
An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions


Abstract
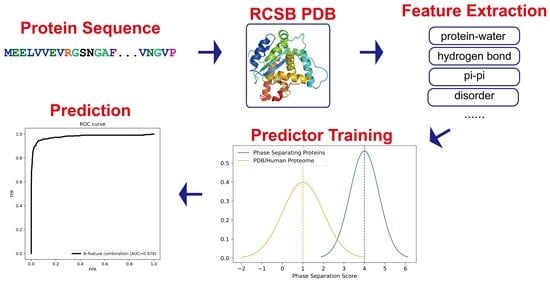
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
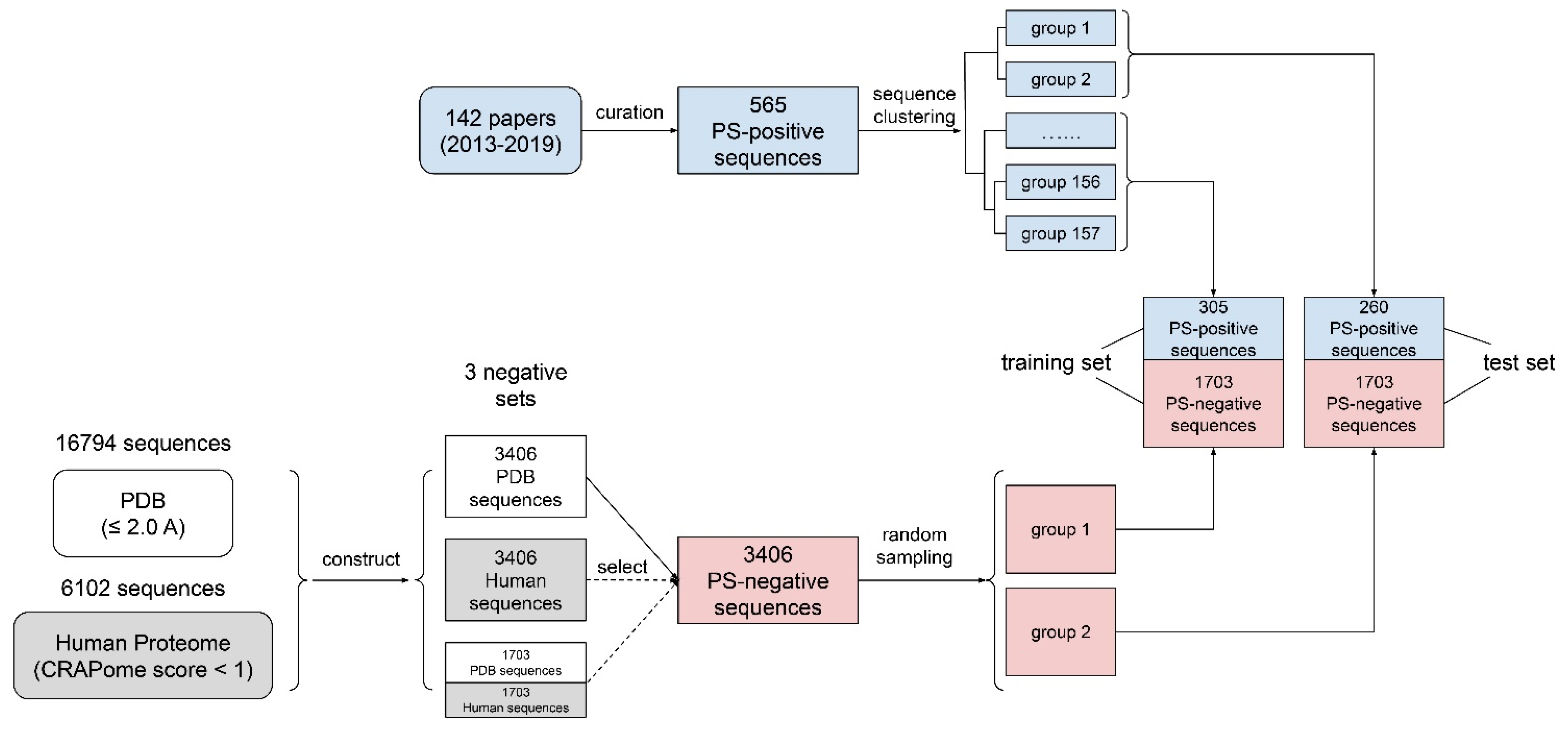
2.1. Data Preparation
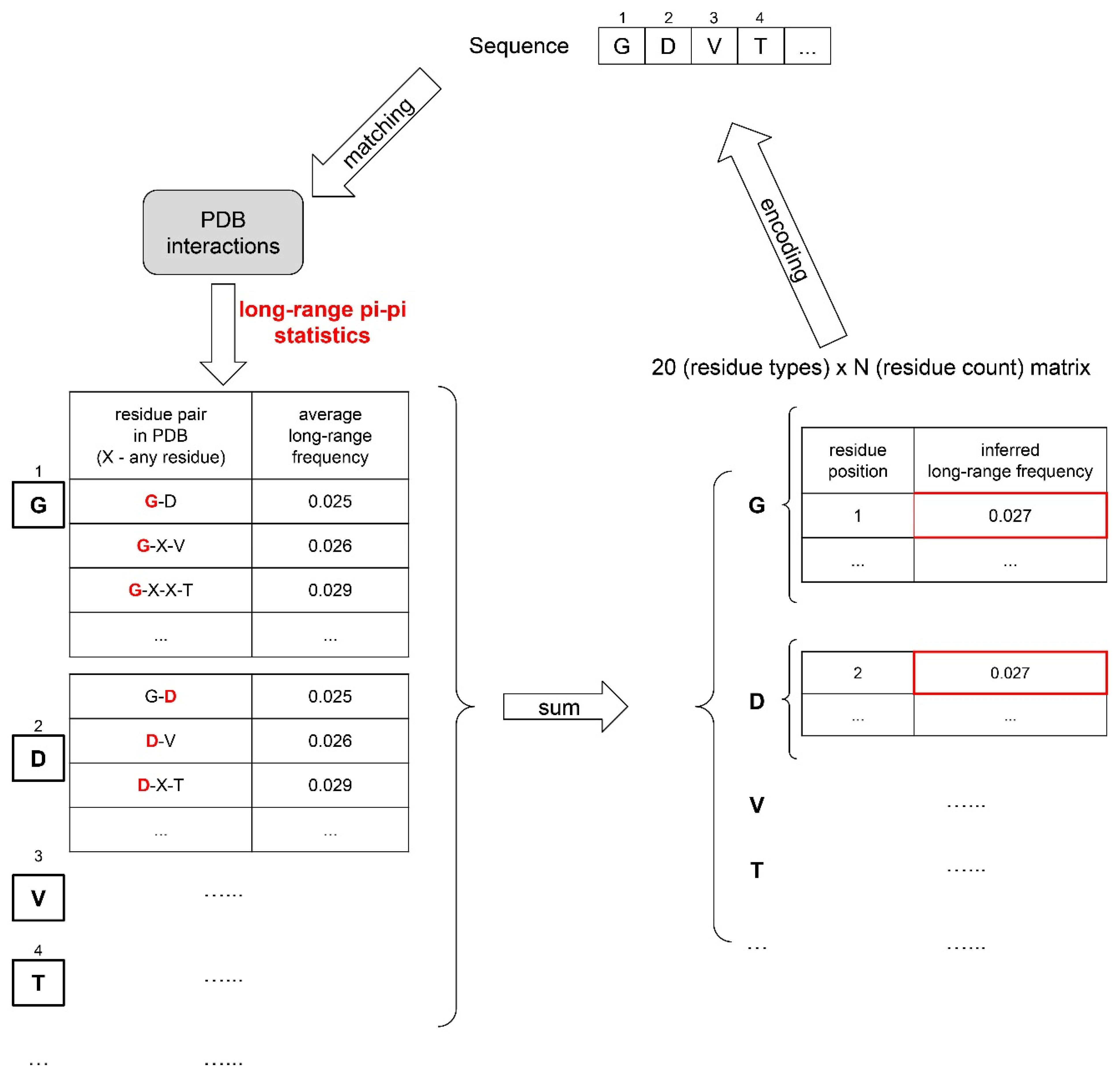
2.2. Construction of Physical-Feature Collection in LLPhyScore
3. Results and Discussion
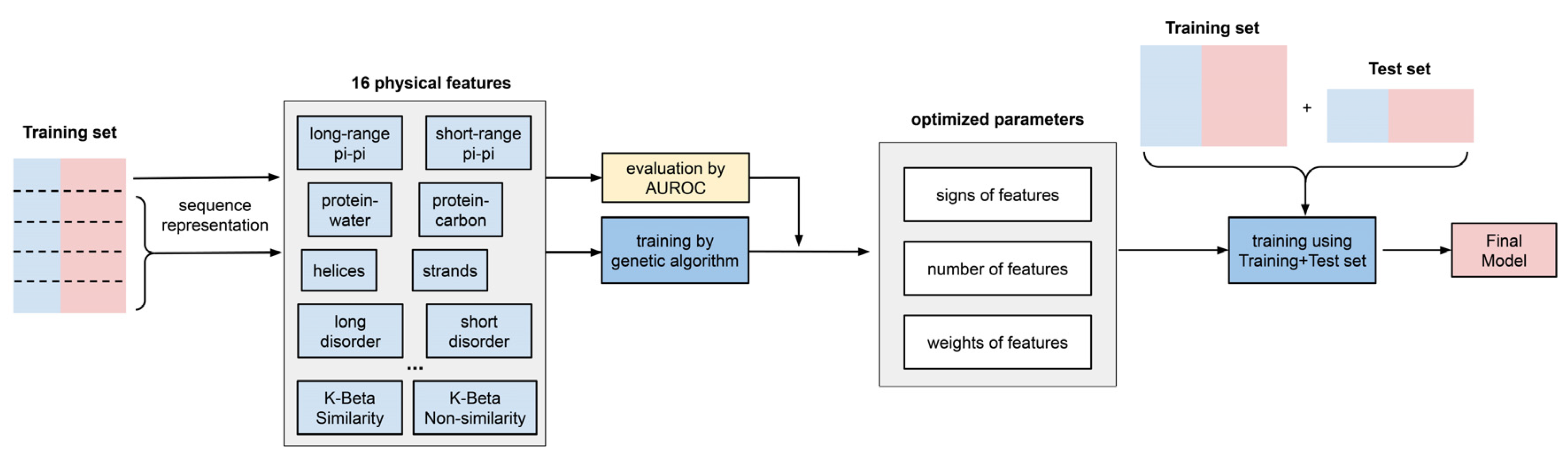
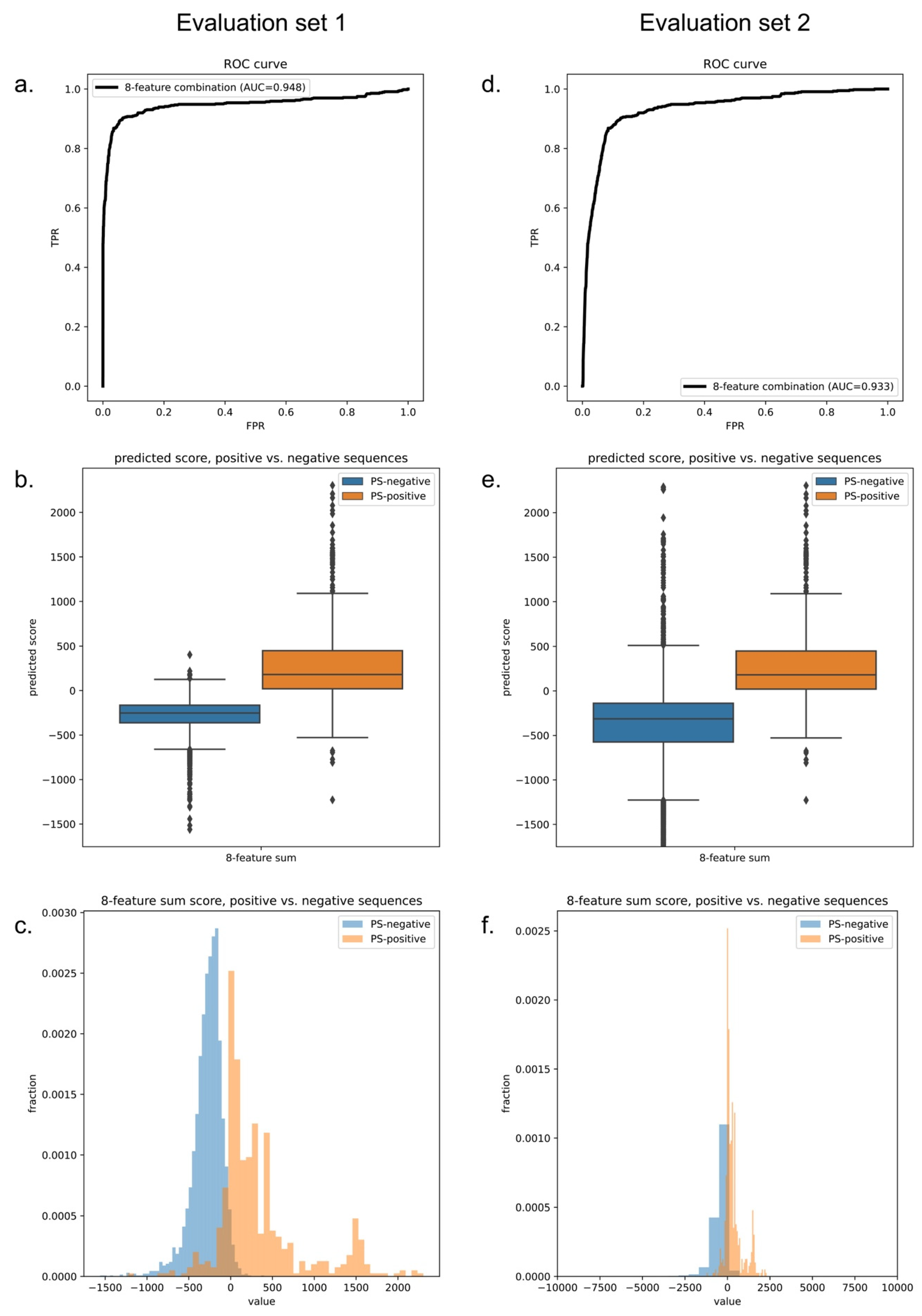
3.1. Predictor Training
3.2. Model Performance Comparison against Different Negative Training Sets
3.3. Predictor Validation
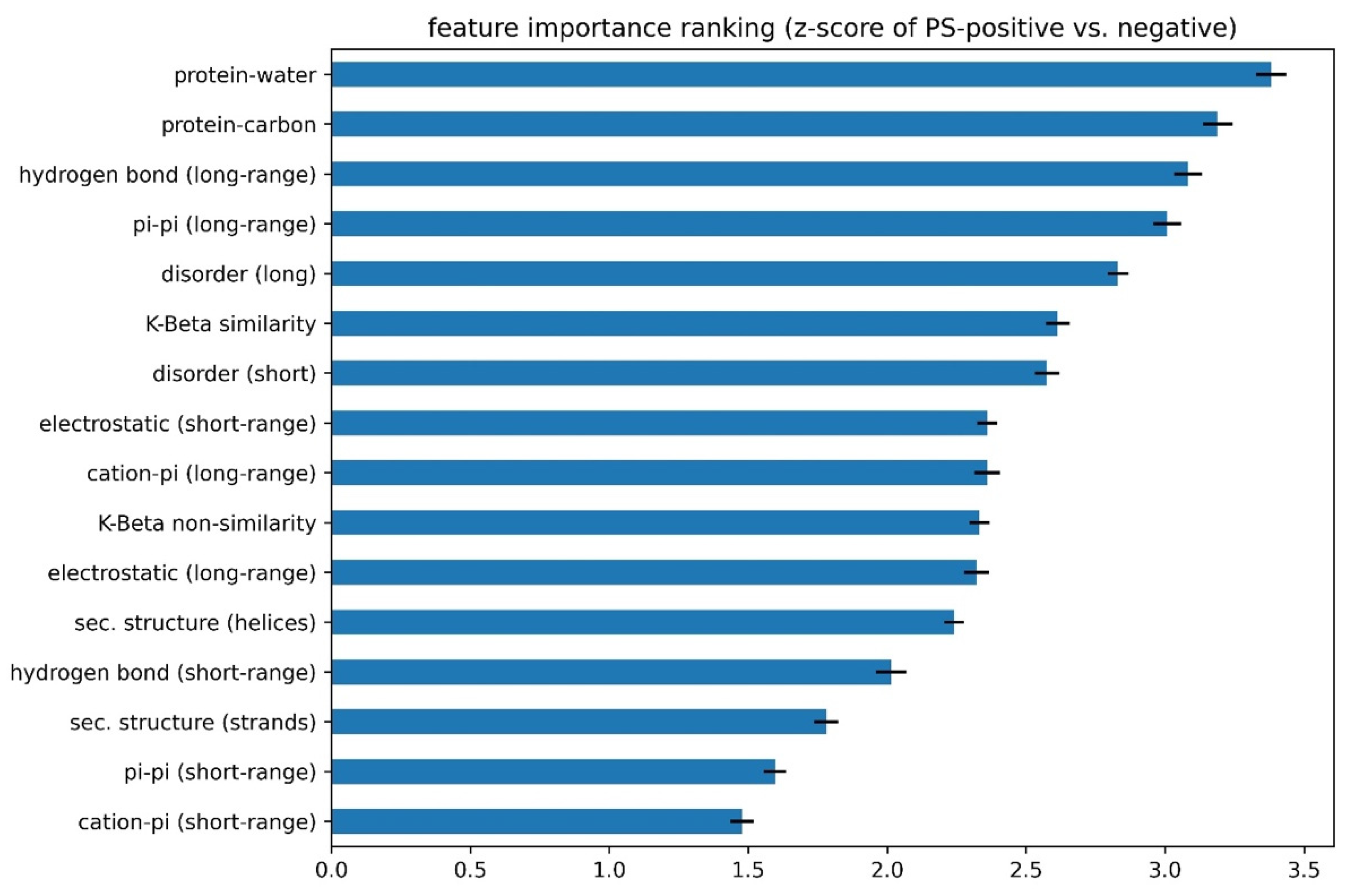
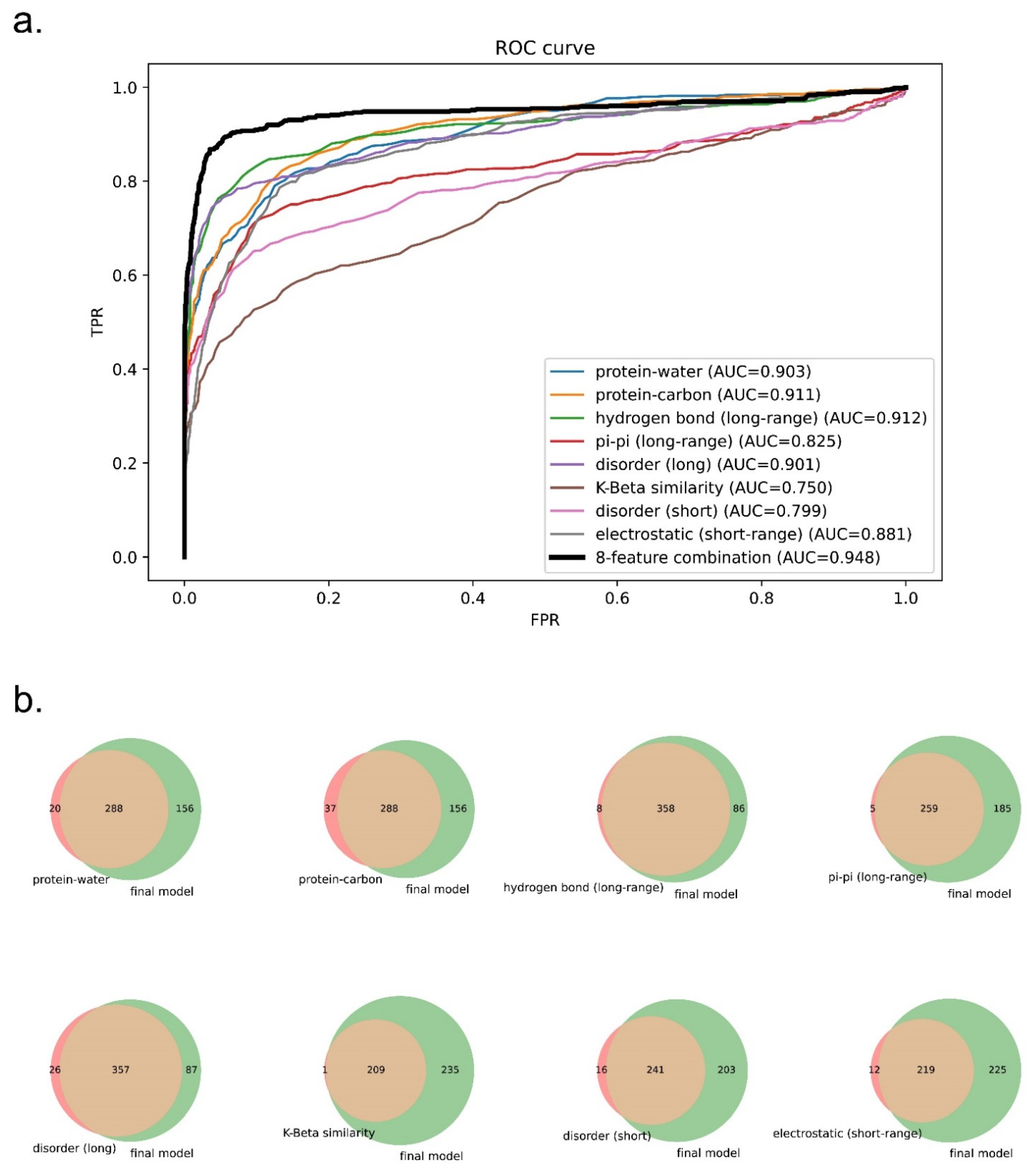
3.4. Comparison of Prediction Using Eight Features or Single Features
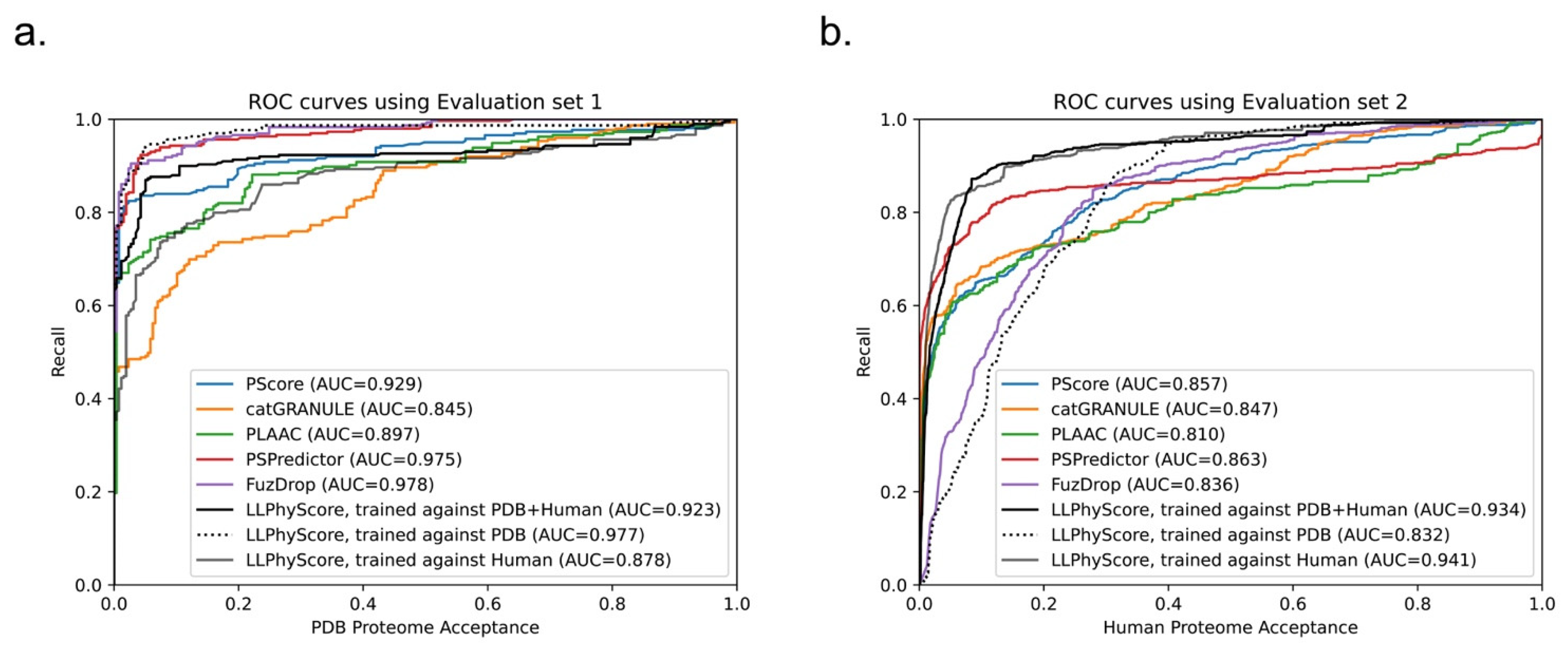
3.5. Comparison between LLPhyScore and Other Phase-Separation Predictors
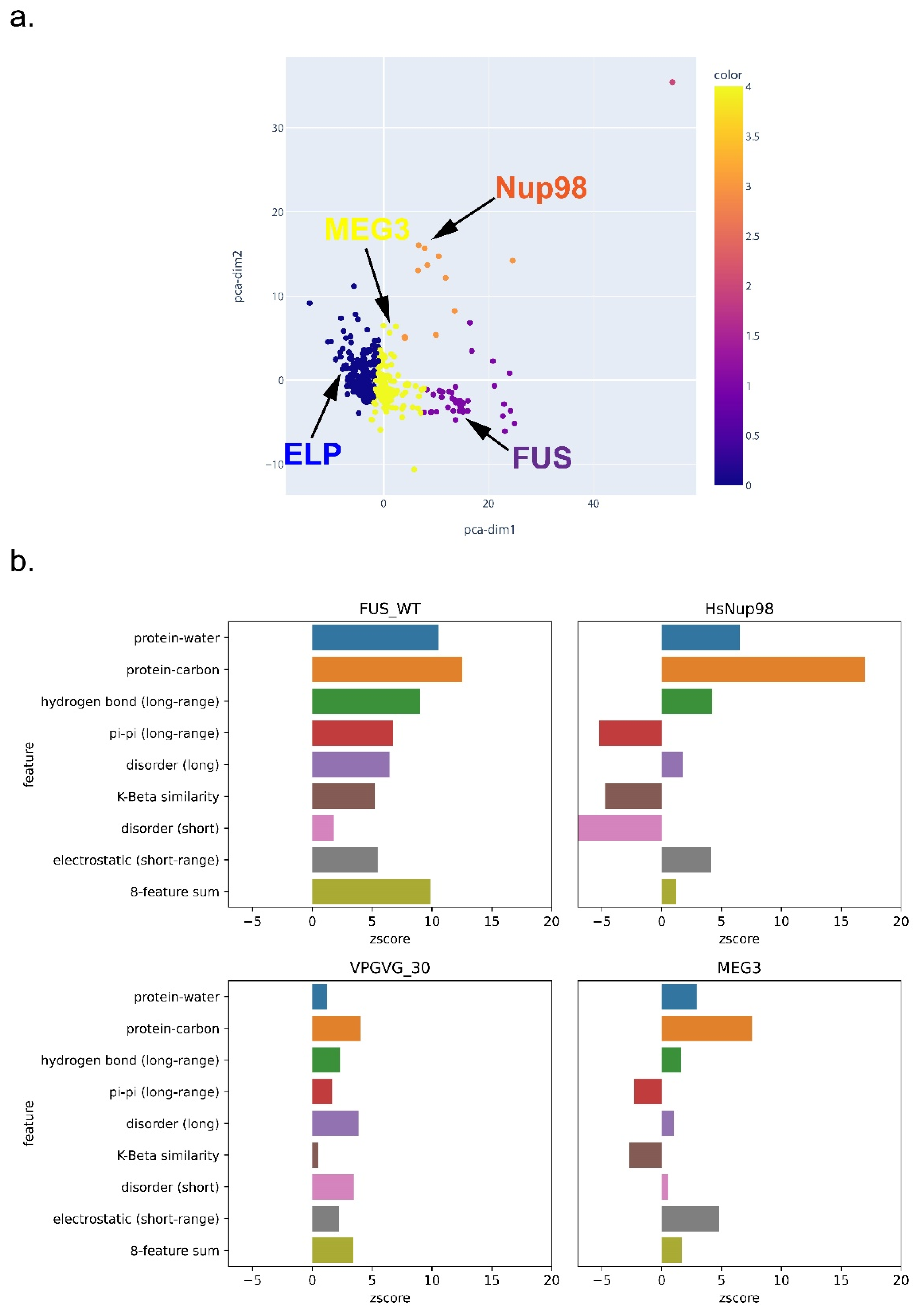
3.6. Feature-Based Breakdown of Scores for Different Sequences
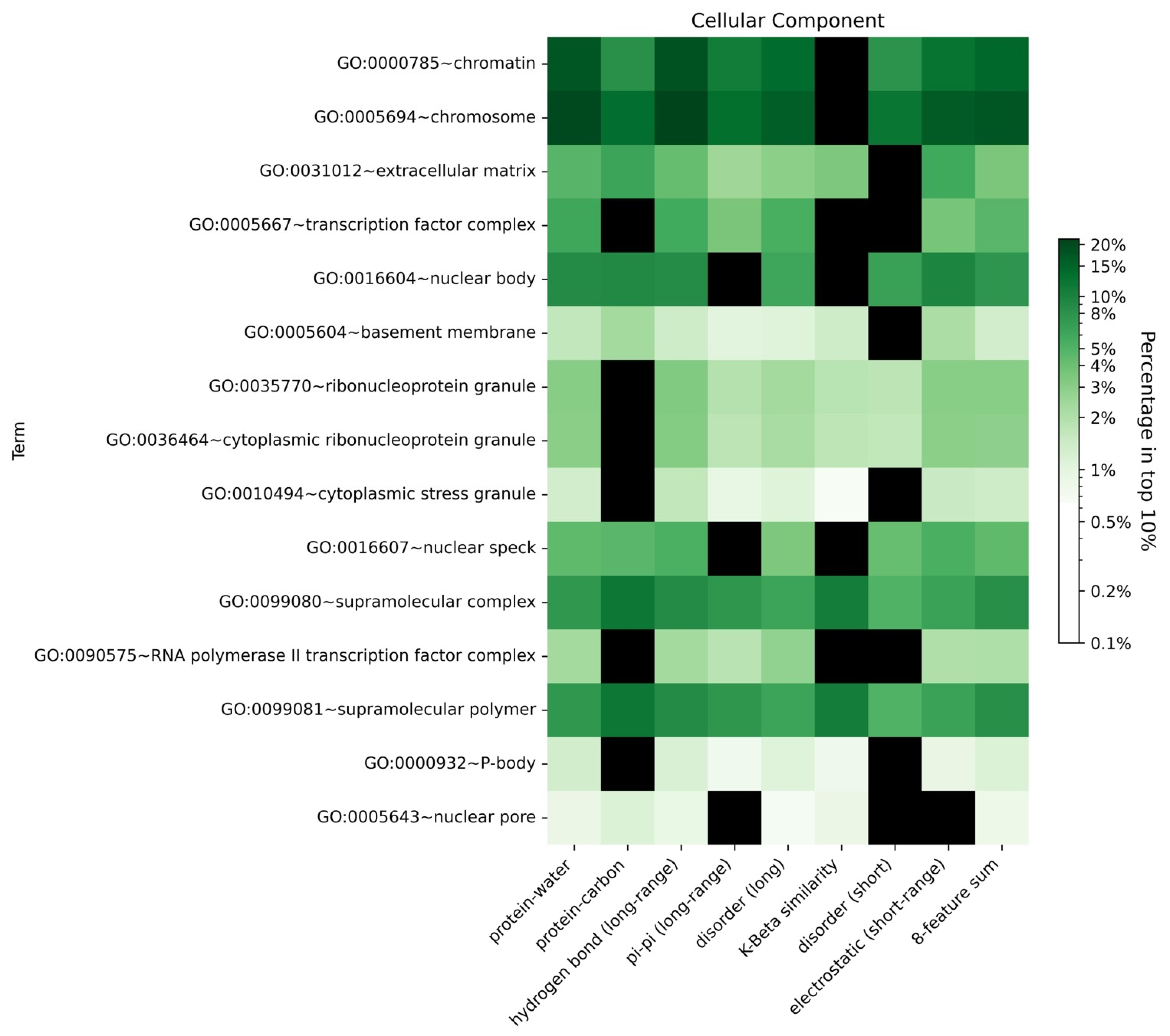
3.7. Gene Ontology Term Enrichment
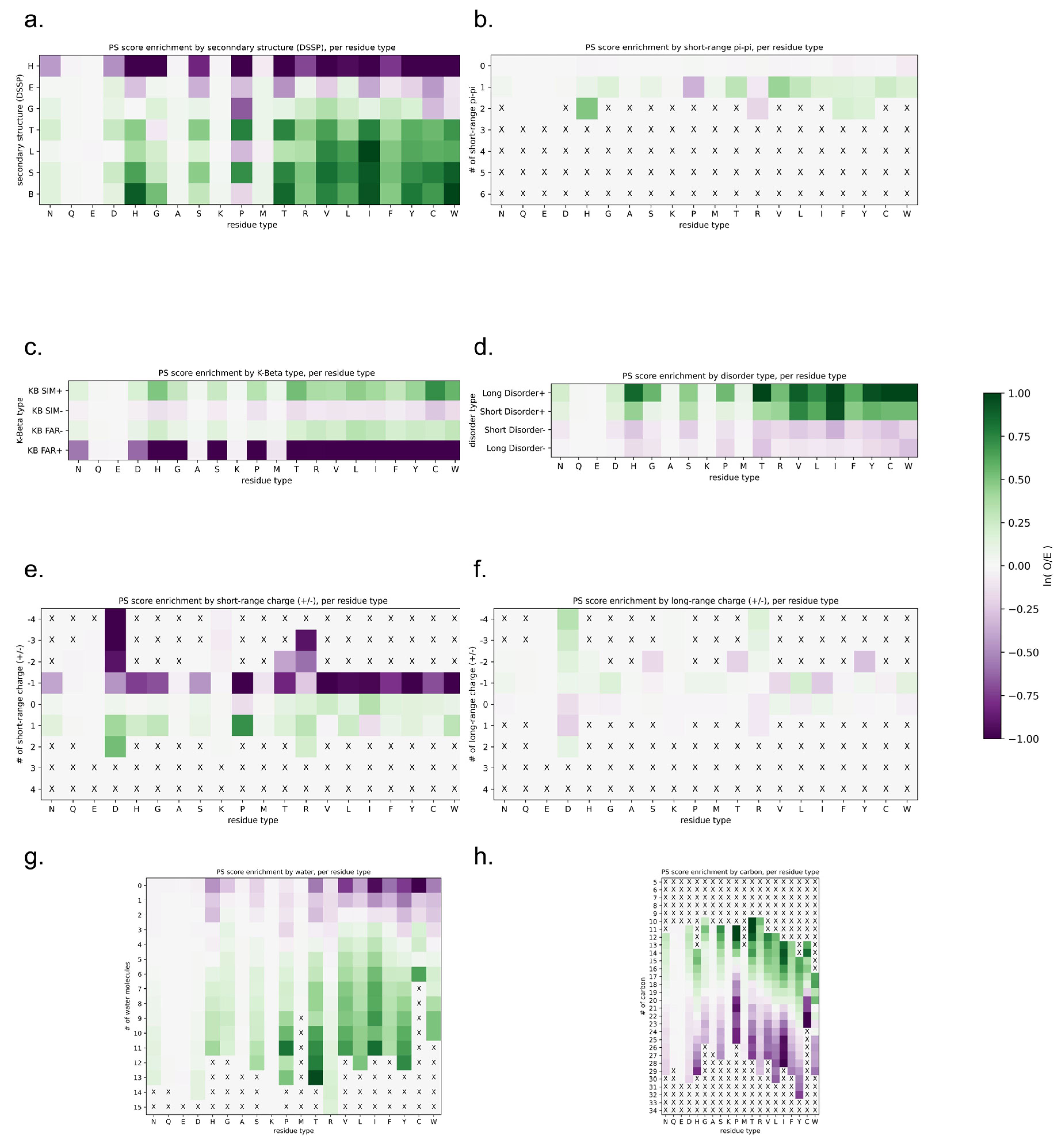
3.8. Physical Insights into Phase Separation Based on LLPhyScores of the PDB Set
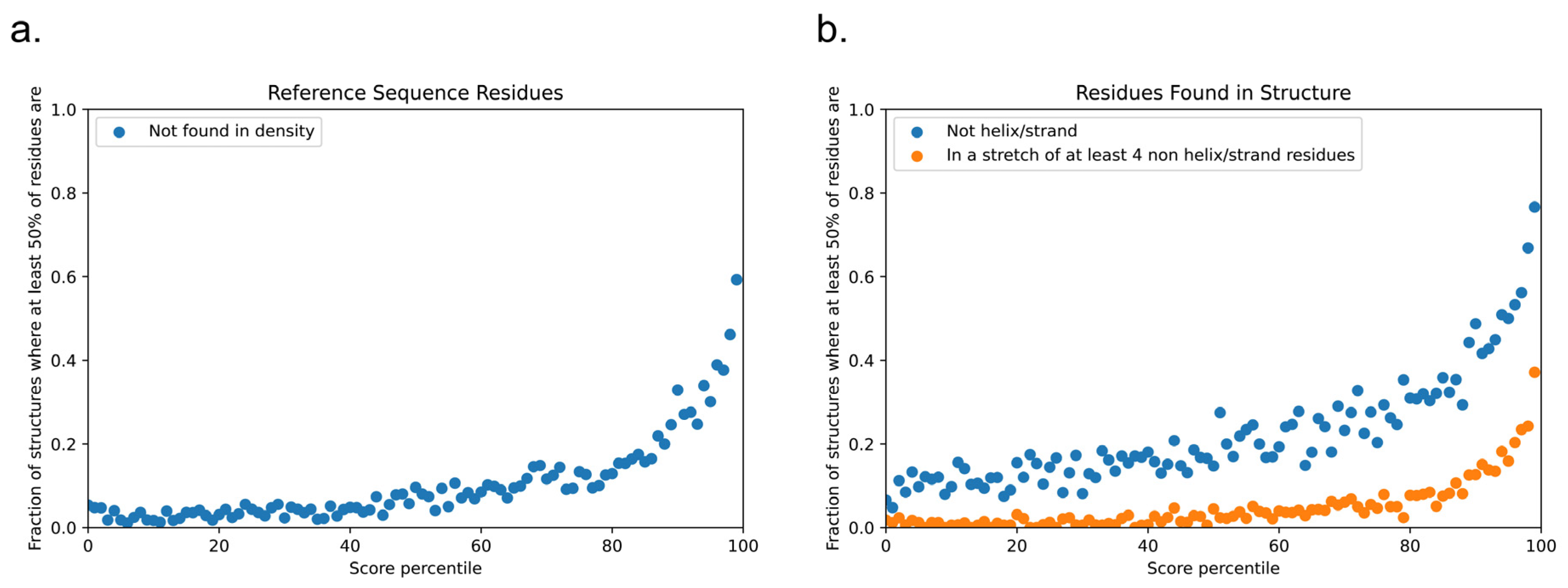
3.9. High-Scoring Structures in the PDB Trend towards Disorder
4. Conclusions
5. Technical Methods
5.1. Curation of PS-Positive Sequences
5.2. Clustering of PS-Positive Sequences
5.3. Preparation of PS-Negative Sequences
5.4. Construction of Training/Test/Evaluation Datasets
5.5. Physical-Feature-Based Sequence Representation
5.6. Predictor Training
5.7. Proteome Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.C. Evidence for and against liquid-liquid phase separation in the nucleus. Non-Coding RNA 2019, 5, 50. [Google Scholar]
- Mittag, T.; Pappu, R.V. A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol. Cell 2022, 82, 2201–2214. [Google Scholar] [CrossRef]
- Harmon, T.S.; Holehouse, A.S.; Rosen, M.K.; Pappu, R.V. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. eLife 2017, 6, e30294. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Brangwynne, C.P. Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Dev. Cell 2011, 21, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef]
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.A.; Forman-Kay, J.D. Liquid–liquid phase separation in cellular signaling systems. Curr. Opin. Struct. Biol. 2016, 41, 180–186. [Google Scholar] [CrossRef]
- Frey, S.; Richter, R.P.; Görlich, D. FG-Rich Repeats of Nuclear Pore Proteins Form a Three-Dimensional Meshwork with Hydrogel-Like Properties. Science 2006, 314, 815–817. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Al-Husini, N.; Tomares, D.T.; Bitar, O.; Childers, W.S.; Schrader, J.M. α-Proteobacterial RNA Degradosomes Assemble Liquid-Liquid Phase-Separated RNP Bodies. Mol. Cell 2018, 71, 1027–1039.e14. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, A.P.; Whitmarsh, A.J.; Ashe, M.P. Ribonucleoprotein bodies are phased in. Biochem. Soc. Trans. 2016, 44, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Muiznieks, L.D.; Sharpe, S.; Pomès, R.; Keeley, F.W. Role of Liquid–Liquid Phase Separation in Assembly of Elastin and Other Extracellular Matrix Proteins. J. Mol. Biol. 2018, 430, 4741–4753. [Google Scholar] [CrossRef]
- Bellingham, C.M.; Woodhouse, K.A.; Robson, P.; Rothstein, S.J.; Keeley, F.W. Self-aggregation characteristics of recombinantly expressed human elastin polypeptides. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 2001, 1550, 6–19. [Google Scholar] [CrossRef]
- Reichheld, S.E.; Muiznieks, L.D.; Keeley, F.W.; Sharpe, S. Direct observation of structure and dynamics during phase separation of an elastomeric protein. Proc. Natl. Acad. Sci. USA 2017, 114, E4408–E4415. [Google Scholar] [CrossRef]
- Wei, W.; Petrone, L.; Tan, Y.; Cai, H.; Israelachvili, J.N.; Miserez, A.; Waite, J.H. An Underwater Surface-Drying Peptide Inspired by a Mussel Adhesive Protein. Adv. Funct. Mater. 2016, 26, 3496–3507. [Google Scholar] [CrossRef]
- Kim, S.; Huang, J.; Lee, Y.; Dutta, S.; Yoo, H.Y.; Jung, Y.M.; Jho, Y.; Zeng, H.; Hwang, D.S. Complexation and coacervation of like-charged polyelectrolytes inspired by mussels. Proc. Natl. Acad. Sci. USA 2016, 113, E847–E853. [Google Scholar] [CrossRef]
- Le Ferrand, H.; Duchamp, M.; Gabryelczyk, B.; Cai, H.; Miserez, A. Time-Resolved Observations of Liquid–Liquid Phase Separation at the Nanoscale Using in Situ Liquid Transmission Electron Microscopy. J. Am. Chem. Soc. 2019, 141, 7202–7210. [Google Scholar] [CrossRef]
- Gabryelczyk, B.; Cai, H.; Shi, X.; Sun, Y.; Swinkels, P.J.M.; Salentinig, S.; Pervushin, K.; Miserez, A. Hydrogen bond guidance and aromatic stacking drive liquid-liquid phase separation of intrinsically disordered histidine-rich peptides. Nat. Commun. 2019, 10, 5465. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Gabryelczyk, B.; Manimekalai, M.S.S.; Grüber, G.; Salentinig, S.; Miserez, A. Self-coacervation of modular squid beak proteins—A comparative study. Soft Matter 2017, 13, 7740–7752. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Hoon, S.; Guerette, P.A.; Wei, W.; Ghadban, A.; Hao, C.; Miserez, A.; Waite, J.H. Infiltration of chitin by protein coacervates defines the squid beak mechanical gradient. Nat. Chem. Biol. 2015, 11, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H.; Forman-Kay, J.D.; Chan, H.S. Sequence-Specific Polyampholyte Phase Separation in Membraneless Organelles. Phys. Rev. Lett. 2016, 117, 178101. [Google Scholar] [CrossRef]
- Pak, C.W.; Kosno, M.; Holehouse, A.S.; Padrick, S.B.; Mittal, A.; Ali, R.; Yunus, A.A.; Liu, D.R.; Pappu, R.V.; Rosen, M.K. Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell 2016, 63, 72–85. [Google Scholar] [CrossRef]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. eLife 2018, 7, e31486. [Google Scholar] [CrossRef]
- Quiroz, F.G.; Chilkoti, A. Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat. Mater. 2015, 14, 1164–1171. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Sherrill, C.D. Energy Component Analysis of π Interactions. Acc. Chem. Res. 2013, 46, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Yeo, G.C.; Keeley, F.W.; Weiss, A.S. Coacervation of tropoelastin. Adv. Colloid Interface Sci. 2011, 167, 94–103. [Google Scholar] [CrossRef]
- Zaslavsky, B.Y.; Uversky, V.N. In aqua veritas: The indispensable yet mostly ignored role of water in phase separation and membrane-less organelles. Biochemistry 2018, 57, 2437–2451. [Google Scholar] [CrossRef]
- Mittag, T.; Parker, R. Multiple modes of protein–protein interactions promote RNP granule assembly. J. Mol. Biol. 2018, 430, 4636–4649. [Google Scholar] [CrossRef]
- Vernon, R.M.; Forman-Kay, J.D. First-generation predictors of biological protein phase separation. Curr. Opin. Struct. Biol. 2019, 58, 88–96. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L. Protein phase separation: A new phase in cell biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of liquid–liquid phase separating proteins using machine learning. BMC Bioinform. 2022, 23, 72. [Google Scholar] [CrossRef]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed representations of words and phrases and their compositionality. In Proceedings of the NIPS’13: 26th International Conference on Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–10 December 2013; pp. 3111–3119. [Google Scholar]
- Orlando, G.; Raimondi, D.; Tabaro, F.; Codicè, F.; Moreau, Y.; Vranken, W.F. Computational identification of prion-like RNA-binding proteins that form liquid phase-separated condensates. Bioinformatics 2019, 35, 4617–4623. [Google Scholar] [CrossRef] [PubMed]
- Paiz, E.A.; Allen, J.H.; Correia, J.J.; Fitzkee, N.C.; Hough, L.E.; Whitten, S.T. Beta turn propensity and a model polymer scaling exponent identify intrinsically disordered phase-separating proteins. J. Biol. Chem. 2021, 297, 101343. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.; Jansen, J.R.; Wang, J.; Poser, I.; van Heeringen, S.J.; Vermeulen, M. Predicting protein condensate formation using machine learning. Cell Rep. 2021, 34, 108705. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Benvenuti, M.; Mangani, S. Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef]
- Bolognesi, B.; Lorenzo Gotor, N.; Dhar, R.; Cirillo, D.; Baldrighi, M.; Tartaglia, G.G.; Lehner, B. A Concentration-Dependent Liquid Phase Separation Can Cause Toxicity upon Increased Protein Expression. Cell Rep. 2016, 16, 222–231. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Li, Q.; Peng, X.; Li, Y.; Tang, W.; Zhu, J.; Huang, J.; Qi, Y.; Zhang, Z. LLPSDB: A database of proteins undergoing liquid–liquid phase separation in vitro. Nucleic Acids Res. 2019, 48, D320–D327. [Google Scholar] [CrossRef]
- You, K.; Huang, Q.; Yu, C.; Shen, B.; Sevilla, C.; Shi, M.; Hermjakob, H.; Chen, Y.; Li, T. PhaSepDB: A database of liquid–liquid phase separation related proteins. Nucleic Acids Res. 2019, 48, D354–D359. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Szabó, B.; Schád, É.; Tantos, Á.; Abukhairan, R.; Horváth, T.; Murvai, N.; Kovács, O.P.; Kovács, M.; et al. PhaSePro: The database of proteins driving liquid–liquid phase separation. Nucleic Acids Res. 2019, 48, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.S.; Samanta, N.; Ebbinghaus, S.; Marcos, J.C. The synergic effect of water and biomolecules in intracellular phase separation. Nat. Rev. Chem. 2019, 3, 552–561. [Google Scholar] [CrossRef]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; Alexandra, M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 α-helical structure tunes liquid–liquid phase separation and function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinform. 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef]
- Mohan, A.; Uversky, V.N.; Radivojac, P. Influence of sequence changes and environment on intrinsically disordered proteins. PLoS Comput. Biol. 2009, 5, e1000497. [Google Scholar] [CrossRef]
- Boyko, S.; Qi, X.; Chen, T.-H.; Surewicz, K.; Surewicz, W.K. Liquid–liquid phase separation of tau protein: The crucial role of electrostatic interactions. J. Biol. Chem. 2019, 294, 11054–11059. [Google Scholar] [CrossRef]
- O’Meara, M.J.; Leaver-Fay, A.; Tyka, M.D.; Stein, A.; Houlihan, K.; DiMaio, F.; Bradley, P.; Kortemme, T.; Baker, D.; Snoeyink, J. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. [Google Scholar] [CrossRef]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid−liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Goldschmidt, L.; Eisenberg, D.S. Prevalence and species distribution of the low-complexity, amyloid-like, reversible, kinked segment structural motif in amyloid-like fibrils. J. Biol. Chem. 2021, 297, 101194. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 2017, 171, 615–627.e16. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed]
- Firman, T.; Ghosh, K. Sequence charge decoration dictates coil-globule transition in intrinsically disordered proteins. J. Chem. Phys. 2018, 148, 123305. [Google Scholar] [CrossRef]
- Enkhbayar, P.; Hikichi, K.; Osaki, M.; Kretsinger, R.H.; Matsushima, N. 310-helices in proteins are parahelices. Proteins Struct. Funct. Bioinform. 2006, 64, 691–699. [Google Scholar] [CrossRef]
- Fiori, W.R.; Miick, S.M.; Millhauser, G.L. Increasing sequence length favors. alpha.-helix over 310-helix in alanine-based peptides: Evidence for a length-dependent structural transition. Biochemistry 1993, 32, 11957–11962. [Google Scholar] [CrossRef]
- Doig, A.J.; Stapley, B.J.; Macarthur, M.W.; Thornton, J.M. Structures of N-termini of helices in proteins. Protein Sci. 1997, 6, 147–155. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. JMLR 2011, 12, 2825–2830. [Google Scholar]
- Pundir, S.; Martin, M.J.; O’Donovan, C. UniProt Protein Knowledgebase. In Protein Bioinformatics: From Protein Modifications and Networks to Proteomics; Wu, C.H., Arighi, C.N., Ross, K.E., Eds.; Springer: New York, NY, USA, 2017; pp. 41–55. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35 (Suppl. S2), W169–W175. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, H.; Vernon, R.M.; Forman-Kay, J.D. An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions. Biomolecules 2022, 12, 1131. https://doi.org/10.3390/biom12081131
Cai H, Vernon RM, Forman-Kay JD. An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions. Biomolecules. 2022; 12(8):1131. https://doi.org/10.3390/biom12081131
Chicago/Turabian StyleCai, Hao, Robert M. Vernon, and Julie D. Forman-Kay. 2022. "An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions" Biomolecules 12, no. 8: 1131. https://doi.org/10.3390/biom12081131
APA StyleCai, H., Vernon, R. M., & Forman-Kay, J. D. (2022). An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions. Biomolecules, 12(8), 1131. https://doi.org/10.3390/biom12081131