
Interaction of the Emerging Mycotoxins Beauvericin, Cyclopiazonic Acid, and Sterigmatocystin with Human Serum Albumin

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Fluorescence Spectroscopic Studies
2.3. Ultrafiltration Studies
2.4. Ultracentrifugation Experiments
2.5. HPLC and LC-MS Analyses
2.6. Modeling Studies
2.7. Statistics
3. Results and Discussion
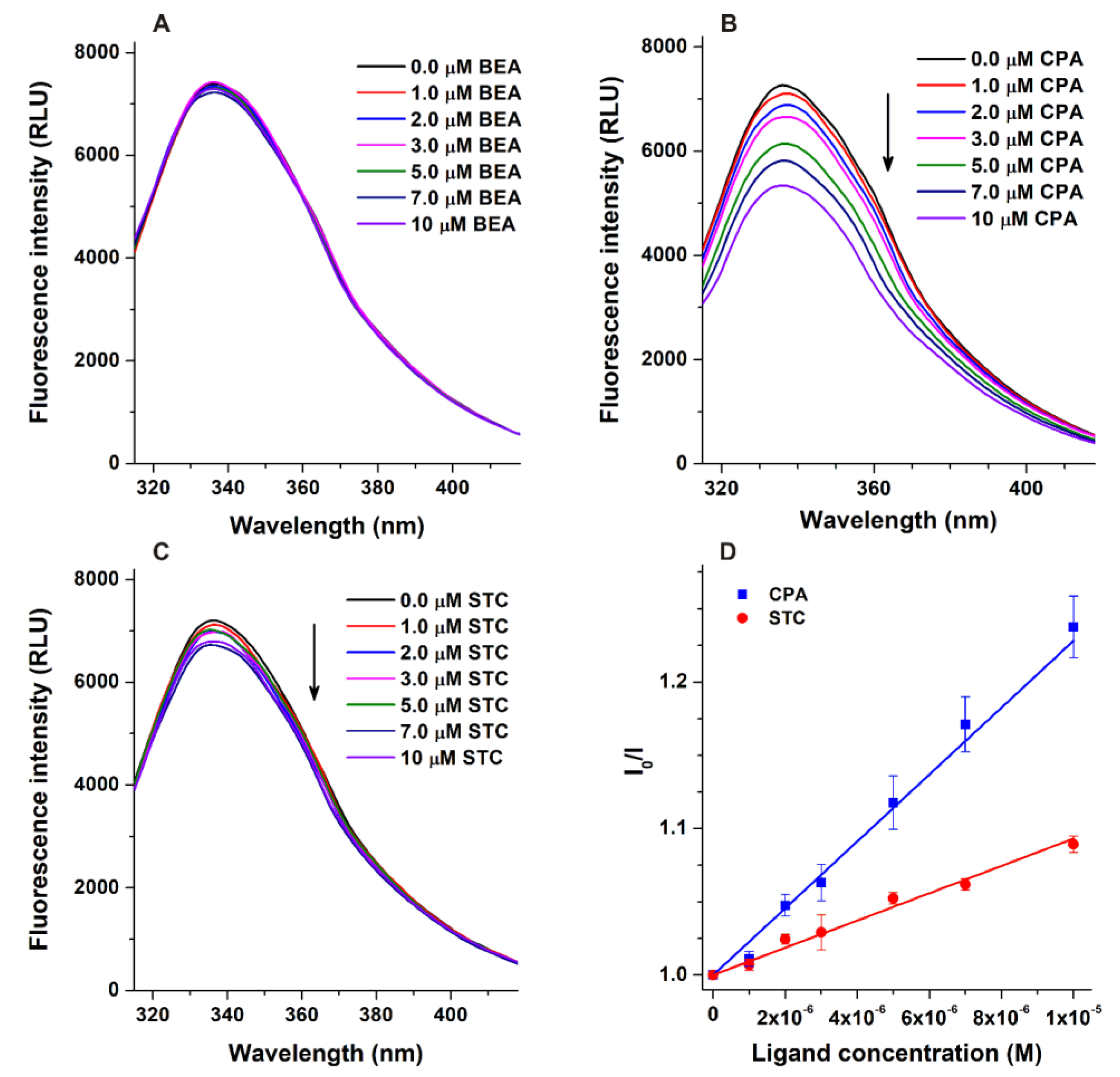
3.1. Fluorescence Quenching Studies
3.2. Ultracentrifugation Studies
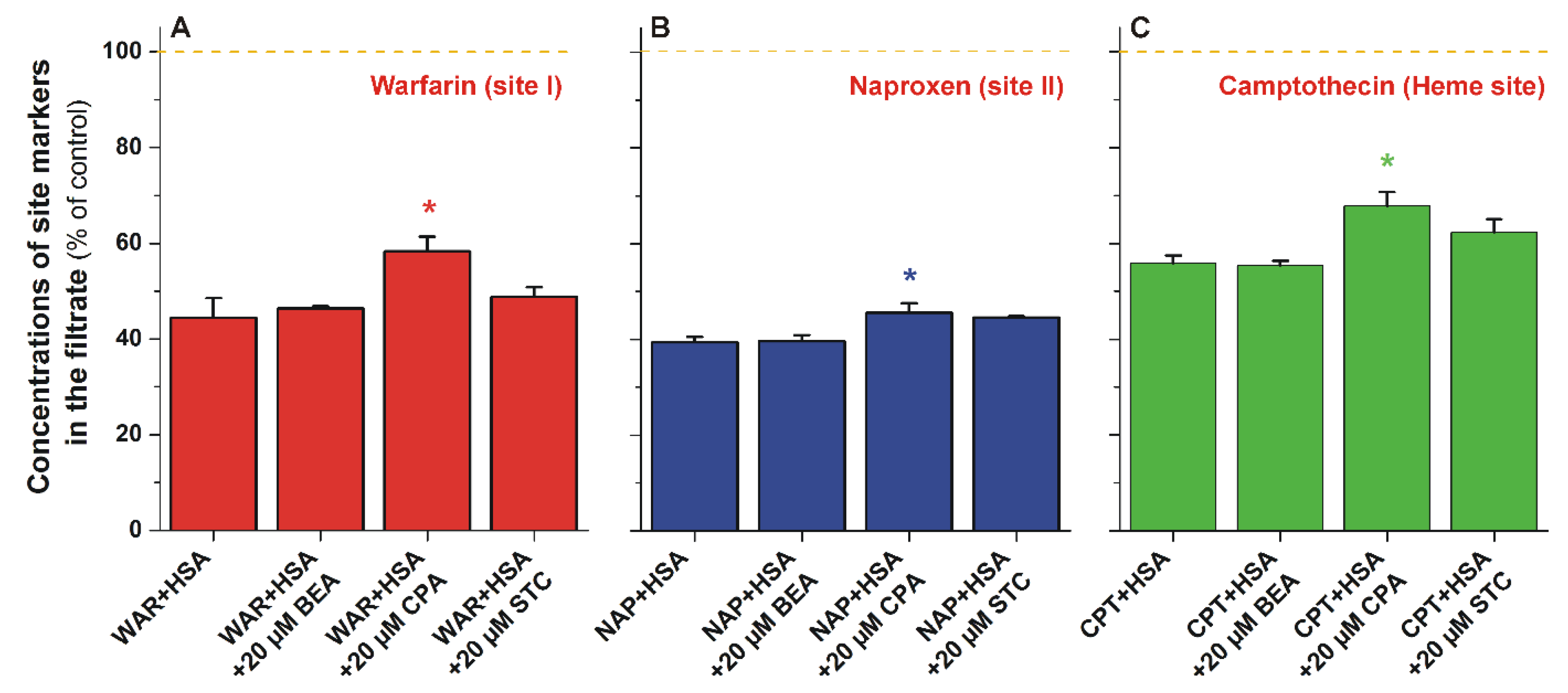
3.3. Ultrafiltration Studies
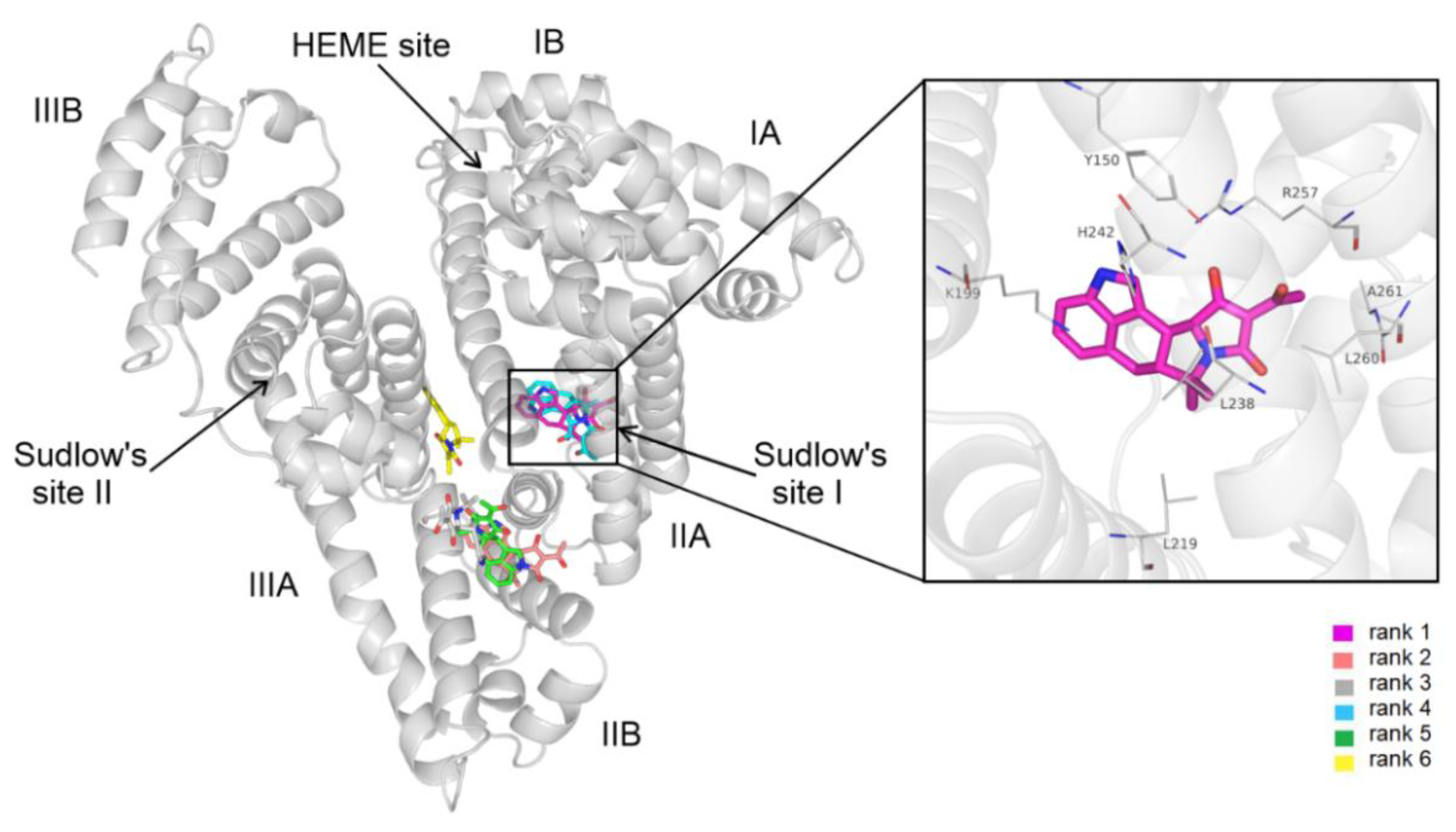
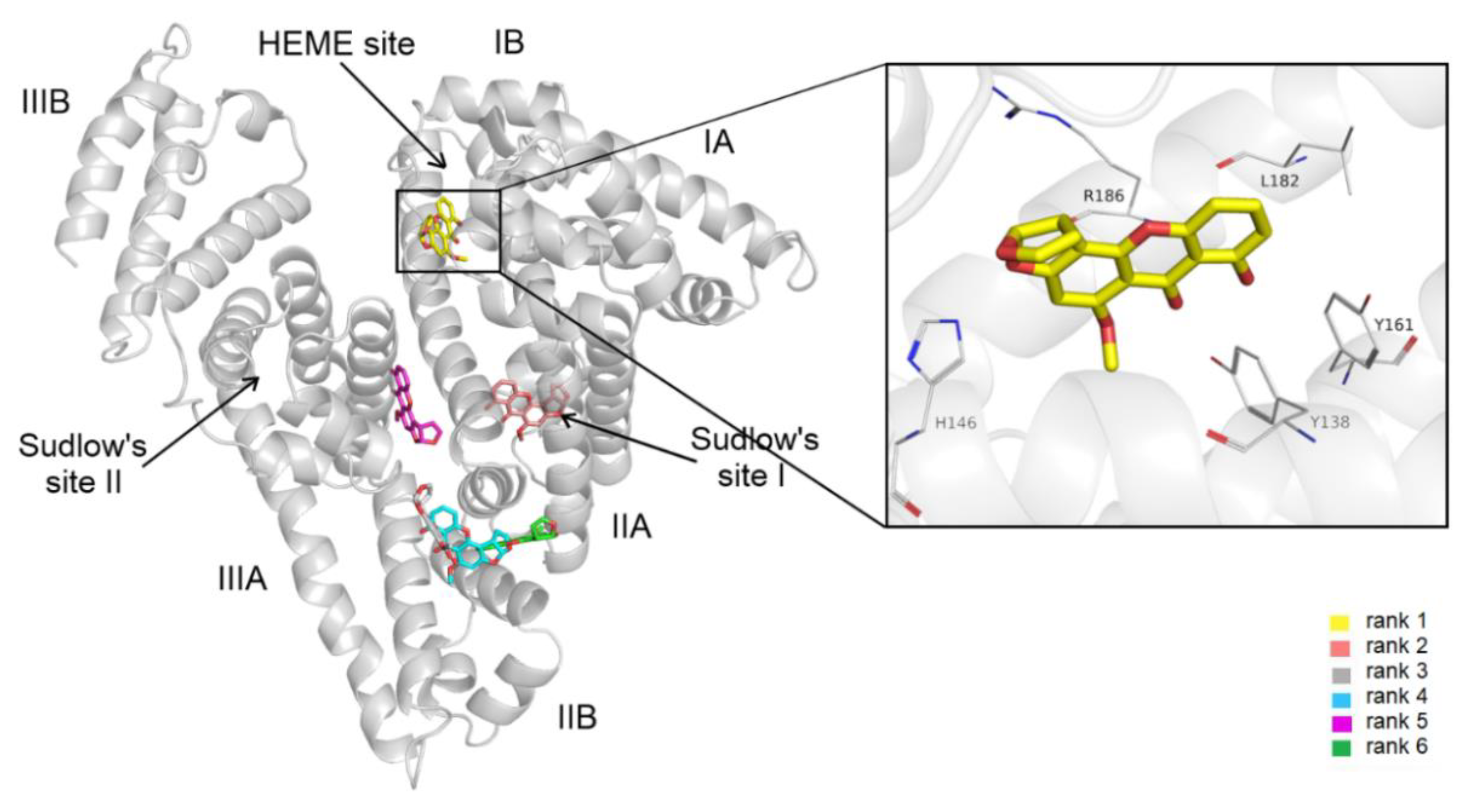
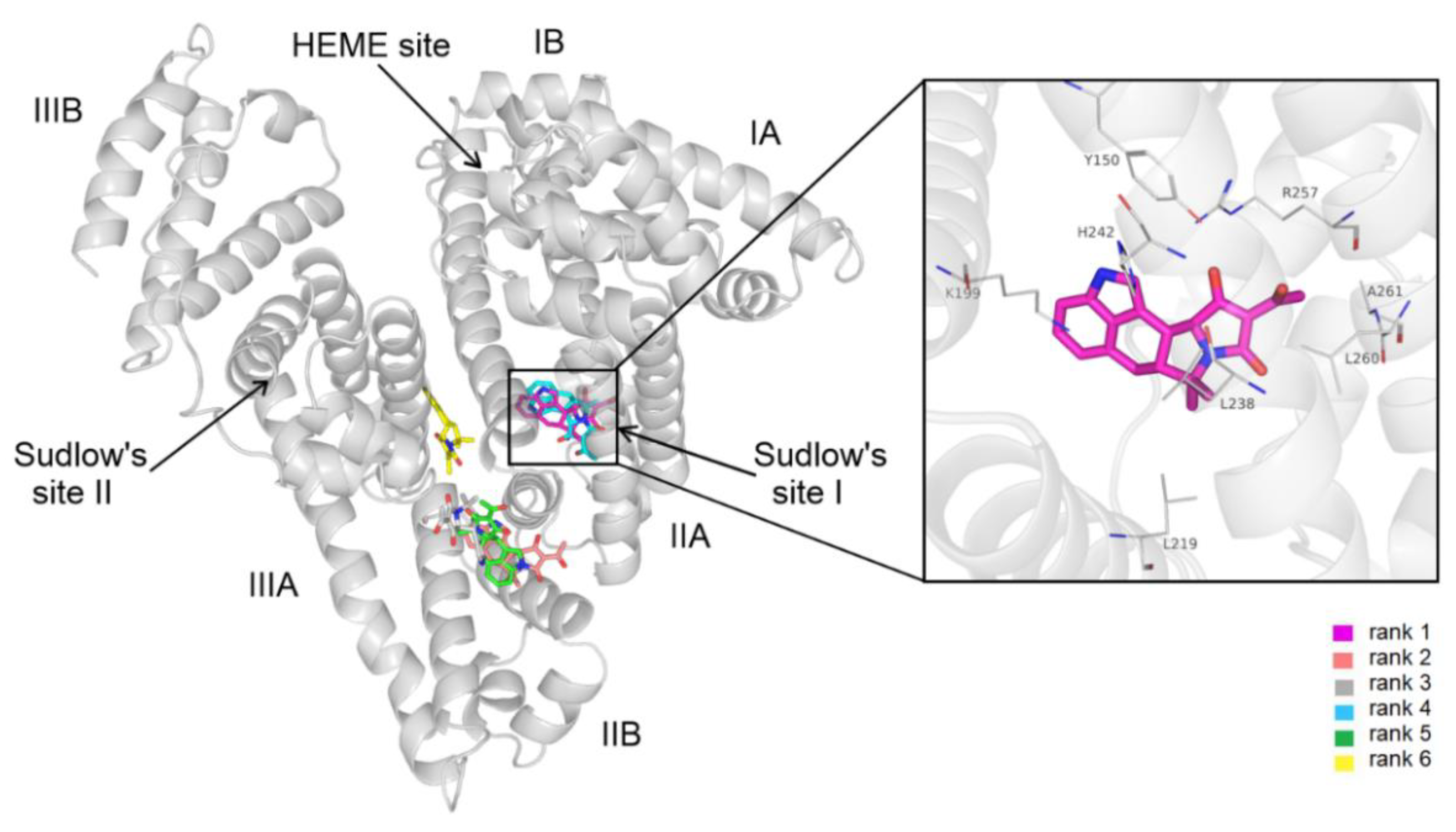
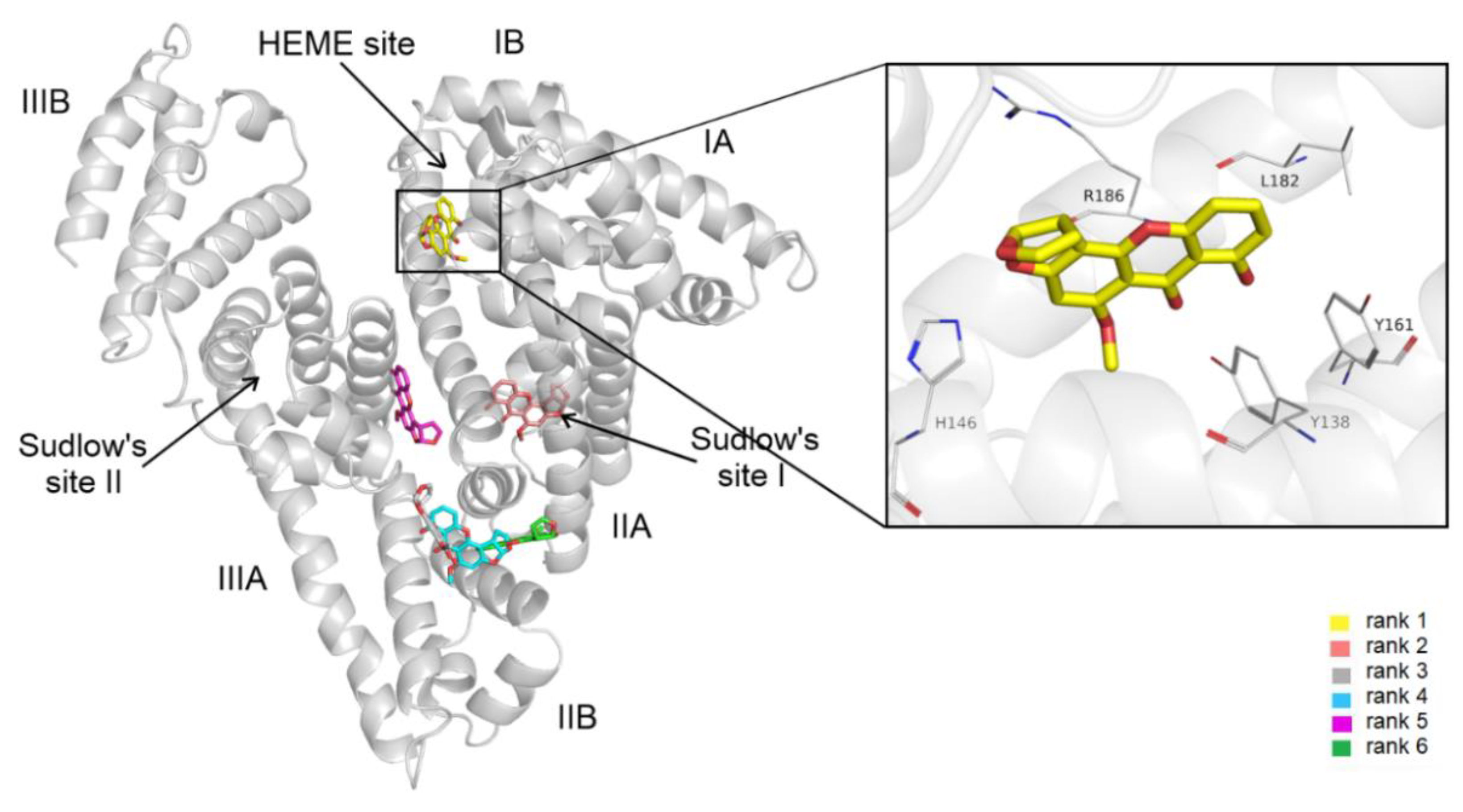
3.4. Modeling Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hussein, H.S.; Brasel, J.M. Toxicity, Metabolism, and Impact of Mycotoxins on Humans and Animals. Toxicology 2001, 167, 101–134. [Google Scholar] [CrossRef]
- Jestoi, M. Emerging Fusarium-Mycotoxins Fusaproliferin, Beauvericin, Enniatins, And Moniliformin—A Review. Crit. Rev. Food Sci. Nutr. 2008, 48, 21–49. [Google Scholar] [CrossRef]
- Tedjiotsop Feudjio, F.; Dornetshuber, R.; Lemmens, M.; Hoffmann, O.; Lemmens-Gruber, R.; Berger, W. Beauvericin and Enniatin: Emerging Toxins and/or Remedies? World Mycotoxin J. 2010, 3, 415–430. [Google Scholar] [CrossRef]
- Santini, A.; Meca, G.; Uhlig, S.; Ritieni, A. Fusaproliferin, Beauvericin and Enniatins: Occurrence in Food—A Review. World Mycotoxin J. 2012, 5, 71–81. [Google Scholar] [CrossRef]
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging Mycotoxins: Beyond Traditionally Determined Food Contaminants. J. Agric. Food Chem. 2017, 65, 7052–7070. [Google Scholar] [CrossRef] [PubMed]
- Caloni, F.; Fossati, P.; Anadón, A.; Bertero, A. Beauvericin: The Beauty and the Beast. Environ. Toxicol. Pharmacol. 2020, 75, 103349. [Google Scholar] [CrossRef]
- Mallebrera, B.; Prosperini, A.; Font, G.; Ruiz, M.J. In Vitro Mechanisms of Beauvericin Toxicity: A Review. Food Chem. Toxicol. 2018, 111, 537–545. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the Risks to Human and Animal Health Related to the Presence of Beauvericin and Enniatins in Food and Feed. EFSA J. 2014, 12, 3802–3976. [Google Scholar] [CrossRef]
- Ostry, V.; Toman, J.; Grosse, Y.; Malir, F. Cyclopiazonic Acid: 50th Anniversary of Its Discovery. World Mycotoxin J. 2018, 11, 135–148. [Google Scholar] [CrossRef]
- Burdock, G.A.; Flamm, W.G. Review Article: Safety Assessment of the Mycotoxin Cyclopiazonic Acid. Int. J. Toxicol. 2000, 19, 195–218. [Google Scholar] [CrossRef]
- Nishie, K.; Cole, R.J.; Dorner, J.W. Toxicity and Neuropharmacology of Cyclopiazonic Acid. Food Chem. Toxicol. 1985, 23, 831–839. [Google Scholar] [CrossRef]
- Veršilovskis, A.; De Saeger, S. Sterigmatocystin: Occurrence in Foodstuffs and Analytical Methods—An Overview. Mol. Nutr. Food Res. 2010, 54, 136–147. [Google Scholar] [CrossRef]
- Yoshinari, T.; Takeuchi, H.; Kosugi, M.; Taniguchi, M.; Waki, M.; Hashiguchi, S.; Fujiyoshi, T.; Shichinohe, Y.; Nakajima, M.; Ohnishi, T.; et al. Determination of Sterigmatocystin in Foods in Japan: Method Validation and Occurrence Data. Food Addit. Contam. Part A 2019, 36, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the Risk for Public and Animal Health Related to the Presence of Sterigmatocystin in Food and Feed. EFSA J. 2013, 11, 3254–3335. [Google Scholar] [CrossRef]
- Díaz Nieto, C.H.; Granero, A.M.; Zon, M.A.; Fernández, H. Sterigmatocystin: A Mycotoxin to Be Seriously Considered. Food Chem. Toxicol. 2018, 118, 460–470. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human Serum Albumin: From Bench to Bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin–Drug Interaction and Its Clinical Implication. Biochim. Biophys. Acta BBA-Gen. Subj. 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef]
- Perry, J.L.; Il’ichev, Y.V.; Kempf, V.R.; McClendon, J.; Park, G.; Manderville, R.A.; Rüker, F.; Dockal, M.; Simon, J.D. Binding of Ochratoxin A Derivatives to Human Serum Albumin. J. Phys. Chem. B 2003, 107, 6644–6647. [Google Scholar] [CrossRef]
- Faisal, Z.; Vörös, V.; Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Csepregi, R.; Kőszegi, T.; Zsila, F.; Poór, M. Probing the Interactions of Ochratoxin B, Ochratoxin C, Patulin, Deoxynivalenol, and T-2 Toxin with Human Serum Albumin. Toxins 2020, 12, 392. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Dellafiora, L.; Dall’Asta, C.; Cruciani, G.; Pethő, G.; Poór, M. Interaction of Mycotoxin Alternariol with Serum Albumin. Int. J. Mol. Sci. 2019, 20, 2352. [Google Scholar] [CrossRef]
- Poór, M.; Lemli, B.; Bálint, M.; Hetényi, C.; Sali, N.; Kőszegi, T.; Kunsági-Máté, S. Interaction of Citrinin with Human Serum Albumin. Toxins 2015, 7, 5155–5166. [Google Scholar] [CrossRef]
- Faisal, Z.; Lemli, B.; Szerencsés, D.; Kunsági-Máté, S.; Bálint, M.; Hetényi, C.; Kuzma, M.; Mayer, M.; Poór, M. Interactions of Zearalenone and Its Reduced Metabolites α-Zearalenol and β-Zearalenol with Serum Albumins: Species Differences, Binding Sites, and Thermodynamics. Mycotoxin Res. 2018, 34, 269–278. [Google Scholar] [CrossRef]
- Hong, C.-Y.; Chen, Y.-C. Selective Enrichment of Ochratoxin A Using Human Serum Albumin Bound Magnetic Beads as the Concentrating Probes for Capillary Electrophoresis/Electrospray Ionization-Mass Spectrometric Analysis. J. Chromatogr. A 2007, 1159, 250–255. [Google Scholar] [CrossRef]
- Janić Hajnal, E.; Čolović, R.; Pezo, L.; Orčić, D.; Vukmirović, Đ.; Mastilović, J. Possibility of Alternaria Toxins Reduction by Extrusion Processing of Whole Wheat Flour. Food Chem. 2016, 213, 784–790. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Szabó, Á.; Szente, L.; Poór, M. Extraction of Mycotoxin Alternariol from Red Wine and from Tomato Juice with Beta-Cyclodextrin Bead Polymer. J. Mol. Liq. 2020, 319, 114180. [Google Scholar] [CrossRef]
- Hu, T.; Liu, Y. Probing the Interaction of Cefodizime with Human Serum Albumin Using Multi-Spectroscopic and Molecular Docking Techniques. J. Pharm. Biomed. Anal. 2015, 107, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Faisal, Z.; Mohos, V.; Derdák, D.; Lemli, B.; Kálai, T.; Sár, C.; Zsidó, B.Z.; Hetényi, C.; Horváth, Á.I.; et al. Interaction of SZV 1287, a Novel Oxime Analgesic Drug Candidate, and Its Metabolites with Serum Albumin. J. Mol. Liq. 2021, 333, 115945. [Google Scholar] [CrossRef]
- Boulton, D.W.; Walle, U.K.; Walle, T. Extensive Binding of the Bioflavonoid Quercetin to Human Plasma Proteins. J. Pharm. Pharmacol. 1998, 50, 243–249. [Google Scholar] [CrossRef]
- Chan, E.; McLachlan, A.; Pegg, M.; MacKay, A.; Cole, R.; Rowland, M. Disposition of Warfarin Enantiomers and Metabolites in Patients during Multiple Dosing with Rac-Warfarin. Br. J. Clin. Pharmacol. 1994, 37, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Forrest, J.A.H.; Clements, J.A.; Prescott, L.F. Clinical Pharmacokinetics of Paracetamol. Clin. Pharmacokinet. 1982, 7, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Peschke, M.; Verkerk, U.H.; Kebarle, P. Features of the ESI Mechanism That Affect the Observation of Multiply Charged Noncovalent Protein Complexes and the Determination of the Association Constant by the Titration Method. J. Am. Soc. Mass Spectrom. 2004, 15, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Mohos, V.; Faisal, Z.; Fliszár-Nyúl, E.; Szente, L.; Poór, M. Testing the Extraction of 12 Mycotoxins from Aqueous Solutions by Insoluble Beta-Cyclodextrin Bead Polymer. Environ. Sci. Pollut. Res. 2022, 29, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-Ray Structures. J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar] [CrossRef]
- Schwab, C.H. Conformations and 3D Pharmacophore Searching. Drug Discov. Today Technol. 2010, 7, e245–e253. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Poór, M.; Bálint, M.; Hetényi, C.; Gődér, B.; Kunsági-Máté, S.; Kőszegi, T.; Lemli, B. Investigation of Non-Covalent Interactions of Aflatoxins (B1, B2, G1, G2, and M1) with Serum Albumin. Toxins 2017, 9, 339. [Google Scholar] [CrossRef]
- Chen, J.; Ohnmacht, C.; Hage, D.S. Studies of Phenytoin Binding to Human Serum Albumin by High-Performance Affinity Chromatography. J. Chromatogr. B 2004, 809, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Amézqueta, S.; Beltrán, J.L.; Bolioli, A.M.; Campos-Vicens, L.; Luque, F.J.; Ràfols, C. Evaluation of the Interactions between Human Serum Albumin (HSA) and Non-Steroidal Anti-Inflammatory (NSAIDs) Drugs by Multiwavelength Molecular Fluorescence, Structural and Computational Analysis. Pharmaceuticals 2021, 14, 214. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | logKSV ± SEM FL Quenching (2 μM HSA) | logKa ± SEM FL Quenching (2 μM HSA) | logKa ± SEM Ultracentrifugation (40 g/L HSA) | Bound Fraction (%) Ultracentrifugation (40 g/L HSA) |
|---|---|---|---|---|
| BEA–HSA | – | – | 3.19 ± 0.02 | 47.9 ± 0.9 |
| CPA–HSA | 4.37 ± 0.05 | 4.38 ± 0.05 | 4.70 ± 0.02 | 96.7 ± 0.2 |
| STC–HSA | 4.32 ± 0.10 | 3.98 ± 0.06 | 4.28 ± 0.04 | 91.8 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fliszár-Nyúl, E.; Faisal, Z.; Skaper, R.; Lemli, B.; Bayartsetseg, B.; Hetényi, C.; Gömbös, P.; Szabó, A.; Poór, M. Interaction of the Emerging Mycotoxins Beauvericin, Cyclopiazonic Acid, and Sterigmatocystin with Human Serum Albumin. Biomolecules 2022, 12, 1106. https://doi.org/10.3390/biom12081106
Fliszár-Nyúl E, Faisal Z, Skaper R, Lemli B, Bayartsetseg B, Hetényi C, Gömbös P, Szabó A, Poór M. Interaction of the Emerging Mycotoxins Beauvericin, Cyclopiazonic Acid, and Sterigmatocystin with Human Serum Albumin. Biomolecules. 2022; 12(8):1106. https://doi.org/10.3390/biom12081106
Chicago/Turabian StyleFliszár-Nyúl, Eszter, Zelma Faisal, Renáta Skaper, Beáta Lemli, Bayarsaikhan Bayartsetseg, Csaba Hetényi, Patrik Gömbös, András Szabó, and Miklós Poór. 2022. "Interaction of the Emerging Mycotoxins Beauvericin, Cyclopiazonic Acid, and Sterigmatocystin with Human Serum Albumin" Biomolecules 12, no. 8: 1106. https://doi.org/10.3390/biom12081106
APA StyleFliszár-Nyúl, E., Faisal, Z., Skaper, R., Lemli, B., Bayartsetseg, B., Hetényi, C., Gömbös, P., Szabó, A., & Poór, M. (2022). Interaction of the Emerging Mycotoxins Beauvericin, Cyclopiazonic Acid, and Sterigmatocystin with Human Serum Albumin. Biomolecules, 12(8), 1106. https://doi.org/10.3390/biom12081106