
Opportunities and Challenges of Data-Driven Virus Discovery

{kind=link}
Abstract
:1. From Technology-Focused to Data-Driven Virus Discovery
2. Opportunities Brought by Data-Driven Virus Discovery
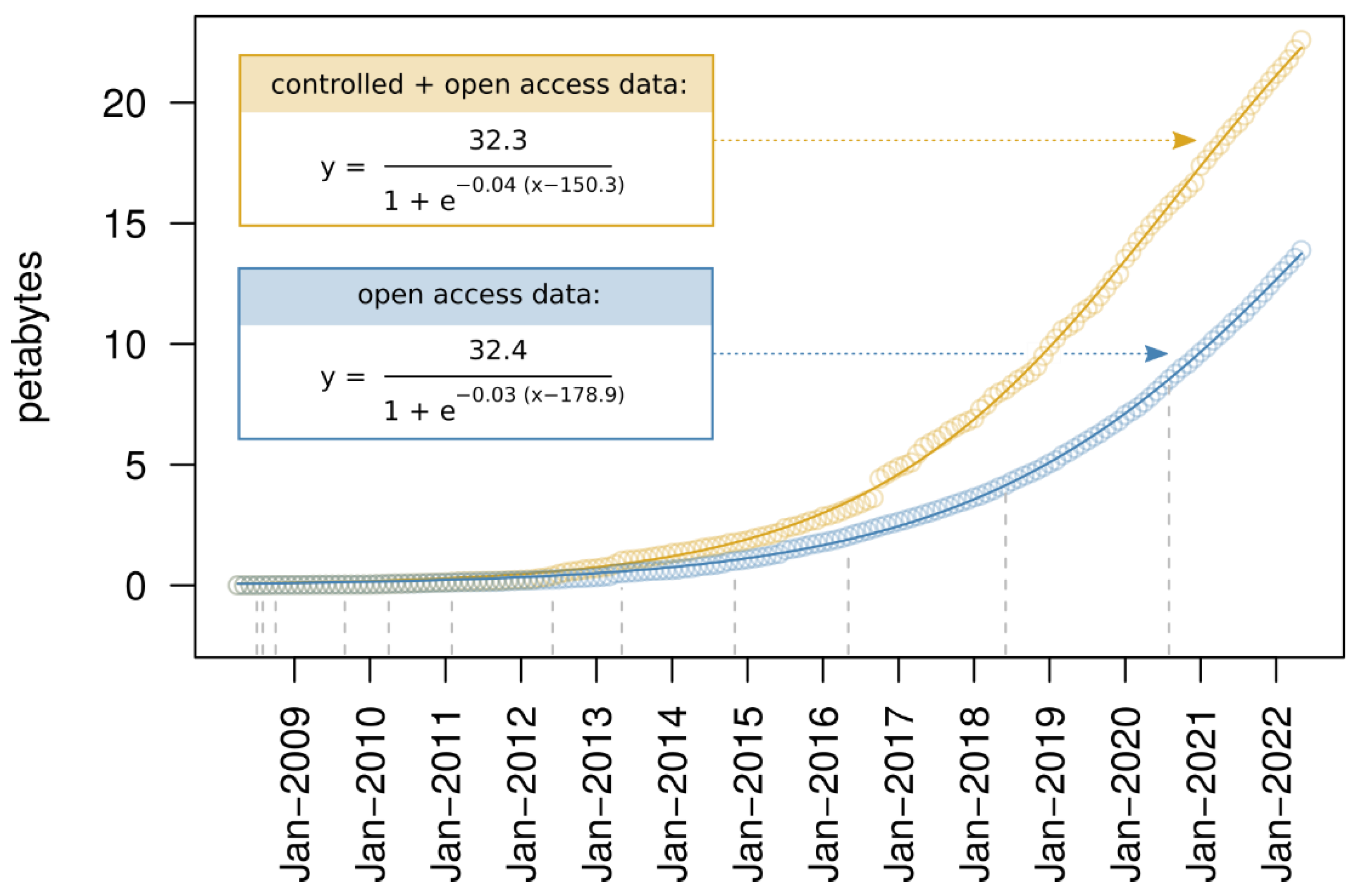
2.1. The SRA as a Unique Source of Viral Sequences
2.2. High-Throughput Mining of Raw Sequencing Data
2.3. Benefits for Biological and Medical Sciences
2.4. Host Assignment
2.5. Data Access and Accuracy
3. Challenges to Be Solved That Facilitate Data-Driven Virus Discovery
3.1. Assembly Quality Standards
3.2. The Value of Incomplete Viral Sequences
3.3. Advancing Assembly Approaches and Tools
3.4. Detection of Highly Divergent Viruses
4. Paradigms for Publication of Data-Driven Virus Discovery Results
4.1. Upgrading the Product of Data-Driven Virus Discovery
4.2. Evidence of Virus Presence and Identity
5. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ivanovsky, D. Über Die Mosaikkrankheit Der Tabakspflanze. Bull. Acad. Imper. Sci. St. Petersburg 1892, 35, 67–70. [Google Scholar]
- Beijerinck, M.W. Über Ein Contagium Vivum Fluidum Als Ursache Der Fleckenkrankheit Der Tabaksblätter. Verh Kon Akad Wetensch 1898, 65, 3–21. [Google Scholar]
- Chamberland, C. A Filter Permitting to Obtain Physiologically Pure Water. Compt. Rend. Acad. Sci. 1884, 99, 247–248. [Google Scholar]
- Löffler, F.; Frosch, P. Summarischer Bericht Über Die Ergebnisse Der Untersuchungen Der Commission Zur Erforschung Der Maul-Und Klauenseuche. Cent. Bakt. Parasit. 1898, 23, 371–391. [Google Scholar]
- Stanley, W.M.; Loring, H.S. The Isolation of Crystalline Tobacco Mosaic Virus Protein from Diseased Tomato Plants. Science 1936, 83, 85. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic Amplification of Beta-Globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Nga, P.T.; Parquet, M.d.C.; Lauber, C.; Parida, M.; Nabeshima, T.; Yu, F.; Thuy, N.T.; Inoue, S.; Ito, T.; Okamoto, K.; et al. Discovery of the First Insect Nidovirus, a Missing Evolutionary Link in the Emergence of the Largest RNA Virus Genomes. PLoS Pathog. 2011, 7, e1002215. [Google Scholar] [CrossRef] [PubMed]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; et al. Re-Assessing the Diversity of Negative Strand RNA Viruses in Insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The Evolutionary History of Vertebrate RNA Viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Hostager, R.; Ryu, D.; Merkel, K.; Angedakin, S.; Arandjelovic, M.; Ayimisin, E.A.; Babweteera, F.; Bessone, M.; Brun-Jeffery, K.J.; et al. Discovery of Novel Herpes Simplexviruses in Wild Gorillas, Bonobos, and Chimpanzees Supports Zoonotic Origin of HSV-2. Mol. Biol. Evol. 2021, 38, 2818–2830. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated Genome and Metagenome Comparative Data Analysis System. Nucleic Acids Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, K.; Mulley, G.; Gulyaeva, A.A.; Zhao, L.; Shu, G.; Jiang, J.; Neuman, B.W. Description and Initial Characterization of Metatranscriptomic Nidovirus-like Genomes from the Proposed New Family Abyssoviridae, and from a Sister Group to the Coronavirinae, the Proposed Genus Alphaletovirus. Virology 2018, 524, 160–171. [Google Scholar] [CrossRef]
- Saberi, A.; Gulyaeva, A.A.; Brubacher, J.L.; Newmark, P.A.; Gorbalenya, A.E. A Planarian Nidovirus Expands the Limits of RNA Genome Size. PLoS Pathog. 2018, 14, e1007314. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-Enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef]
- Lauber, C.; Seifert, M.; Bartenschlager, R.; Seitz, S. Discovery of Highly Divergent Lineages of Plant-Associated Astro-Like Viruses Sheds Light on the Emergence of Potyviruses. Virus Res. 2019, 260, 38–48. [Google Scholar] [CrossRef]
- Tisza, M.J.; Buck, C.B. A Catalog of Tens of Thousands of Viruses from Human Metagenomes Reveals Hidden Associations with Chronic Diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Roux, S.; Paez-Espino, D.; Jungbluth, S.; Walsh, D.A.; Denef, V.J.; McMahon, K.D.; Konstantinidis, K.T.; Eloe-Fadrosh, E.A.; Kyrpides, N.C.; et al. Giant Virus Diversity and Host Interactions through Global Metagenomics. Nature 2020, 578, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Zayed, A.A.; Wainaina, J.M.; Dominguez-Huerta, G.; Pelletier, E.; Guo, J.; Mohssen, M.; Tian, F.; Pratama, A.A.; Bolduc, B.; Zablocki, O.; et al. Cryptic and Abundant Marine Viruses at the Evolutionary Origins of Earth’s RNA Virome. Science 2022, 376, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-Scale Sequence Alignment Catalyses Viral Discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Vaas, J.; Klingler, F.; Mutz, P.; Gorbalenya, A.E.; Bartenschlager, R.; Seitz, S. Deep Mining of the Sequence Read Archive Reveals Bipartite Coronavirus Genomes and Inter-Family Spike Glycoprotein Recombination. bioRxiv 2021. [Google Scholar]
- Neri, U.; Wolf, Y.I.; Roux, S.; Camargo, A.P.; Lee, B.; Kazlauskas, D.; Chen, I.M.; Ivanova, N.; Allen, L.Z.; Paez-Espino, D.; et al. A Five-Fold Expansion of the Global RNA Virome Reveals Multiple New Clades of RNA Bacteriophages. bioRxiv 2022. [Google Scholar] [CrossRef]
- Blackwell, G.A.; Hunt, M.; Malone, K.M.; Lima, L.; Horesh, G.; Alako, B.T.F.; Thomson, N.R.; Iqbal, Z. Exploring Bacterial Diversity via a Curated and Searchable Snapshot of Archived DNA Sequences. PLoS Biol. 2021, 19, e3001421. [Google Scholar] [CrossRef]
- Karasikov, M.; Mustafa, H.; Danciu, D.; Zimmermann, M.; Barber, C.; Rätsch, G.; Kahles, A. MetaGraph: Indexing and Analysing Nucleotide Archives at Petabase-Scale. bioRxiv 2020. [Google Scholar]
- Coclet, C.; Roux, S. Global Overview and Major Challenges of Host Prediction Methods for Uncultivated Phages. Curr. Opin. Virol. 2021, 49, 117–126. [Google Scholar] [CrossRef]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating Viral Sequences in High-Throughput Sequencing Viromics: A Linkage Study of 700 Sequencing Libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef]
- Mitra, A.; Skrzypczak, M.; Ginalski, K.; Rowicka, M. Strategies for Achieving High Sequencing Accuracy for Low Diversity Samples and Avoiding Sample Bleeding Using Illumina Platform. PLoS ONE 2015, 10, e0120520. [Google Scholar] [CrossRef]
- Cobbin, J.C.; Charon, J.; Harvey, E.; Holmes, E.C.; Mahar, J.E. Current Challenges to Virus Discovery by Meta-Transcriptomics. Curr. Opin. Virol. 2021, 51, 48–55. [Google Scholar] [CrossRef]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. ICTV Report Consortium ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F. Complementary DNA Sequencing: Expressed Sequence Tags and Human Genome Project. Science 1991, 252, 1651–1656. [Google Scholar] [CrossRef]
- Steinegger, M.; Mirdita, M.; Söding, J. Protein-Level Assembly Increases Protein Sequence Recovery from Metagenomic Samples Manyfold. Nat. Methods 2019, 16, 603–606. [Google Scholar] [CrossRef]
- Gulyaeva, A.A.; Sigorskih, A.I.; Ocheredko, E.S.; Samborskiy, D.V.; Gorbalenya, A.E. LAMPA, LArge Multidomain Protein Annotator, and Its Application to RNA Virus Polyproteins. Bioinformatics 2020, 36, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef] [PubMed]
- Soding, J. Protein Homology Detection by HMM-HMM Comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-Fast Iterative Protein Sequence Searching by HMM-HMM Alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure Is Three to Ten Times More Conserved than Sequence—A Study of Structural Response in Protein Cores. Proteins 2009, 77, 499–508. [Google Scholar] [CrossRef]
- Beck, J.; Seitz, S.; Lauber, C.; Nassal, M. Conservation of the HBV RNA Element Epsilon in Nackednaviruses Reveals Ancient Origin of Protein-Primed Reverse Transcription. Proc. Natl. Acad. Sci. USA 2021, 118, e2022373118. [Google Scholar] [CrossRef] [PubMed]
- Oberhuber, M.; Schopf, A.; Hennrich, A.A.; Santos-Mandujano, R.; Huhn, A.G.; Seitz, S.; Riedel, C.; Conzelmann, K.-K. Glycoproteins of Predicted Amphibian and Reptile Lyssaviruses Can Mediate Infection of Mammalian and Reptile Cells. Viruses 2021, 13, 1726. [Google Scholar] [CrossRef] [PubMed]
- Bergner, L.M.; Orton, R.J.; Broos, A.; Tello, C.; Becker, D.J.; Carrera, J.E.; Patel, A.H.; Biek, R.; Streicker, D.G. Diversification of Mammalian Deltaviruses by Host Shifting. Proc. Natl. Acad. Sci. USA 2021, 118, e2019907118. [Google Scholar] [CrossRef]
- Feschotte, C.; Gilbert, C. Endogenous Viruses: Insights into Viral Evolution and Impact on Host Biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Gilbert, C.; Feschotte, C. Endogenous Viral Elements: Evolution and Impact. Virologie 2016, 20, 158–173. [Google Scholar] [CrossRef]
- Suh, A.; Weber, C.C.; Kehlmaier, C.; Braun, E.L.; Green, R.E.; Fritz, U.; Ray, D.A.; Ellegren, H. Early Mesozoic Coexistence of Amniotes and Hepadnaviridae. PLoS Genet. 2014, 10, e1004559. [Google Scholar] [CrossRef] [PubMed]
- Barreat, J.G.N.; Katzourakis, A. Paleovirology of the DNA Viruses of Eukaryotes. Trends Microbiol. 2022, 30, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.-Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of Several Thousand Highly Diverse Circular DNA Viruses. eLife 2020, 9, e51971. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauber, C.; Seitz, S. Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules 2022, 12, 1073. https://doi.org/10.3390/biom12081073
Lauber C, Seitz S. Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules. 2022; 12(8):1073. https://doi.org/10.3390/biom12081073
Chicago/Turabian StyleLauber, Chris, and Stefan Seitz. 2022. "Opportunities and Challenges of Data-Driven Virus Discovery" Biomolecules 12, no. 8: 1073. https://doi.org/10.3390/biom12081073
APA StyleLauber, C., & Seitz, S. (2022). Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules, 12(8), 1073. https://doi.org/10.3390/biom12081073