
GraphSite: Ligand Binding Site Classification with Deep Graph Learning

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets of Ligand Binding Pockets
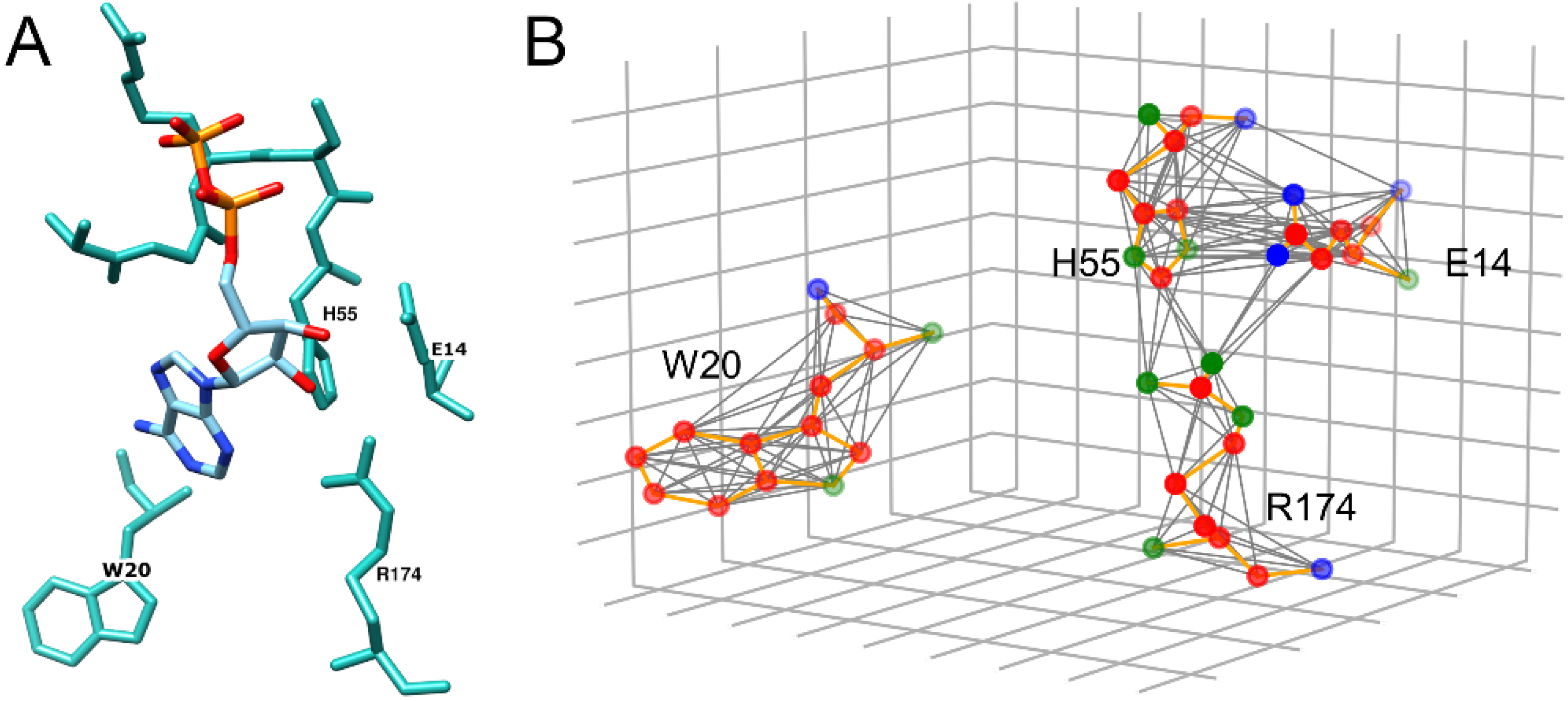
2.2. Graph Representation of Binding Sites
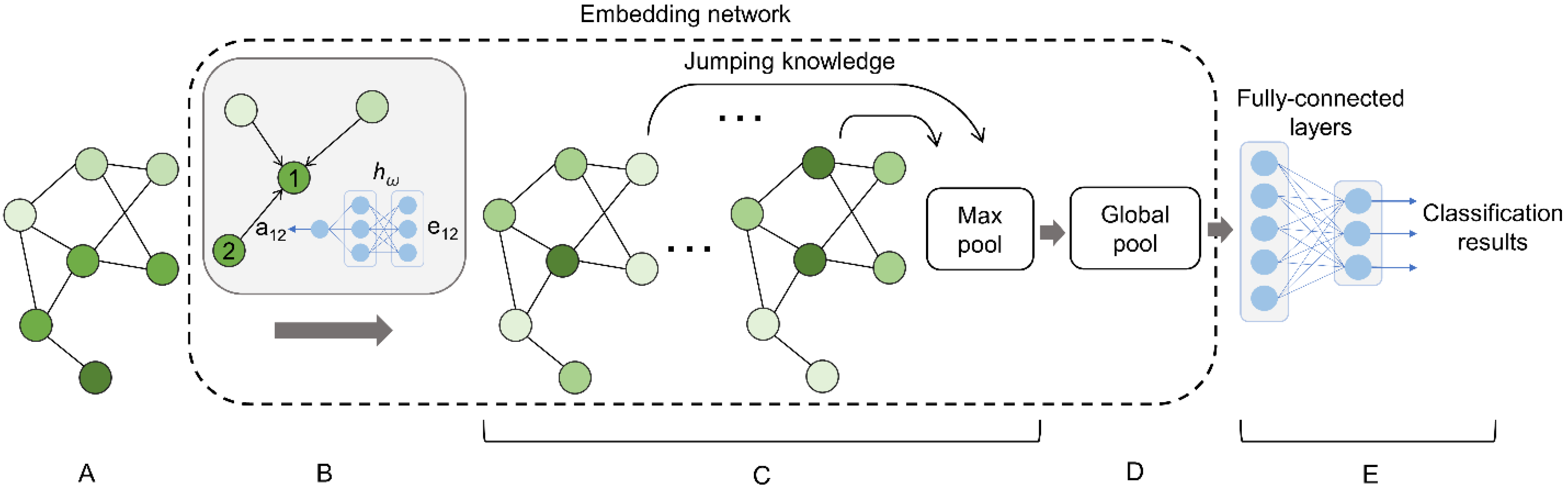
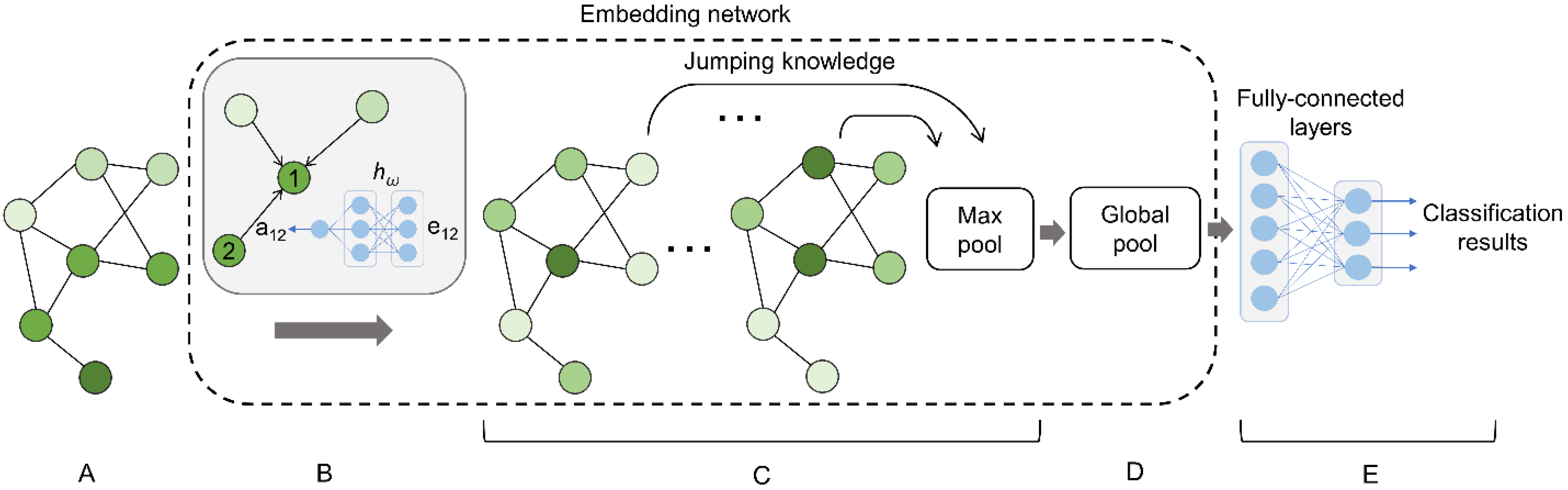
2.3. Graph Neural Network
2.3.1. Message Passing
2.3.2. Graph Readout
2.3.3. Loss Function
2.4. Other Methods to Classify Pockets
3. Results
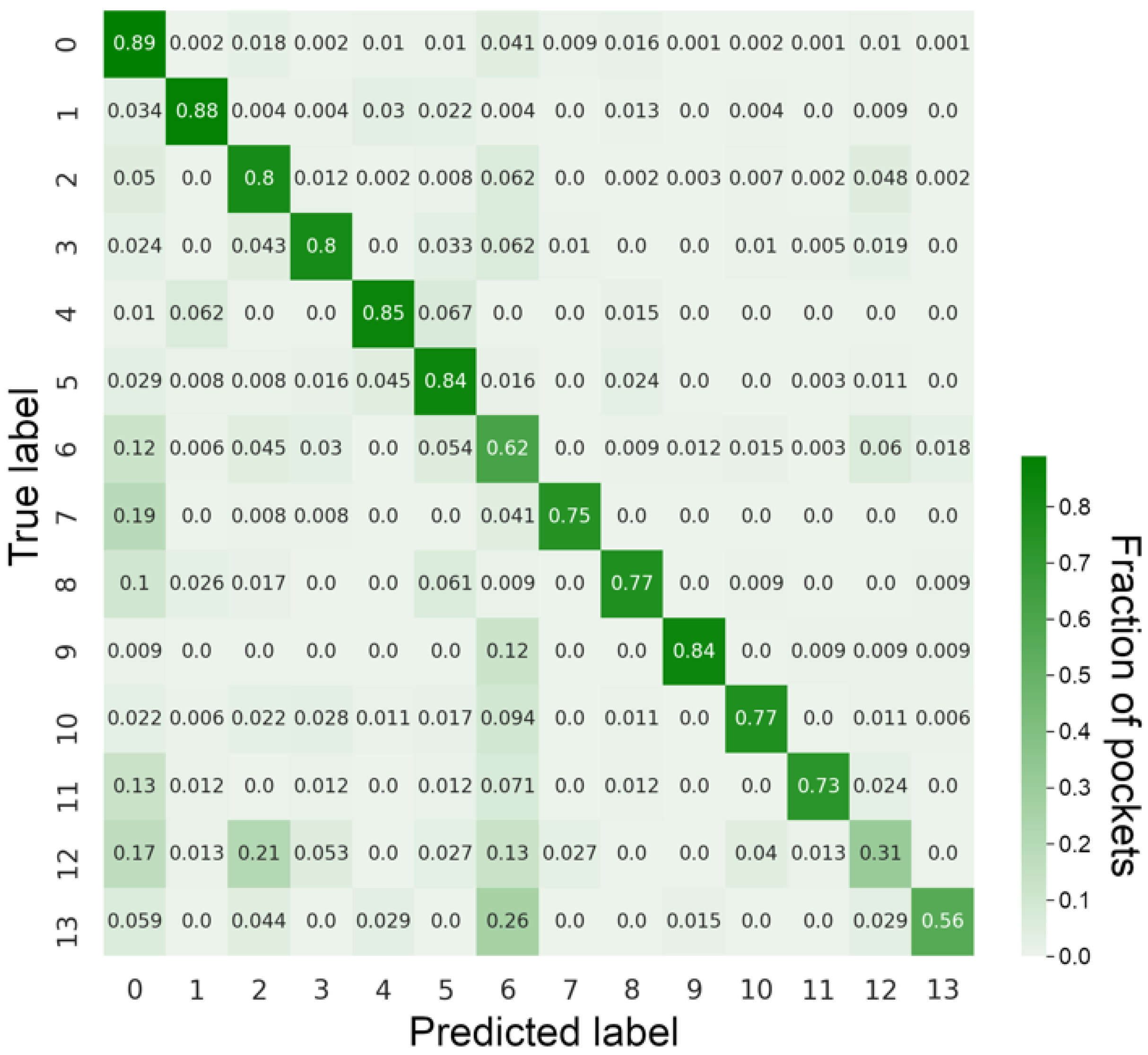
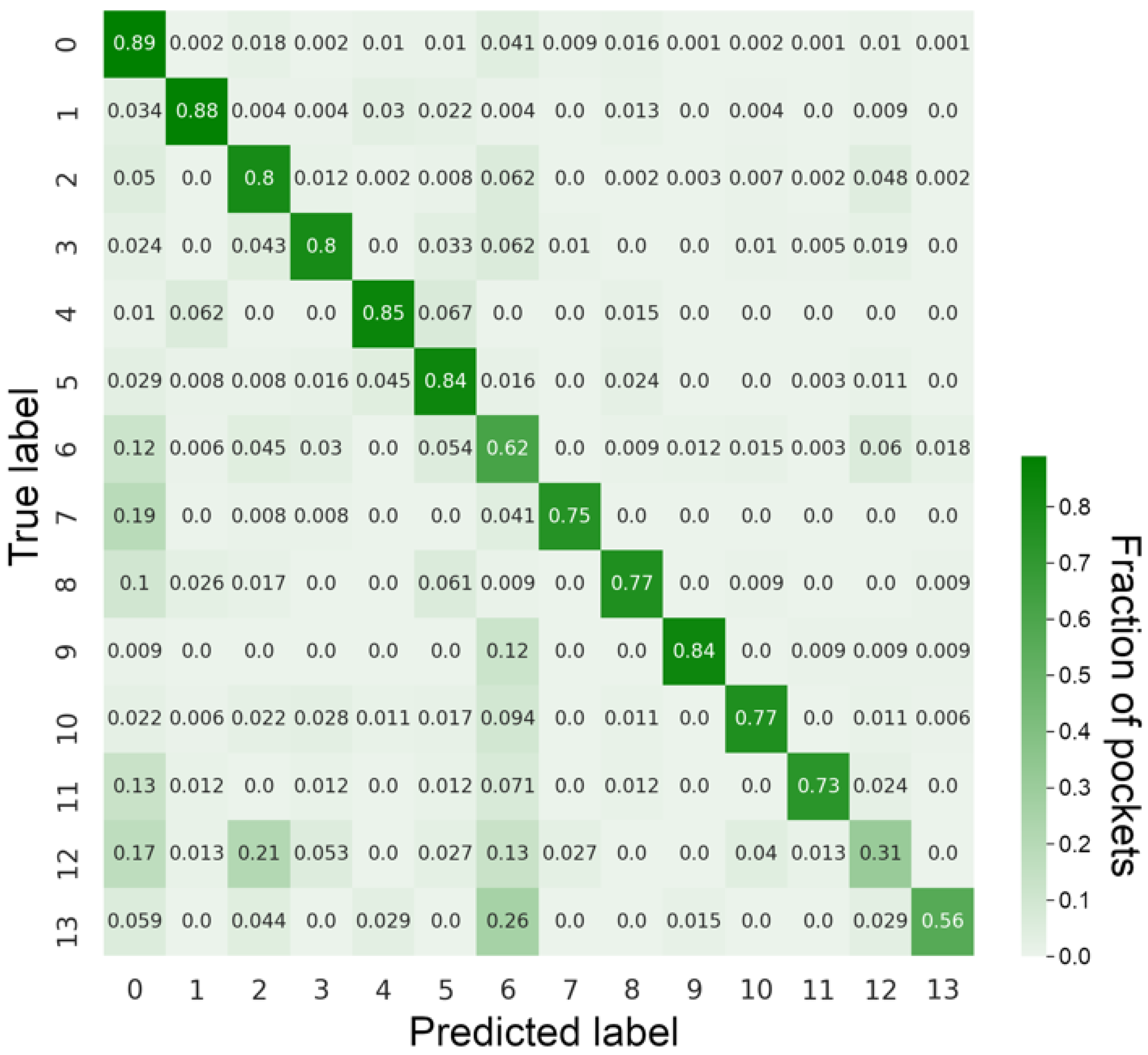
3.1. Classification Performance against the Benchmarking Dataset
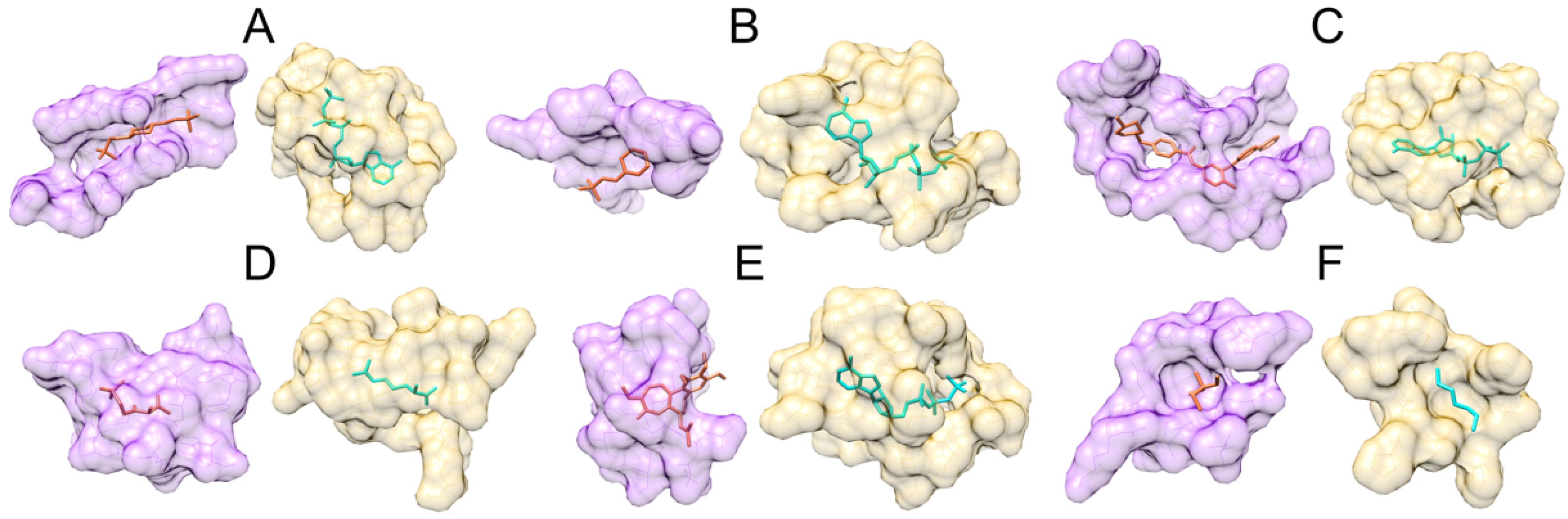
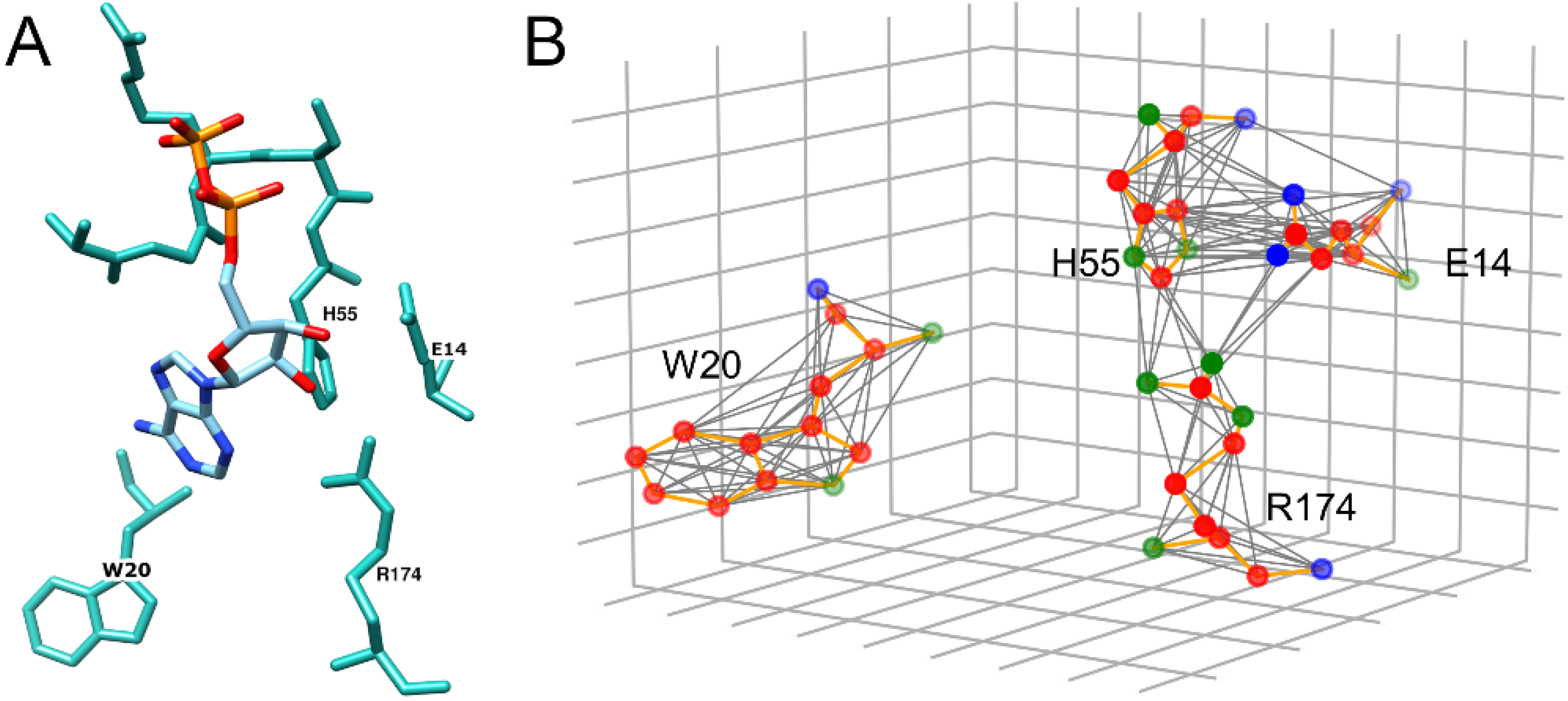
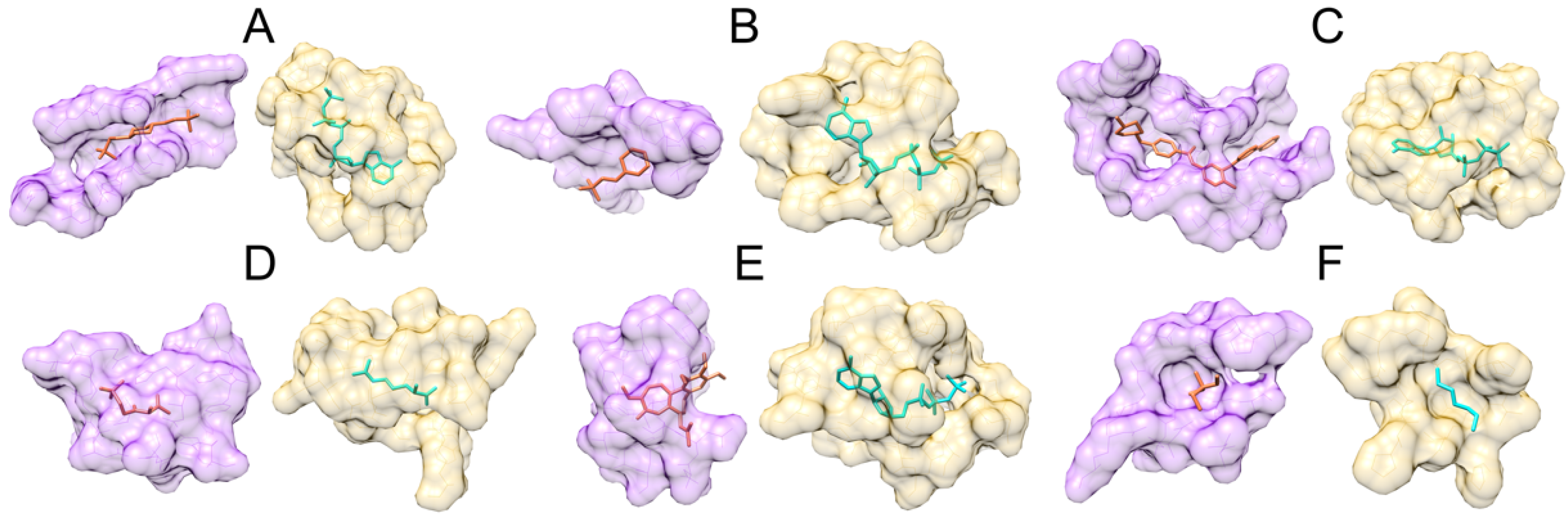
3.2. Examples of Misclassified Pockets
3.3. Performance on Unseen Data
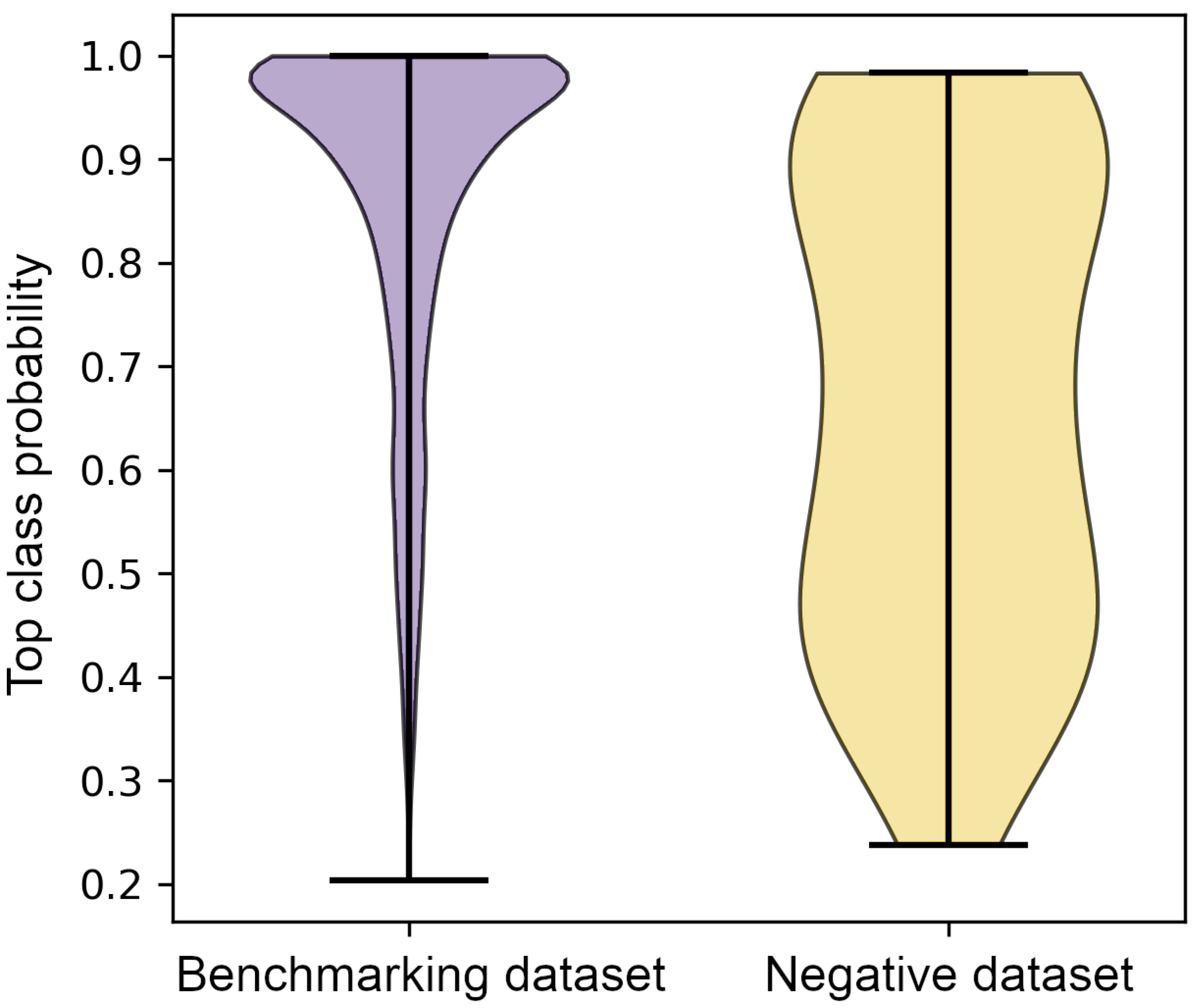
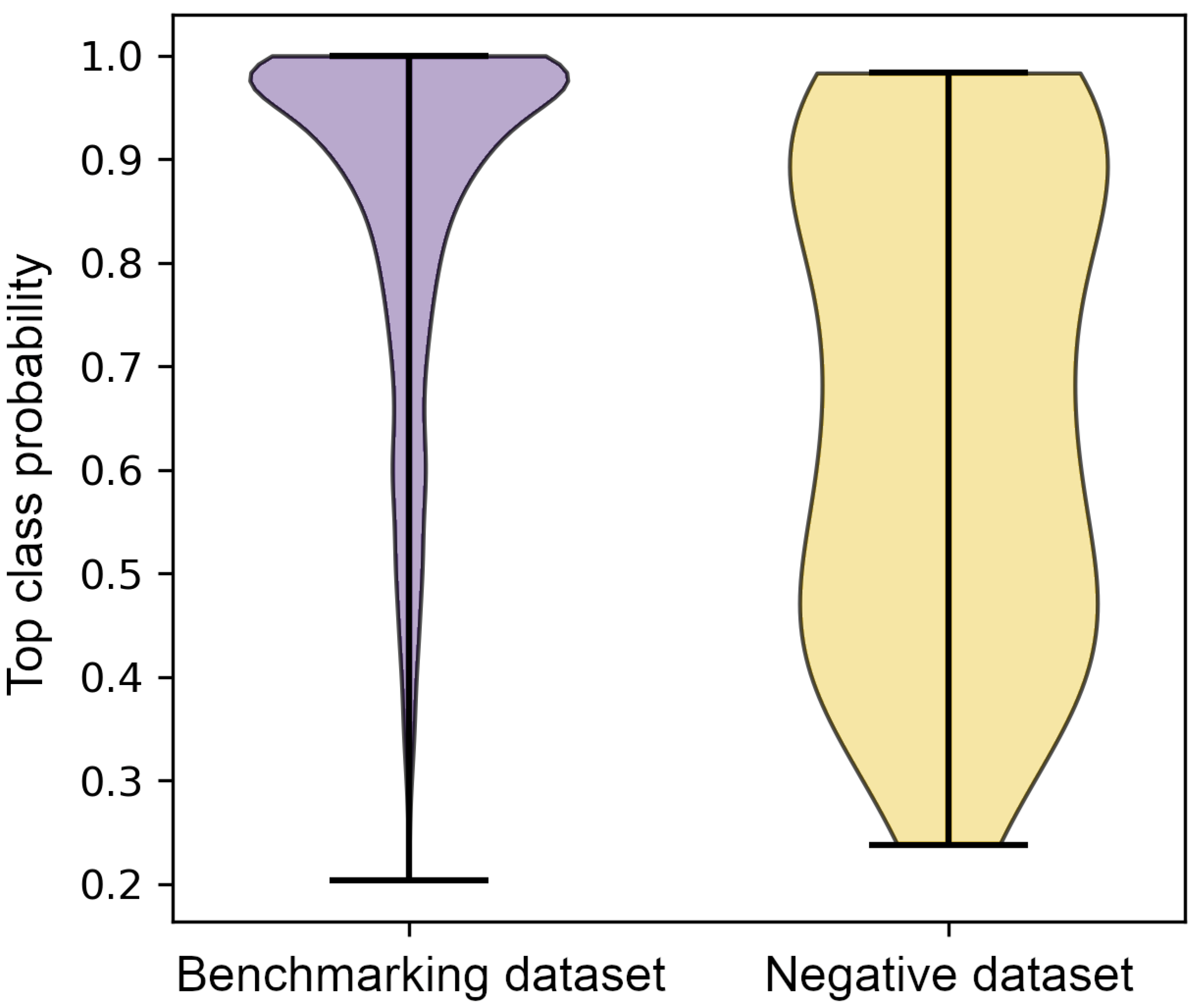
3.4. Classification of the Negative Dataset
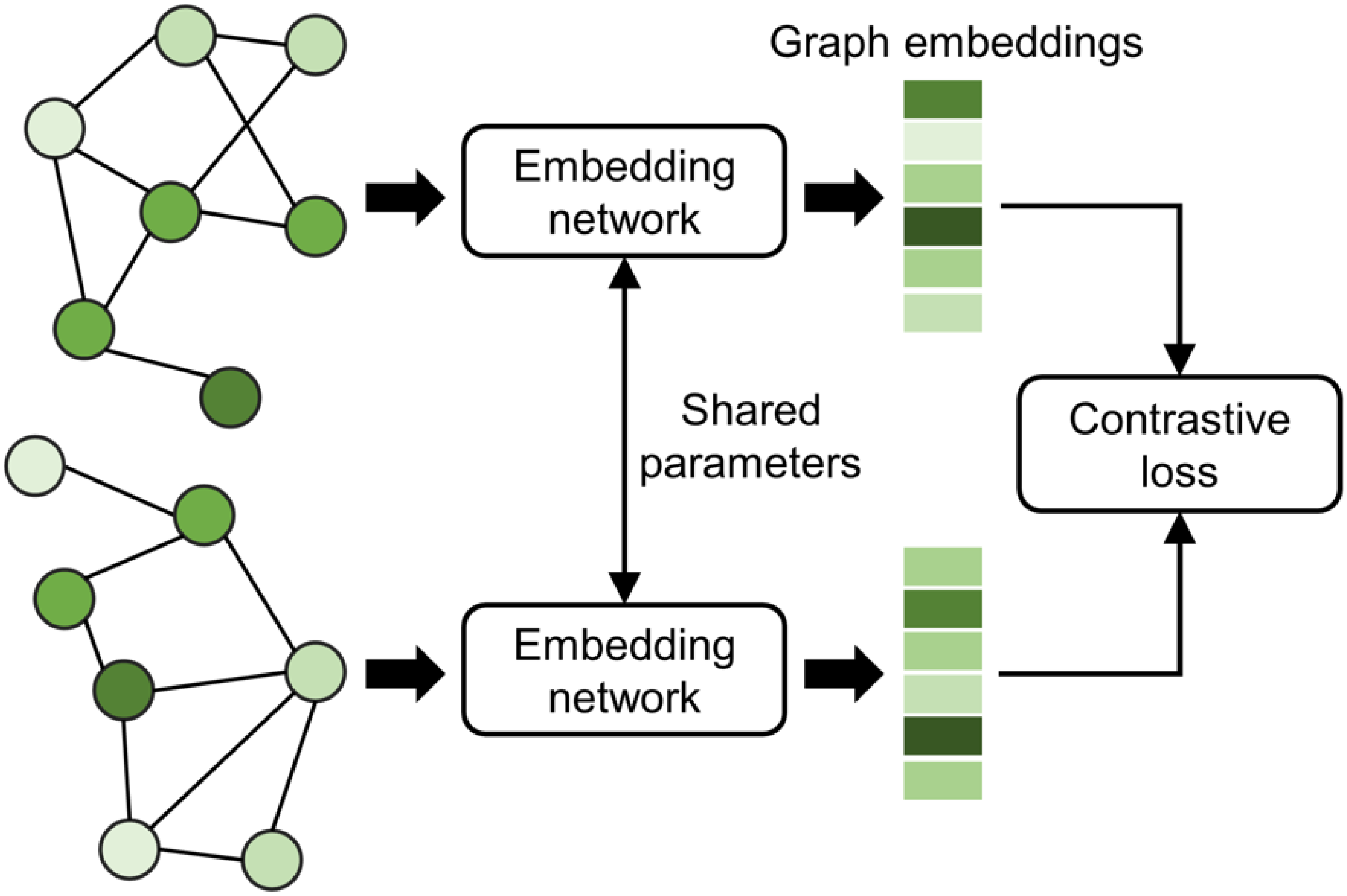
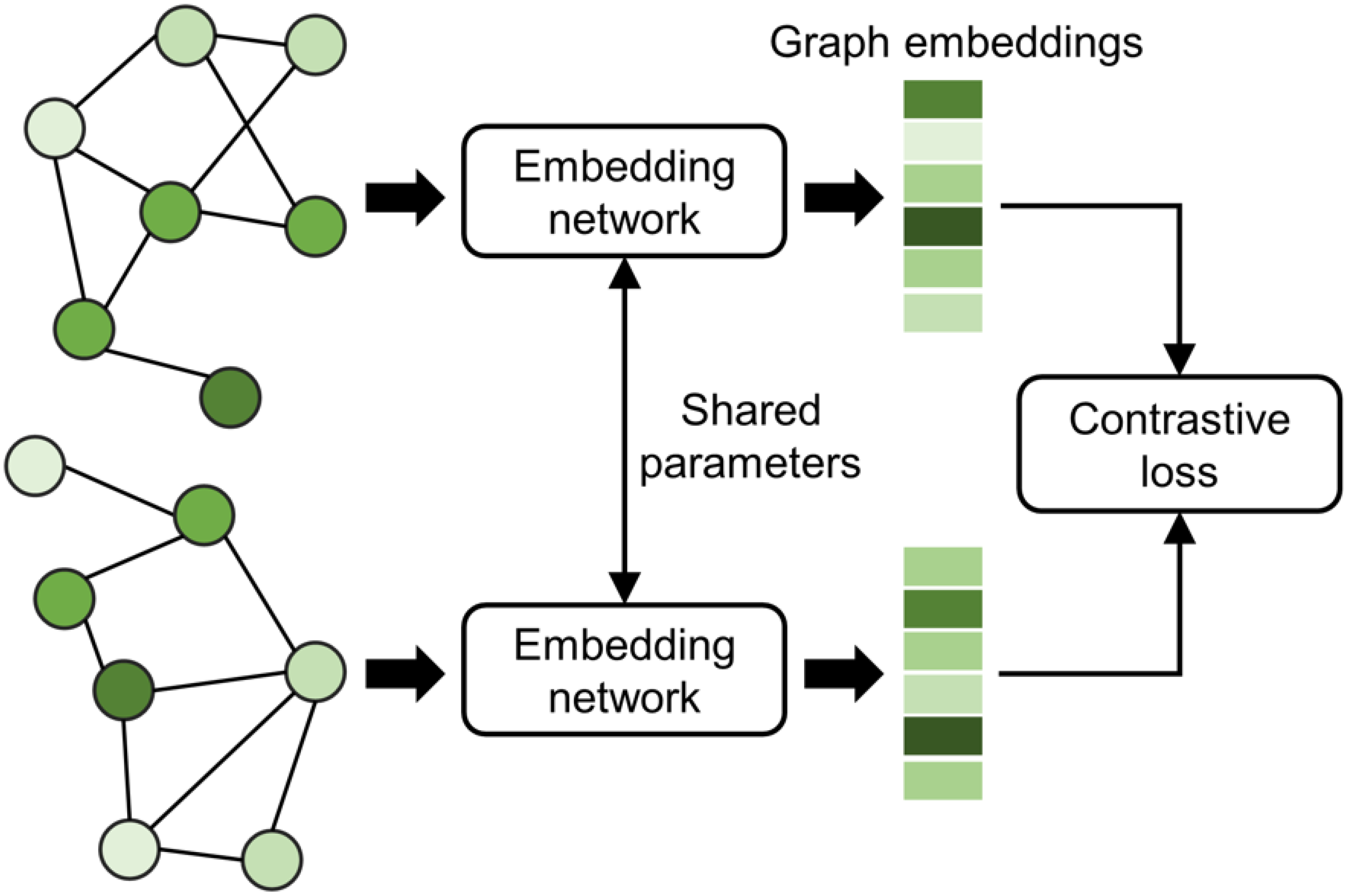
3.5. Siamese-GraphSite Extension
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armstrong, J.D.; Hubbard, R.E.; Farrell, T.; Maiguashca, B. (Eds.) Structure-Based Drug Discovery: An Overview; The Royal Society of Chemistry: Cambridge, UK, 2006. [Google Scholar]
- Roche, D.B.; Brackenridge, D.A.; McGuffin, L.J. Proteins and Their Interacting Partners: An Introduction to Protein–Ligand Binding Site Prediction Methods. Int. J. Mol. Sci. 2015, 16, 29829–29842. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Naderi, M.; Singha, M.; Lemoine, J.; Brylinski, M. Large-scale computational drug repositioning to find treatments for rare diseases. npj Syst. Biol. Appl. 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Ngan, C.-H.; Hall, D.R.; Zerbe, B.S.; Grove, L.E.; Kozakov, D.; Vajda, S. FTSite: High accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2011, 28, 286–287. [Google Scholar] [CrossRef]
- Brylinski, M.; Feinstein, W.P. eFindSite: Improved prediction of ligand binding sites in protein models using meta-threading, machine learning and auxiliary ligands. J. Comput. Mol. Des. 2013, 27, 551–567. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Yoo, S.; Sanchez, R. SiteComp: A server for ligand binding site analysis in protein structures. Bioinformatics 2012, 28, 1172–1173. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Iwata, H.; Ma, B.; Fujita, A.; Terayama, K.; Sagae, Y.; Ono, F.; Tsuda, K.; Kamiya, N.; Okuno, Y. Improving the Accuracy of Protein-Ligand Binding Mode Prediction Using a Molecular Dynamics-Based Pocket Generation Approach. J. Comput. Chem. 2018, 39, 2679–2689. [Google Scholar] [CrossRef]
- Kana, O.; Brylinski, M. Elucidating the druggability of the human proteome with eFindSite. J. Comput. Mol. Des. 2019, 33, 509–519. [Google Scholar] [CrossRef]
- Ngan, C.H.; Bohnuud, T.; Mottarella, S.E.; Beglov, D.; Villar, E.A.; Hall, D.R.; Kozakov, D.; Vajda, S. FTMAP: Extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 2012, 40, W271–W275. [Google Scholar] [CrossRef]
- Skolnick, J.; Gao, M.; Roy, A.; Srinivasan, B.; Zhou, H. Implications of the small number of distinct ligand binding pockets in proteins for drug discovery, evolution and biochemical function. Bioorganic Med. Chem. Lett. 2015, 25, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Brylinski, M. Local Alignment of Ligand Binding Sites in Proteins for Polypharmacology and Drug Repositioning. In Protein Function Prediction; Humana Press: New York, NY, USA, 2017; Volume 1611, pp. 109–122. [Google Scholar] [CrossRef] [Green Version]
- Ehrt, C.; Brinkjost, T.; Koch, O. Impact of Binding Site Comparisons on Medicinal Chemistry and Rational Molecular Design. J. Med. Chem. 2016, 59, 4121–4151. [Google Scholar] [CrossRef]
- Naderi, M.; Lemoine, J.M.; Govindaraj, R.G.; Kana, O.Z.; Feinstein, W.P.; Brylinski, M. Binding site matching in rational drug design: Algorithms and applications. Briefings Bioinform. 2018, 20, 2167–2184. [Google Scholar] [CrossRef]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.-C.; Brylinski, M. DeepDrug3D: Classification of ligand-binding pockets in proteins with a convolutional neural network. PLOS Comput. Biol. 2019, 15, e1006718. [Google Scholar] [CrossRef] [Green Version]
- Konc, J.; Janežič, D. ProBiS-ligands: A web server for prediction of ligands by examination of protein binding sites. Nucleic Acids Res. 2014, 42, W215–W220. [Google Scholar] [CrossRef] [Green Version]
- Panwar, B.; Gupta, S.; Raghava, G.P.S. Prediction of vitamin interacting residues in a vitamin binding protein using evolutionary information. BMC Bioinform. 2013, 14, 44. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Mizianty, M.J.; Kurgan, L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics 2011, 28, 331–341. [Google Scholar] [CrossRef]
- Liu, R.; Hu, J. HemeBIND: A novel method for heme binding residue prediction by combining structural and sequence information. BMC Bioinform. 2011, 12, 207. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.K.; Raghava, G.P. Prediction of FAD interacting residues in a protein from its primary sequence using evolutionary information. BMC Bioinform. 2010, 11, S48. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, J.S.; Mishra, N.K.; Raghava, G.P. Prediction of GTP interacting residues, dipeptides and tripeptides in a protein from its evolutionary information. BMC Bioinform. 2010, 11, 301. [Google Scholar] [CrossRef] [Green Version]
- Ansari, H.R.; Raghava, G.P. Identification of NAD interacting residues in proteins. BMC Bioinform. 2010, 11, 160. [Google Scholar] [CrossRef] [Green Version]
- Horst, J.A.; Samudrala, R. A protein sequence meta-functional signature for calcium binding residue prediction. Pattern Recognit. Lett. 2010, 31, 2103–2112. [Google Scholar] [CrossRef] [Green Version]
- Shu, N.; Zhou, T.; Hovmöller, S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics 2008, 24, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Gkioxari, G.; Dollár, P.; Girshick, R. Mask r-cnn. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017. [Google Scholar]
- Schroff, F.; Kalenichenko, D.; Philbin, J. Facenet: A unified embedding for face recognition and clustering. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015. [Google Scholar]
- Wei, S.E.; Ramakrishna, V.; Kanade, T.; Sheikh, Y. Convolutional pose machines. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016. [Google Scholar]
- Bronstein, M.M.; Bruna, J.; LeCun, Y.; Szlam, A.; Vandergheynst, P. Geometric Deep Learning: Going beyond Euclidean data. IEEE Signal Process. Mag. 2017, 34, 18–42. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Lemoine, J.M.; Shawky, A.-E.; Singha, M.; Pu, L.; Yang, S.; Ramanujam, J.; Brylinski, M. BionoiNet: Ligand-binding site classification with off-the-shelf deep neural network. Bioinformatics 2020, 36, 3077–3083. [Google Scholar] [CrossRef] [PubMed]
- Simonovsky, M.; Meyers, J. DeeplyTough: Learning Structural Comparison of Protein Binding Sites. J. Chem. Inf. Model. 2020, 60, 2356–2366. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [Green Version]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Hamilton, W.; Ying, Z.; Leskovec, J. Inductive representation learning on large graphs. In Proceedings of the 31st Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Li, Y.; Yu, R.; Shahabi, C.; Liu, Y. Diffusion convolutional recurrent neural network: Data-driven traffic forecasting. arXiv 2017, arXiv:1707.01926. [Google Scholar]
- Sanchez-Gonzalez, A.; Godwin, J.; Pfaff, T.; Ying, R.; Leskovec, J.; Battaglia, P.W. Learning to simulate complex physics with graph networks. In International Conference on Machine Learning; PMLR: London, UK, 2020. [Google Scholar]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 1263–1272. [Google Scholar]
- Duvenaud, D.K.; Maclaurin, D.; Iparraguirre, J.; Bombarell, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional networks on graphs for learning molecular fingerprints. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: New York, NY, USA, 2015. [Google Scholar]
- Fout, A.; Byrd, J.; Shariat, B.; Ben-Hur, A. Protein interface prediction using graph convolutional networks. In Proceedings of the 31st Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef]
- Yuan, Q.; Chen, S.; Rao, J.; Zheng, S.; Zhao, H.; Yang, Y. AlphaFold2-aware protein–DNA binding site prediction using graph transformer. Briefings Bioinform. 2022, 23, bbab564. [Google Scholar] [CrossRef]
- Feinstein, W.P.; Brylinski, M. eFindSite: Enhanced Fingerprint-Based Virtual Screening Against Predicted Ligand Binding Sites in Protein Models. Mol. Inform. 2014, 33, 135–150. [Google Scholar] [CrossRef]
- Voigt, J.H.; Bienfait, B.; Wang, S.; Nicklaus, M.C. Comparison of the NCI Open Database with Seven Large Chemical Structural Databases. J. Chem. Inf. Comput. Sci. 2001, 41, 702–712. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.C.A.; Nassif, H.; Page, D.; Muggleton, S.H.; Sternberg, M.J.E. Automated identification of protein-ligand interaction features using Inductive Logic Programming: A hexose binding case study. BMC Bioinform. 2012, 13, 162. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, V.; Sorokin, A.; Prilusky, J.; Abola, E.E.; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999, 15, 327–332. [Google Scholar] [CrossRef]
- Feinstein, J.; Shi, W.; Ramanujam, J.; Brylinski, M. Bionoi: A Voronoi Diagram-Based Representation of Ligand-Binding Sites in Proteins for Machine Learning Applications. Methods Mol. Biol. 2021, 2266, 299–312. [Google Scholar] [CrossRef]
- Xu, K.; Li, C.; Tian, Y.; Sonobe, T.; Kawarabayashi, K.-I.; Jegelka, S. Representation learning on graphs with jumping knowledge networks. arXiv 2018, arXiv:1806.03536. [Google Scholar]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How powerful are graph neural networks? arXiv 2018, arXiv:1810.00826. [Google Scholar]
- Corso, G.; Cavalleri, L.; Beaini, D.; Liò, P.; Veličković, P. Principal neighbourhood aggregation for graph nets. arXiv 2020, arXiv:2004.05718. [Google Scholar]
- Vinyals, O.; Bengio, S.; Kudlur, M. Order matters: Sequence to sequence for sets. arXiv 2015, arXiv:1511.06391. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Goyal, P.; Girshick, R.; He, K.; Dollár, P. Focal loss for dense object detection. In Proceedings of the IEEE International Conference on Computer Vision, 23–27 July 2018. [Google Scholar]
- Koes, D.R.; Baumgartner, M.; Camacho, C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Lee, H.S.; Im, W. G-LoSA: An efficient computational tool for local structure-centric biological studies and drug design. Protein Sci. 2016, 25, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Good, N.E.; Winget, G.D.; Winter, W.; Connolly, T.N.; Izawa, S.; Singh, R.M.M. Hydrogen Ion Buffers for Biological Research. Biochemistry 1966, 5, 467–477. [Google Scholar] [CrossRef]
- Garcia-Saez, I.; Yen, T.; Wade, R.H.; Kozielski, F. Crystal Structure of the Motor Domain of the Human Kinetochore Protein CENP-E. J. Mol. Biol. 2004, 340, 1107–1116. [Google Scholar] [CrossRef]
- Velankar, S.S.; Best, C.; Beuth, B.; Boutselakis, C.H.; Cobley, N.; da Silva, A.W.S.; Dimitropoulos, D.; Golovin, A.; Hirshberg, M.; John, M.; et al. PDBe: Protein Data Bank in Europe. Nucleic Acids Res. 2009, 38, D308–D317. [Google Scholar] [CrossRef] [PubMed]
- Yeturu, K.; Chandra, N. PocketAlign A Novel Algorithm for Aligning Binding Sites in Protein Structures. J. Chem. Inf. Model. 2011, 51, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Parkin, A.; Oswald, I.D.; Parsons, S. Structures of piperazine, piperidine and morpholine. Acta. Cryst. B 2004, 60, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Song, Y.; Liu, H.; Yang, Y.; Wang, S.; Yi, C.; Chen, P.R. Allosteric histidine switch for regulation of intracellular zinc(II) fluctuation. Proc. Natl. Acad. Sci. USA 2017, 114, 13661–13666. [Google Scholar] [CrossRef] [Green Version]
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Moulton, J.D. Morpholino Antisense Oligos. Available online: https://www.gene-tools.com/morpholino_antisense_oligos (accessed on 28 February 2022).
- Zask, A.; Kaplan, J.; Verheijen, J.C.; Richard, D.J.; Curran, K.; Brooijmans, N.; Bennett, E.M.; Toral-Barza, L.; Hollander, I.; Ayral-Kaloustian, S.; et al. Morpholine Derivatives Greatly Enhance the Selectivity of Mammalian Target of Rapamycin (mTOR) Inhibitors. J. Med. Chem. 2009, 52, 7942–7945. [Google Scholar] [CrossRef]
- Avendaño, C.; Menendez, J.C. Drugs That Inhibit Signalling Pathways for Tumor Cell Growth and Proliferation. Med. Chem. Anticancer. Drugs 2008, 251–305. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, W.-Z.; Li, Y.; Zhou, G.-L.; Liu, X.-G. Recent development of ATP-competitive small molecule phosphatidylinostitol-3-kinase inhibitors as anticancer agents. Oncotarget 2016, 8, 7181–7200. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.J. Chapter 1—Heterocycles and Medicine: A Survey of the Heterocyclic Drugs Approved by the U.S. FDA from 2000 to Present. In Progress in Heterocyclic Chemistry; Gordon, W.G., John, A.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 24, pp. 1–53. [Google Scholar]
- Wilson, C.; Agafonov, R.V.; Hoemberger, M.; Kutter, S.; Zorba, A.; Halpin, J.; Buosi, V.; Otten, R.; Waterman, D.; Theobald, D.L.; et al. Using ancient protein kinases to unravel a modern cancer drug’s mechanism. Science 2015, 347, 882–886. [Google Scholar] [CrossRef]
- Marques, J.C.; Oh, I.K.; Ly, D.C.; Lamosa, P.; Ventura, M.R.; Miller, S.T.; Xavier, K.B. LsrF, a coenzyme A-dependent thiolase, catalyzes the terminal step in processing the quorum sensing signal autoinducer-2. Proc. Natl. Acad. Sci. USA 2014, 111, 14235–14240. [Google Scholar] [CrossRef] [Green Version]
- Humm, A.E.A. Crystal structure and mechanism of human L-arginine:glycine amidinotransferase: A mitochondrial enzyme involved in creatine biosynthesis. EMBO J. 1997, 16, 3373–3385. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Stryer, L. Amino acids are made from intermediates of the citric acid cycle and other major pathways. In Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Purine bases can be synthesized de novo or recycled by salvage pathways. In Biochemistry; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Probenecid and Colchicine Tablets. USP Rx Only. Available online: https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=842dd93d-54e6-43b8-8bd5-d135fc5a3400&type=display (accessed on 28 February 2022).
- Yokoyama, T.; Matsumoto, K.; Ostermann, A.; Schrader, T.E.; Nabeshima, Y.; Mizuguchi, M. Structural and thermodynamic characterization of the binding of isoliquiritigenin to the first bromodomain of BRD4. FEBS J. 2018, 286, 1656–1667. [Google Scholar] [CrossRef]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef]
- Denis, G.V.; Vaziri, C.; Guo, N.; Faller, U.V. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 2000, 11, 417–424. [Google Scholar]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zada, B.; Joo, S.; Wang, C.; Tseten, T.; Jeong, S.-H.; Seo, H.; Sohn, J.-H.; Kim, K.-J.; Kim, S.-W. Metabolic engineering of Escherichia coli for production of non-natural acetins from glycerol. Green Chem. 2020, 22, 7788–7802. [Google Scholar] [CrossRef]
- Brand, B.; Boos, W. Maltose transacetylase of Escherichia coli. Mapping and cloning of its structural, gene, mac, and characterization of the enzyme as a dimer of identical polypeptides with a molecular weight of 20,000. J. Biol. Chem. 1991, 266, 14113–14118. [Google Scholar] [CrossRef]
- Kelly, D.J.; Hughes, N.J. The citric acid cycle and fatty acid biosynthesis. In Helicobacter Pylori: Physiology and Genetics; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Kuzin, A.; Su, M.; Seetharaman, J.; Mao, M.; Xiao, R.; Ciccosanti, C.; Lee, D.; Everett, J.K.; Nair, R.; Acton, T.B.; et al. Northeast Structural Genomics Consortium Target BtR318A; Northeast Structural Genomics Consortium (NESG), National Institutes of Health (NIH): Bethesda, MD, USA, 2010.
- Hadsell, R.; Chopra, S.; LeCun, Y. Dimensionality reduction by learning an invariant mapping. In Proceedings of the 2006 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’06), New York, NY, USA, 17–22 June 2006. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Fix, E.; Hodges, J.L. Discriminatory analysis. Nonparametric discrimination: Consistency properties. Int. Stat. Rev./Rev. Int. Stat. 1989, 57, 238–247. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
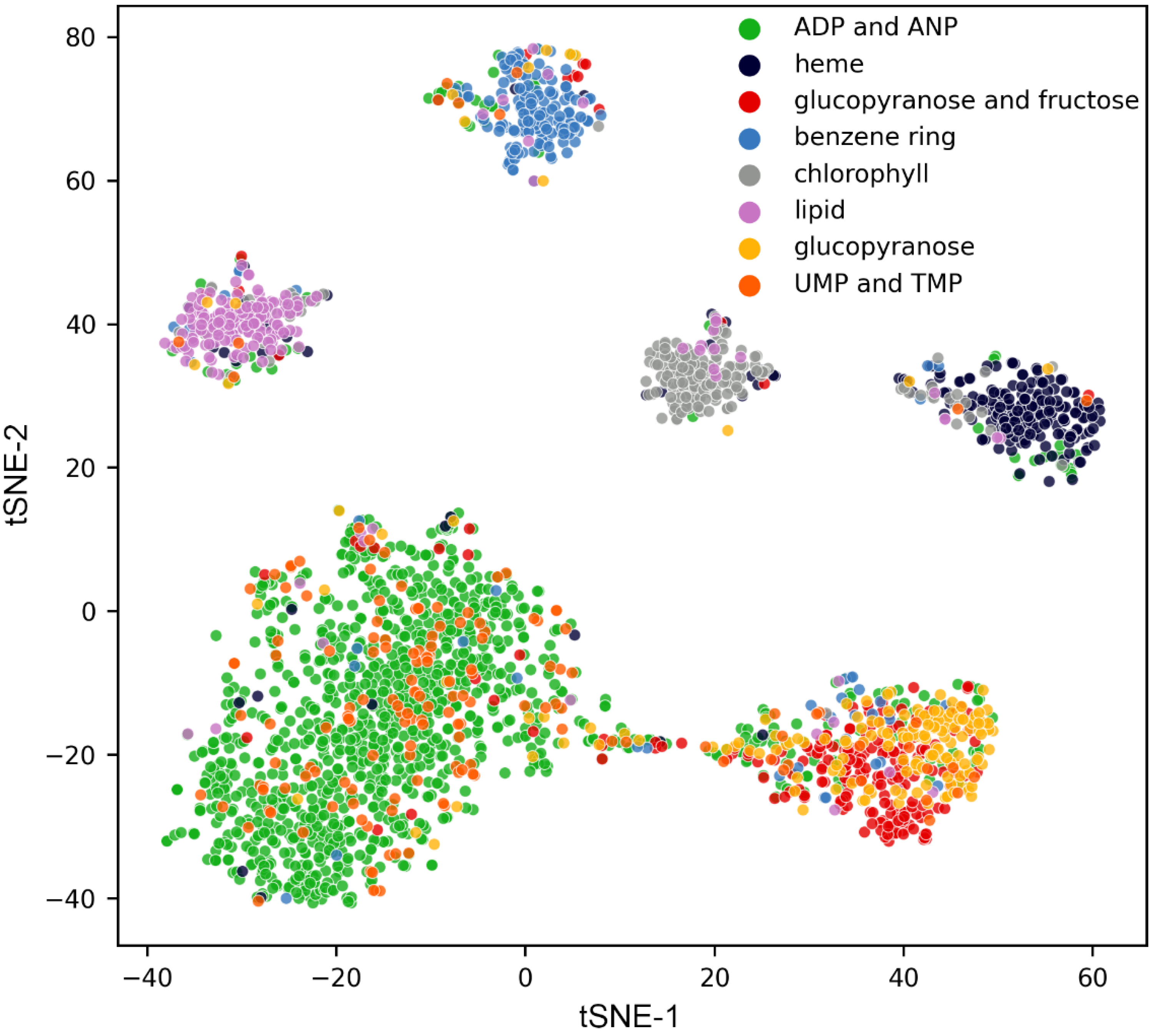{kind=link}
| Class | Binding Ligands | Support |
|---|---|---|
| 0 | nucleotide | 7625 |
| 1 | heme | 1158 |
| 2 | carbohydrate | 3001 |
| 3 | benzene ring | 1054 |
| 4 | chlorophyll | 968 |
| 5 | lipid | 1890 |
| 6 | essential amino/citric/tartaric acids | 1663 |
| 7 | S-adenosyl-L-homocysteine | 602 |
| 8 | coenzyme A | 573 |
| 9 | pyridoxal phosphate | 566 |
| 10 | benzoic acid | 897 |
| 11 | flavin mononucleotide | 417 |
| 12 | morpholine ring | 374 |
| 13 | phosphate | 337 |
| Method | Recall | Precision | F1-Score |
|---|---|---|---|
| GraphSite | 81.3% | 82. 3% | 81.7% |
| GIN | 75.1% | 74.3% | 74.3% |
| Smina | 16.7% | 43.4% | 16.1% |
| G-LoSA | 14.8% | 34.4% | 15.9% |
| Random | 17.8% | 17.7% | 17.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, W.; Singha, M.; Pu, L.; Srivastava, G.; Ramanujam, J.; Brylinski, M. GraphSite: Ligand Binding Site Classification with Deep Graph Learning. Biomolecules 2022, 12, 1053. https://doi.org/10.3390/biom12081053
Shi W, Singha M, Pu L, Srivastava G, Ramanujam J, Brylinski M. GraphSite: Ligand Binding Site Classification with Deep Graph Learning. Biomolecules. 2022; 12(8):1053. https://doi.org/10.3390/biom12081053
Chicago/Turabian StyleShi, Wentao, Manali Singha, Limeng Pu, Gopal Srivastava, Jagannathan Ramanujam, and Michal Brylinski. 2022. "GraphSite: Ligand Binding Site Classification with Deep Graph Learning" Biomolecules 12, no. 8: 1053. https://doi.org/10.3390/biom12081053
APA StyleShi, W., Singha, M., Pu, L., Srivastava, G., Ramanujam, J., & Brylinski, M. (2022). GraphSite: Ligand Binding Site Classification with Deep Graph Learning. Biomolecules, 12(8), 1053. https://doi.org/10.3390/biom12081053