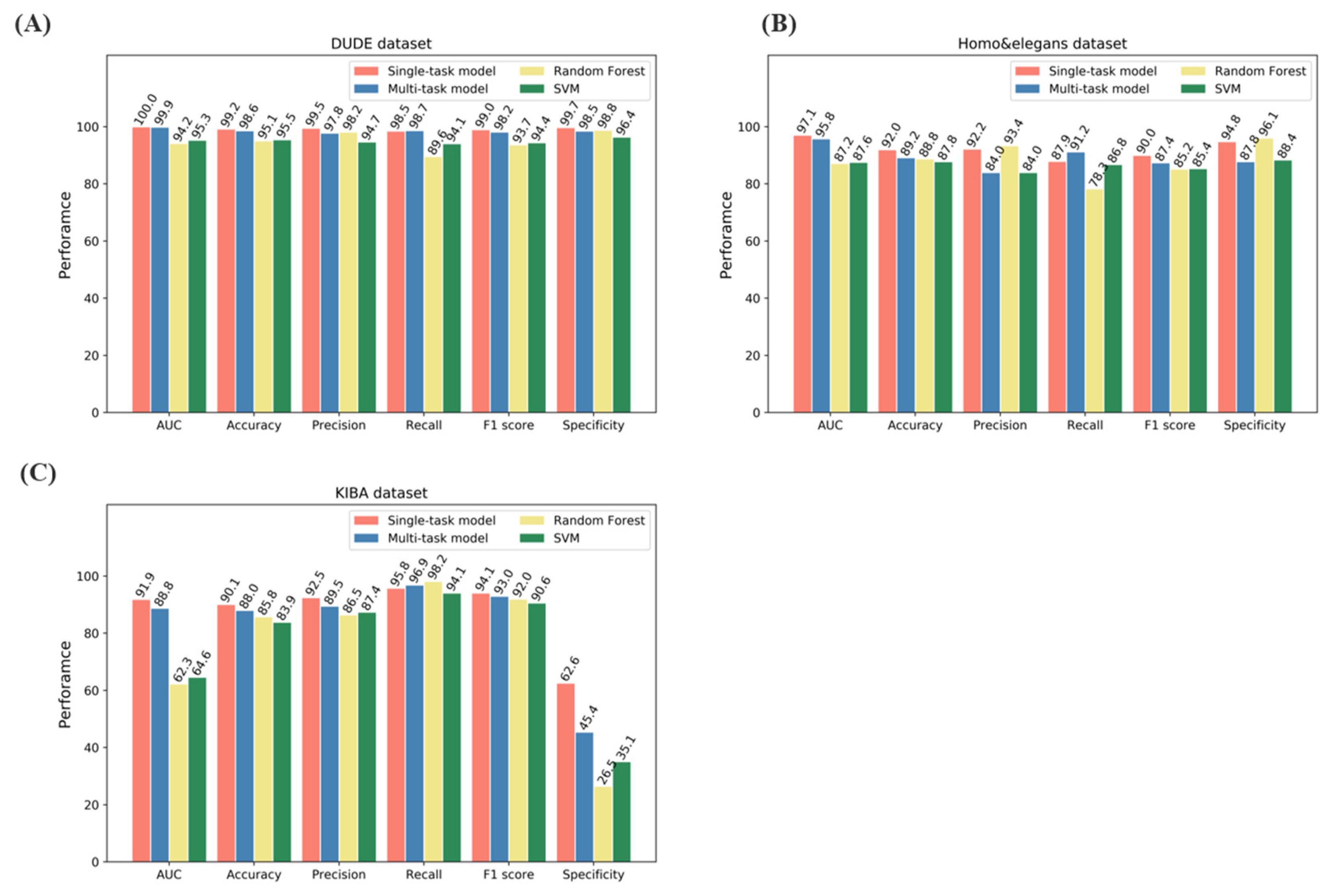
2.1. Model Performance on Benchmarks
2.2. Screening of SARS-CoV-2 Inhibitors
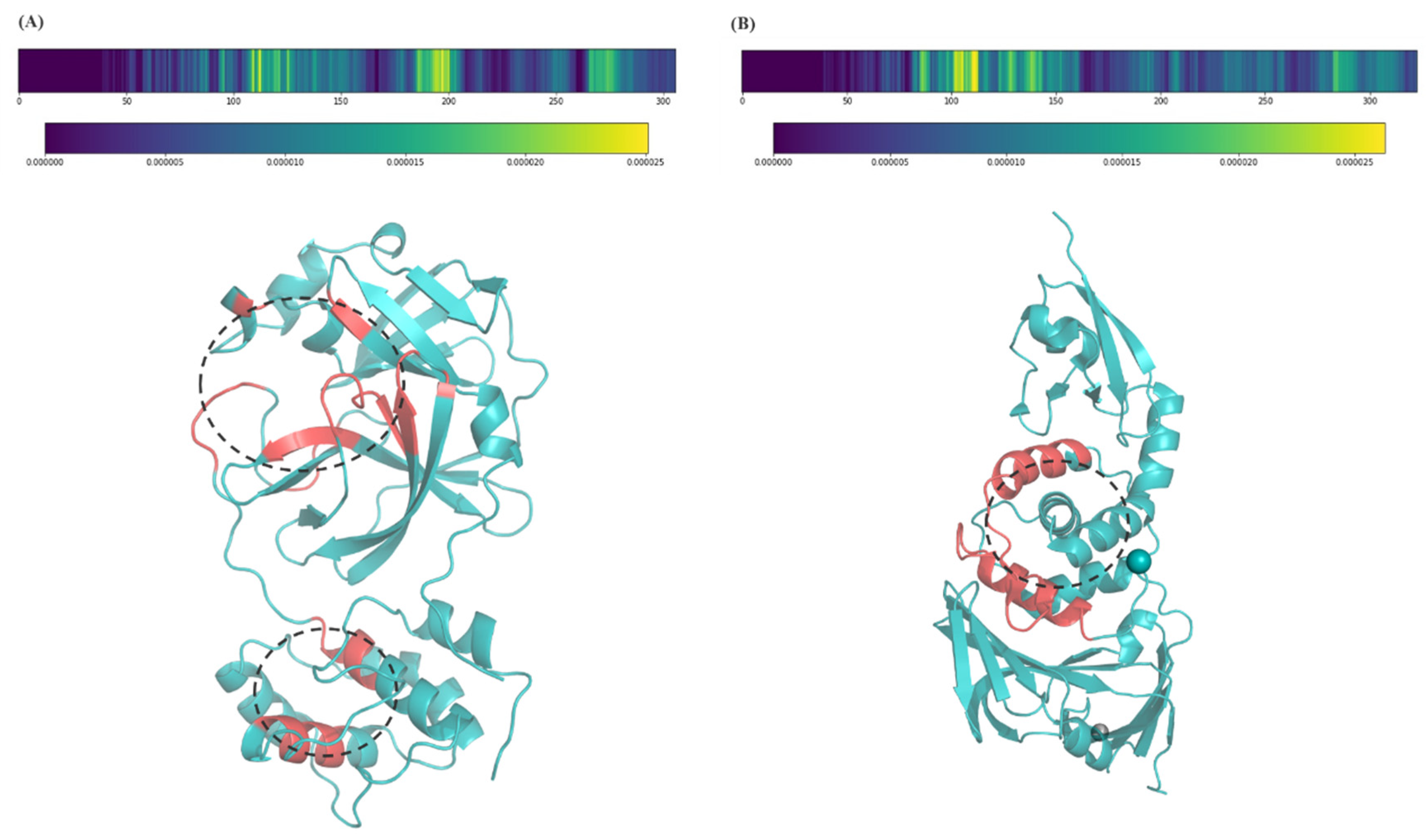
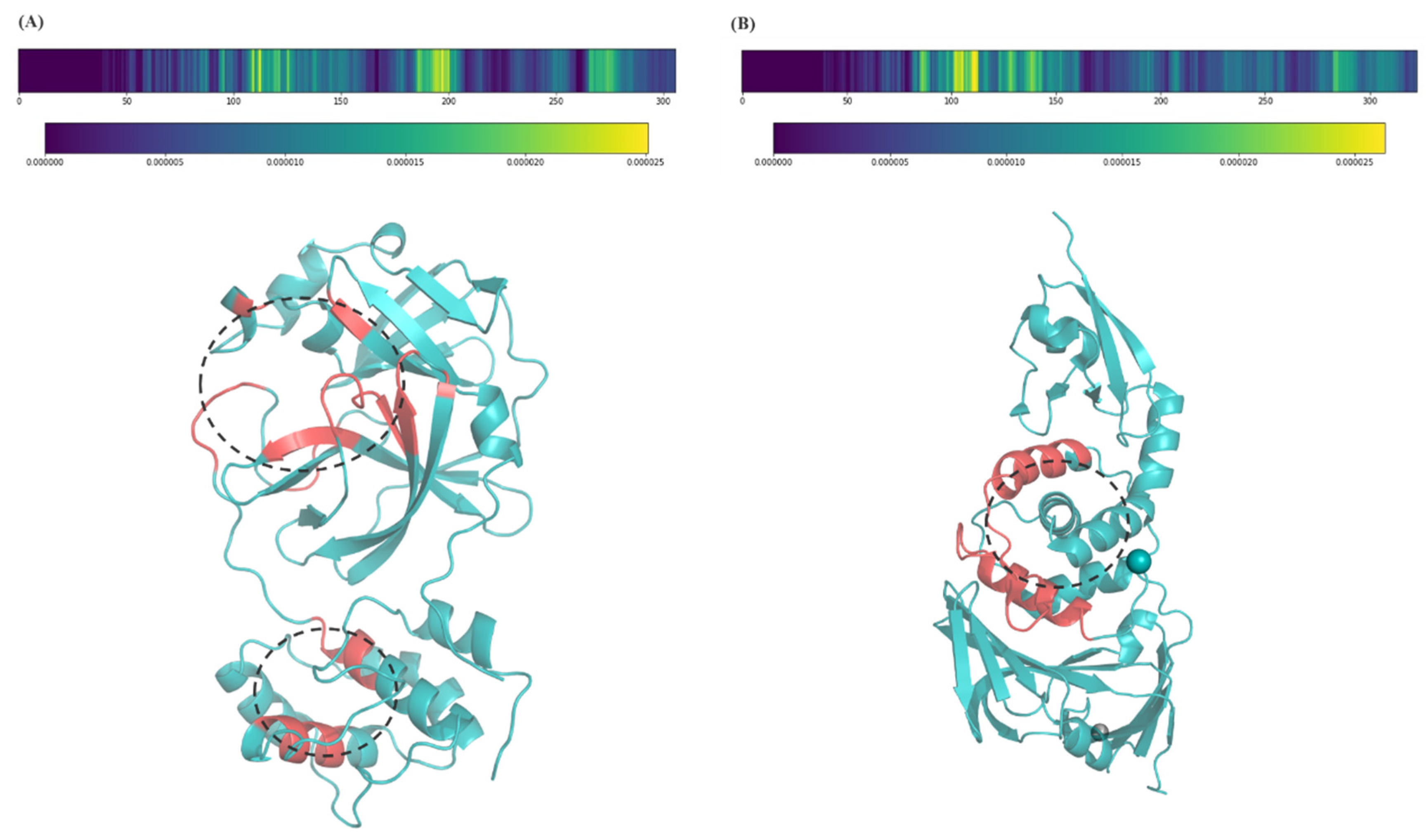
2.3. Model Interpretation
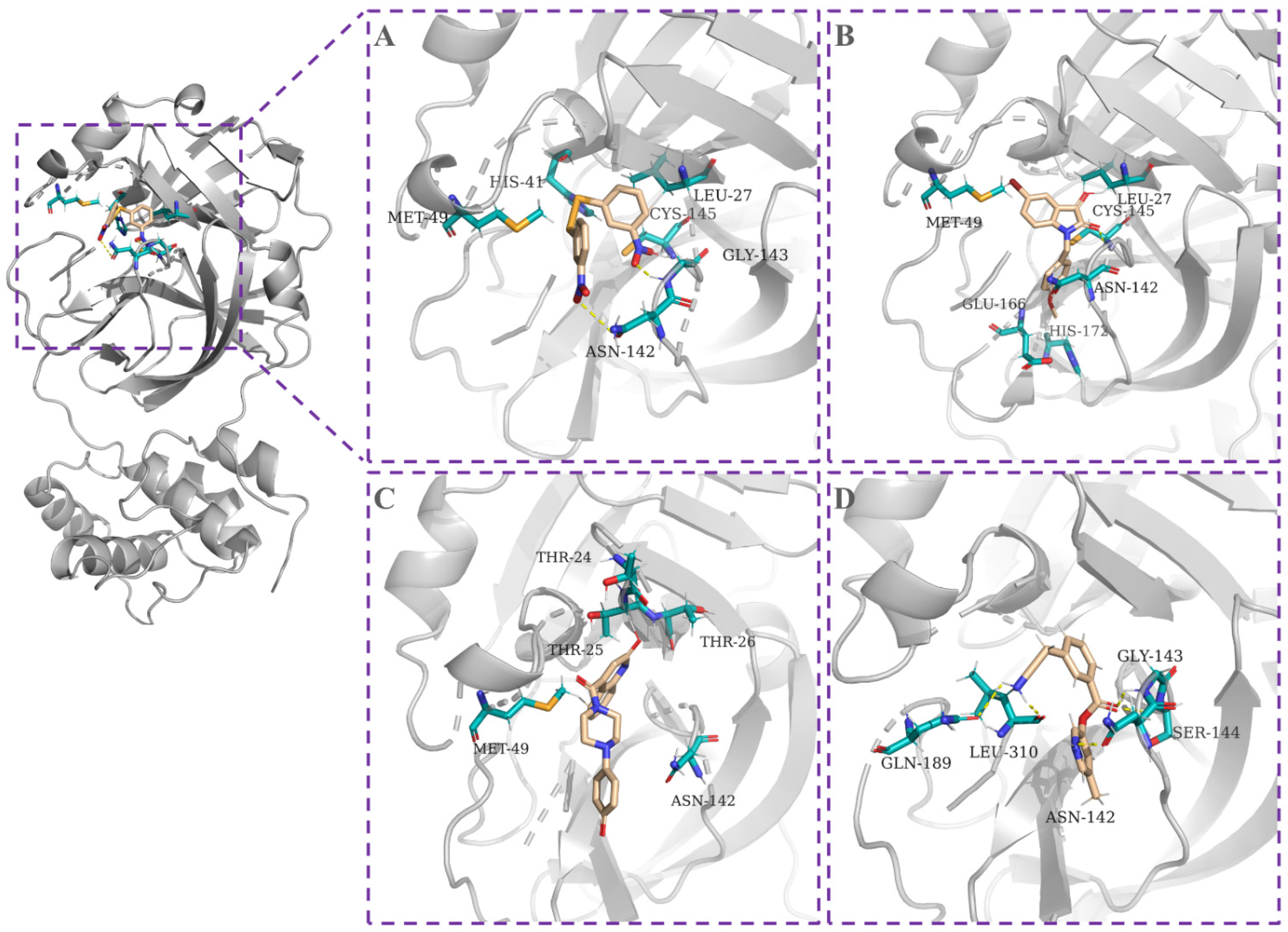
2.4. Molecular Docking
3. Conclusions
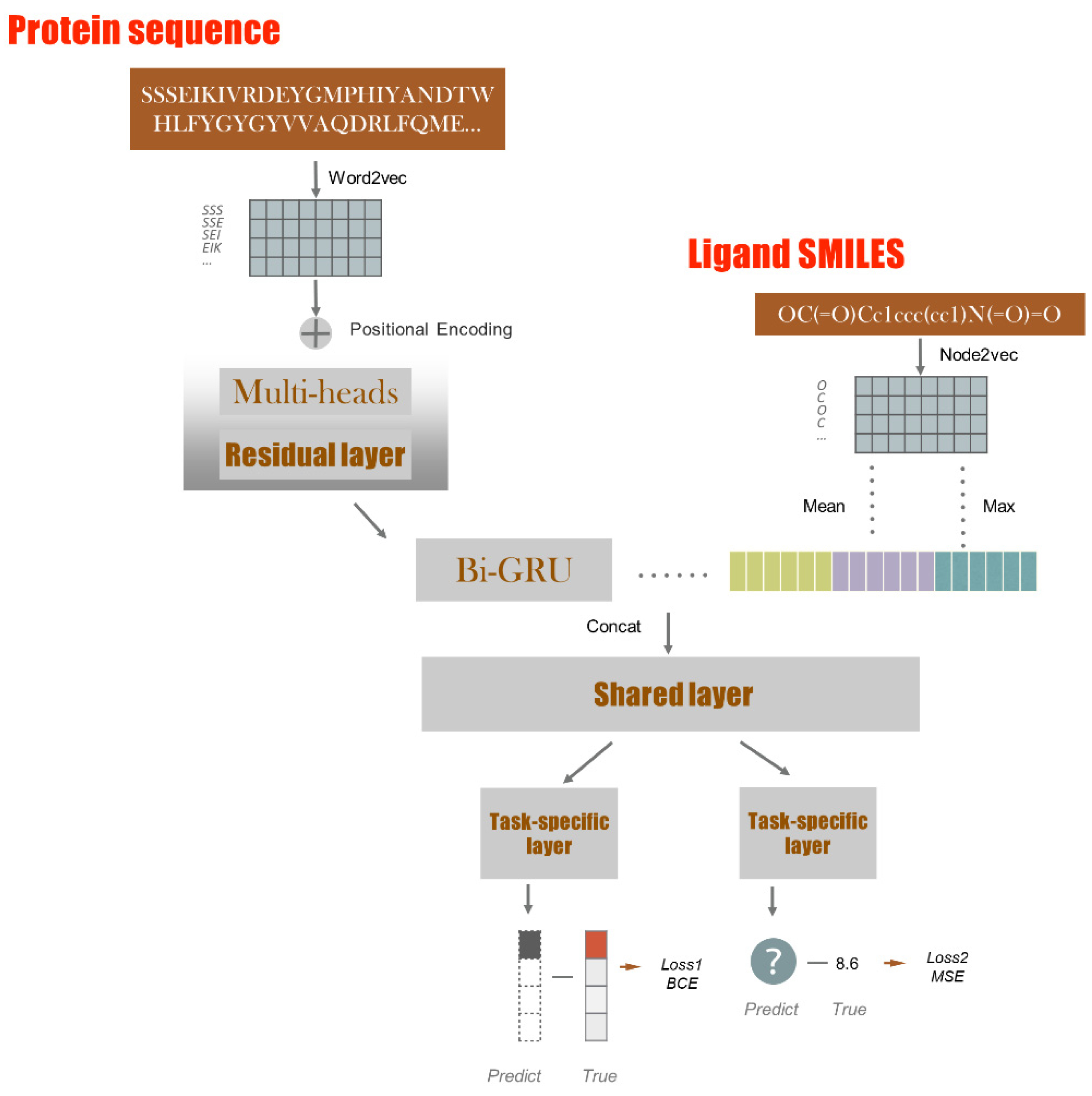
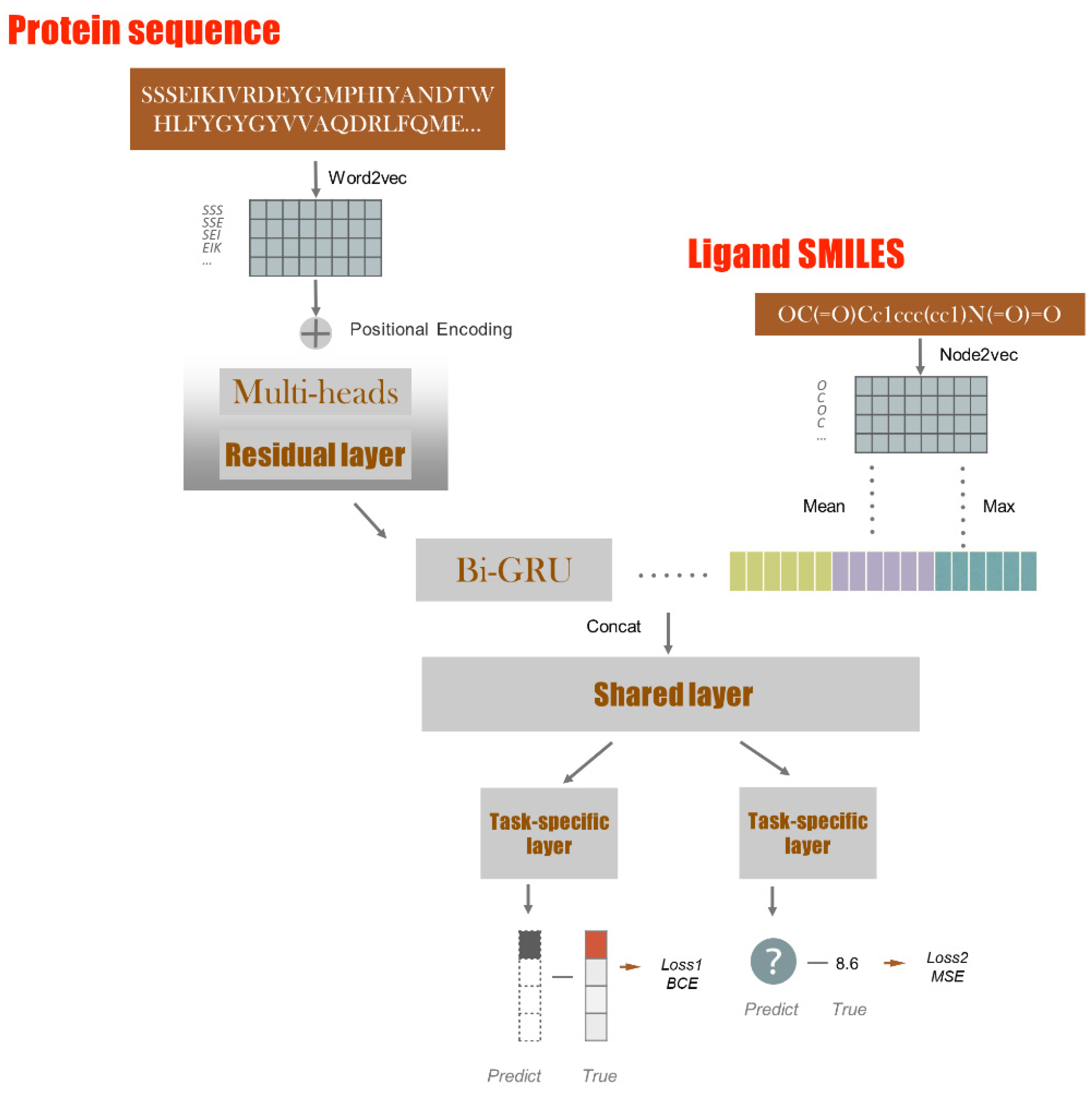
4. Methods
4.1. Data
4.2. Model
4.3. Biological Interpretation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. Severe Acute Respiratory Syndrome-Related Coronavirus: The Species and Its Viruses-a Statement of the Coronavirus Study Group. bioRxiv 2020, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; De Clercq, E. Therapeutic Options for the 2019 Novel Coronavirus (2019-NCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.J.; Won, J.J.; Graham, R.L.; Dinnon, K.H.; Sims, A.C.; Feng, J.Y.; Cihlar, T.; Denison, M.R.; Baric, R.S.; Sheahan, T.P. Broad Spectrum Antiviral Remdesivir Inhibits Human Endemic and Zoonotic Deltacoronaviruses with a Highly Divergent RNA Dependent RNA Polymerase. Antivir. Res. 2019, 169, 104541. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative Therapeutic Efficacy of Remdesivir and Combination Lopinavir, Ritonavir, and Interferon Beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchesnokov, E.; Feng, J.; Porter, D.; Götte, M. Mechanism of Inhibition of Ebola Virus RNA-Dependent RNA Polymerase by Remdesivir. Viruses 2019, 11, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep Drug–Target Binding Affinity Prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [Green Version]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound-Protein Interaction Prediction with End-to-End Learning of Neural Networks for Graphs and Sequences. Bioinformatics 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Hu, F.; Jiang, J.; Wang, D.; Zhu, M.; Yin, P. Multi-PLI: Interpretable Multi-task Deep Learning Model for Unifying Protein–Ligand Interaction Datasets. J. Cheminform. 2021, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Grover, A.; Leskovec, J. Node2vec. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: New York, NY, USA, 2016; pp. 855–864. [Google Scholar]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind Database: Collection of Binding Affinities for Protein−Ligand Complexes with Known Three-Dimensional Structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and Evaluation of a Deep Learning Model for Protein–Ligand Binding Affinity Prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Molecular Docking Simulations with ArgusLab. In Docking Screens for Drug Discovery; Humana: New York, NY, USA, 2019; pp. 203–220. [Google Scholar]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef]
- Hu, F.; Wang, L.; Hu, Y.; Wang, D.; Wang, W.; Jiang, J.; Li, N.; Yin, P. A Novel Framework Integrating AI Model and Enzymological Experiments Promotes Identification of SARS-CoV-2 3CL Protease Inhibitors and Activity-Based Probe. Brief. Bioinform. 2021, 22, bbab301. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, J.; Guan, J.; Zheng, J.; Zhou, S. Improving Compound-Protein Interaction Prediction by Building up Highly Credible Negative Samples. Bioinformatics 2015, 31, i221–i229. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making Sense of Large-Scale Kinase Inhibitor Bioactivity Data Sets: A Comparative and Integrative Analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 Efficaciously Inhibit SARS-CoV-2 by Targeting Its Main Protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Dong, Q.-W.; Wang, X.-L.; Lin, L. Application of Latent Semantic Analysis to Protein Remote Homology Detection. Bioinformatics 2006, 22, 285–290. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | CAS | Target | Predicted Affinity (nM) |
|---|---|---|---|
| Abacavir (sulfate) | 188062-50-2 | RdRp helicase | 3.03 3.06 |
| Darunavir | 206361-99-1 | 3CLpro PLpro | 57.30 46.16 |
| Darunavir (ethanolate) | 635728-49-3 | 3CLpro PLpro | 44.51 35.86 |
| Itraconazole | 84625-61-6 | PLpro RdRp | 127.98 16.90 |
| Almitrine mesylate | 29608-49-9 | 3CLpro | 29.31 |
| Daclatasvir | 1009119-64-5 | RdRp | 15.03 |
| Daclatasvir (dihydrochloride) | 1009119-65-6 | RdRp | 19.87 |
| Metoprolol tartrate | 56392-17-7 | PLpro | 153.23 |
| Fiboflapon sodium | 1196070-26-4 | PLpro | 197.63 |
| Roflumilast | 162401-32-3 | 3CLpro | 248.89 |
| ID | SMILES | Probability |
|---|---|---|
| Z56899184 | O=[N+]([O-])c1cccc(SSc2cccc([N+](=O)[O-])c2)c1 | 0.969 |
| Z229622170 | N#Cc1cccc(CN2C(=O)C(=O)c3cccc(Br)c32)c1 | 0.947 |
| Z57728899 | COc1ccc(CN2C(=O)C(=O)c3cc(Br)ccc32)cc1 | 0.918 |
| Z90667629 | [Na+].[O-][n+]1ccccc1[S-] | 0.863 |
| Z1238998507 | N#Cc1cc([N+](=O)[O-])ccc1Oc1cncc(Cl)c1 | 0.782 |
| Z56833036 | O=C1c2ccccc2C(=O)c2c1cccc2S(=O)(=O)N1CCOCC1 | 0.724 |
| Z57013003 | COc1ccc(N2CCN(C(=O)c3cc(=O)[nH]c4ccccc34)CC2)cc1 | 0.711 |
| Z1245218850 | N#CCc1cccc(C(=O)Oc2cncc(Cl)c2)c1 | 0.697 |
| Z56785091 | c1nc(SSc2nc[nH]n2)n[nH]1 | 0.682 |
| Z1776036493 | Cc1cc(Br)cc2c1N(CCBr)C(=O)C2=O | 0.676 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, F.; Jiang, J.; Yin, P. Prediction of Potential Commercially Available Inhibitors against SARS-CoV-2 by Multi-Task Deep Learning Model. Biomolecules 2022, 12, 1156. https://doi.org/10.3390/biom12081156
Hu F, Jiang J, Yin P. Prediction of Potential Commercially Available Inhibitors against SARS-CoV-2 by Multi-Task Deep Learning Model. Biomolecules. 2022; 12(8):1156. https://doi.org/10.3390/biom12081156
Chicago/Turabian StyleHu, Fan, Jiaxin Jiang, and Peng Yin. 2022. "Prediction of Potential Commercially Available Inhibitors against SARS-CoV-2 by Multi-Task Deep Learning Model" Biomolecules 12, no. 8: 1156. https://doi.org/10.3390/biom12081156
APA StyleHu, F., Jiang, J., & Yin, P. (2022). Prediction of Potential Commercially Available Inhibitors against SARS-CoV-2 by Multi-Task Deep Learning Model. Biomolecules, 12(8), 1156. https://doi.org/10.3390/biom12081156