
α-Synuclein Conformational Plasticity: Physiologic States, Pathologic Strains, and Biotechnological Applications



Abstract
1. Introduction
2. Physiologic States
2.1. Monomer
2.1.1. Structure
2.1.2. Function
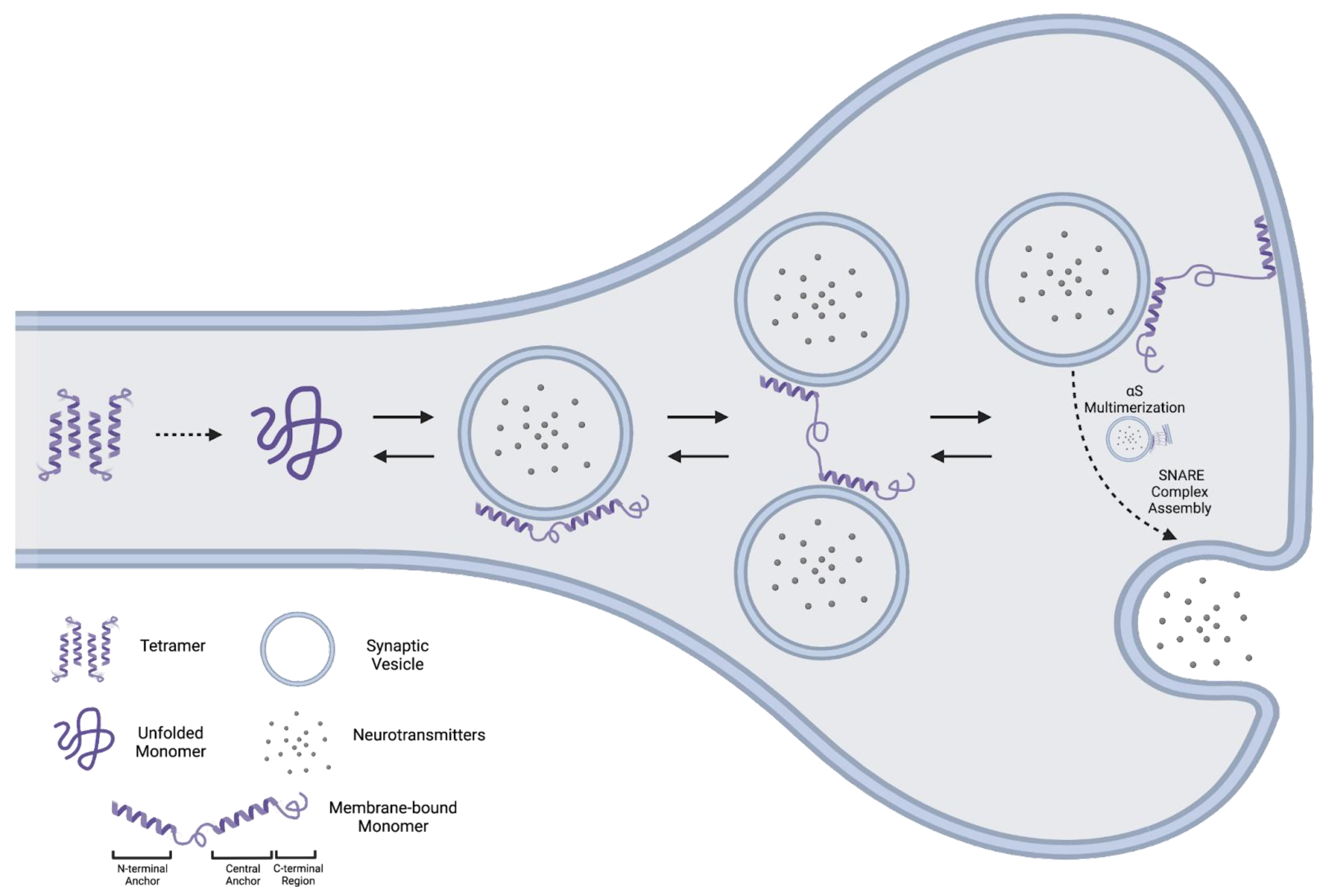
2.2. Tetramer
2.2.1. Evidence for an αS Tetramer
2.2.2. Evidence against an αS Tetramer
3. Pathologic αS
3.1. Prion-like Properties of αS
3.2. Formation of the αS Fibril
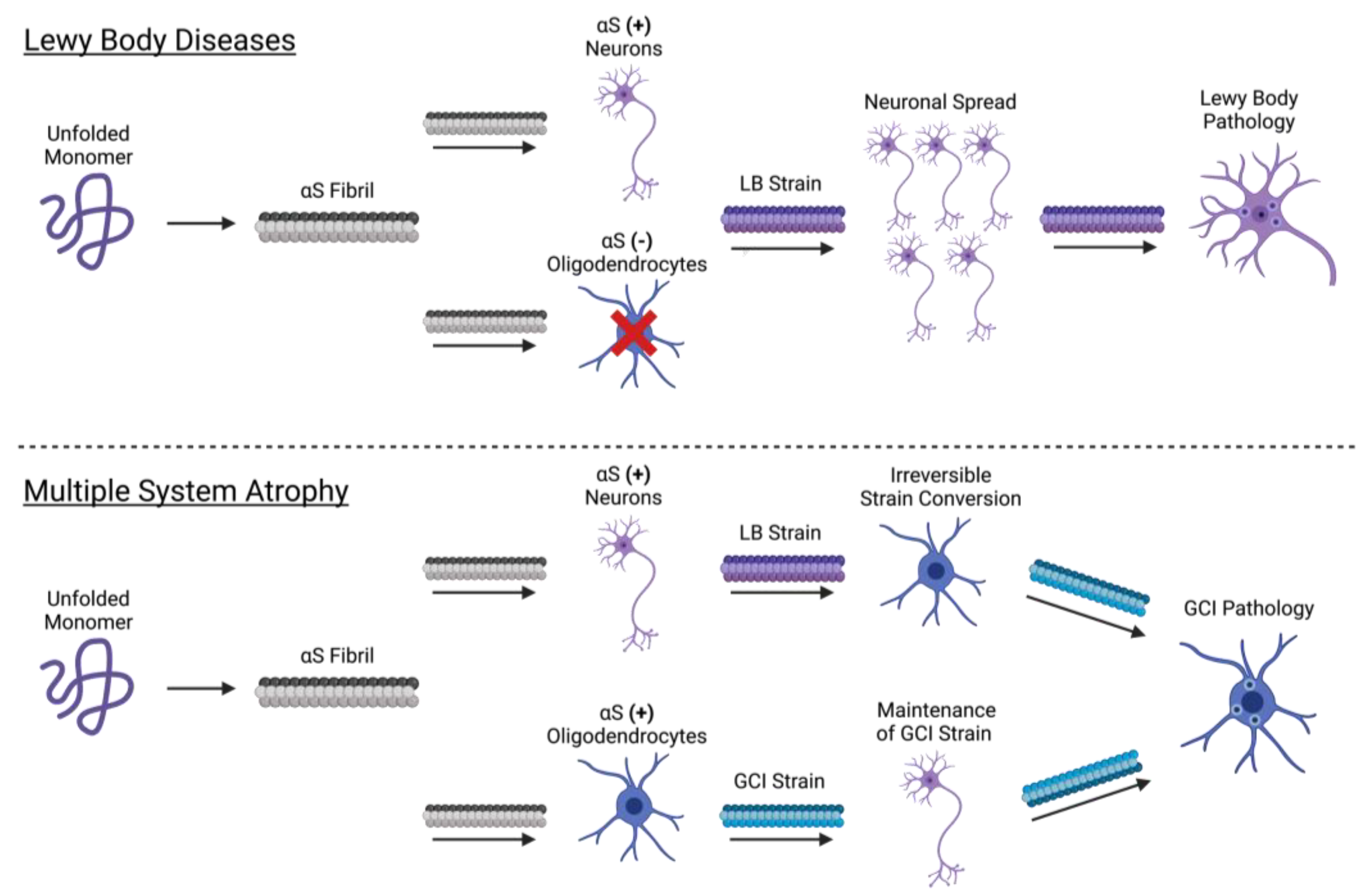
3.3. Propagation of the αS Fibril
3.4. Cellular Determinants of Strain Formation
4. Applications of Strain-Amplification Techniques
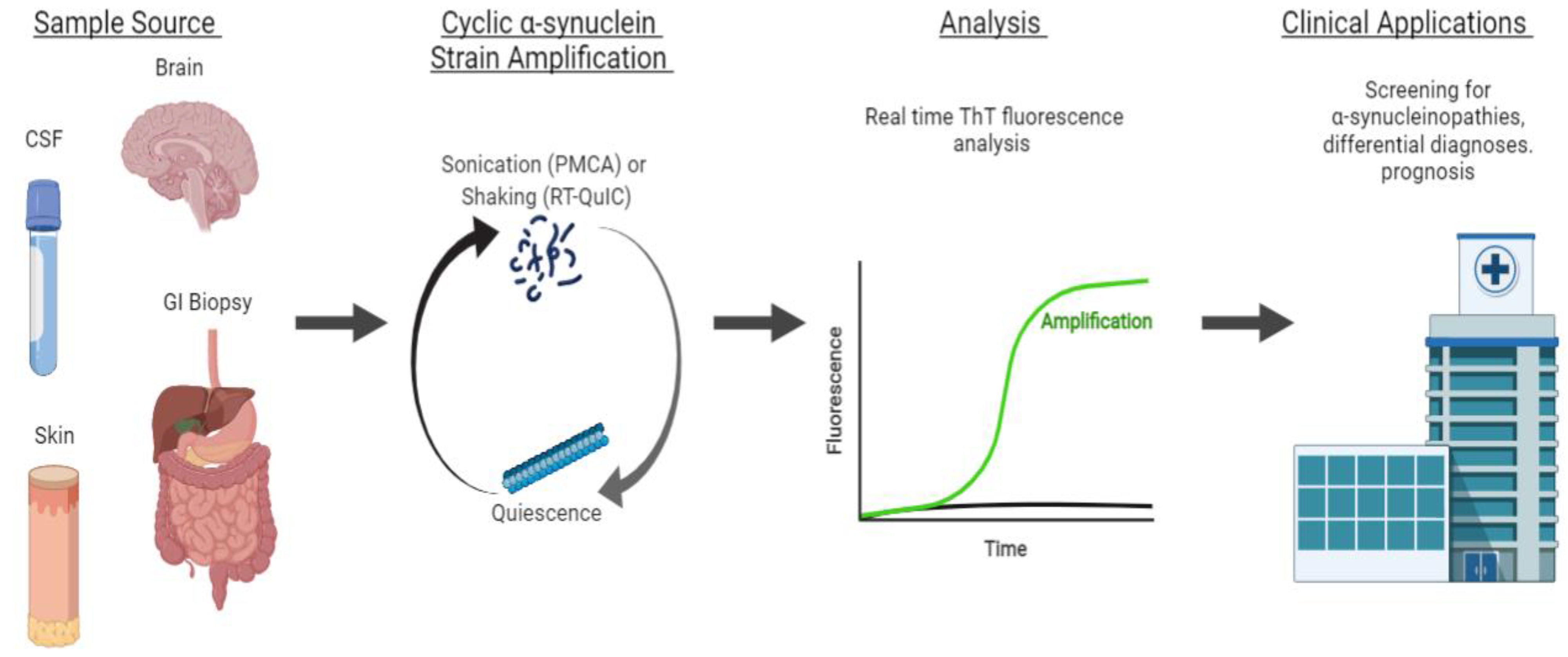
4.1. αS Detection through Strain Amplification Assays
4.1.1. Detection of αS in CSF and Brain
4.1.2. Detecting αS with Seeding Aggregation Assays in Non-CSF Samples
4.1.3. Strain Amplification Assays in Differentiating Synucleinopathies

{kind=link}
{kind=link}
{kind=link}
| Sample Source | Application | Technique | Sensitivity | Specificity | Reference |
|---|---|---|---|---|---|
| CSF | Differentiation of PD from non-synucleinopathy controls | PMCA | 88.5% | 96.6% | Shahnawaz et al., 2017 [3] |
| CSF | Differentiation of DLB from controls | RT-QuIC | 92% | 95% | Fairfoul et al., 2016 [93] |
| Differentiation of PD from controls | 95% | 95% | |||
| CSF | Differentiation of PD from controls | PMCA | 95.2% | 89.9% | Kang et al., 2019 [91] |
| RT-QuIC | 96.2% | 82.3% | |||
| PMCA and RT-QuIC | 97.1% | 92.5% | |||
| CSF | Differentiation of IRBD participants from controls | RT-QuIC | 90% | 90% | Iranzo et al., 2021 [94] |
| GI biopsies | Differentiation of PD from controls | PMCA | 55.56% | 81.81% | Fenyi et al., 2019 [97] |
| Autopsy skin biopsies | Differentiation of PD from controls | PMCA | 82% | 96% | Wang et al., 2020 [98] |
| RT-QuIC | 94% | 98% | |||
| Living skin biopsies | PMCA | 80% | 90% | ||
| RT-QuIC | 95% | 95% | |||
| CSF | Differentiation of MSA from PD | PMCA | 95.4% | 100% | Shahnawaz et al., 2020 [99] |
| Differentiation of PD from non-synucleinopathy controls | 93.6% | 100% | |||
| Differentiation of MSA from controls | 84.6% | 100% | |||
| CSF | Differentiation of MSA from PD/DLB (maximum ThT fluorescence cutoff of <2000 AU) | PMCA | 100% | 83% | Singer et al., 2020 [101] |
| Differentiation of MSA from controls (maximum ThT fluorescence cutoff of >150 AU) | 97% | 100% |
4.2. Comparing PMCA and RT-QuIC Detection of αS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-Synuclein Transmission Initiated by Binding Lymphocyte-Activation Gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017, 74, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural Disorder of Monomeric α-Synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. Folding and Misfolding of Alpha-Synuclein on Membranes. Biochim. Biophys. Acta 2012, 1818, 1013–1018. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct Observation of the Three Regions in α-Synuclein That Determine Its Membrane-Bound Behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Arosio, P.; Vendruscolo, M.; Veglia, G.; Dobson, C.M. Structural Ensembles of Membrane-Bound α-Synuclein Reveal the Molecular Determinants of Synaptic Vesicle Affinity. Sci. Rep. 2016, 6, 27125. [Google Scholar] [CrossRef]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural Basis of Synaptic Vesicle Assembly Promoted by α-Synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef]
- Man, W.K.; Tahirbegi, B.; Vrettas, M.D.; Preet, S.; Ying, L.; Vendruscolo, M.; De Simone, A.; Fusco, G. The Docking of Synaptic Vesicles on the Presynaptic Membrane Induced by α-Synuclein Is Modulated by Lipid Composition. Nat. Commun. 2021, 12, 927. [Google Scholar] [CrossRef]
- Engelman, D.M. Membranes Are More Mosaic than Fluid. Nature 2005, 438, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.S.; Eichmann, C.; Dema, A.; Mercadante, D.; Selenko, P. α-Synuclein Plasma Membrane Localization Correlates with Cellular Phosphatidylinositol Polyphosphate Levels. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Kaur, U.; Lee, J.C. Unroofing Site-Specific α-Synuclein-Lipid Interactions at the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 18977–18983. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rizo, J. Synaptic Vesicle Exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef] [PubMed]
- Rizo, J. Molecular Mechanisms Underlying Neurotransmitter Release. Annu. Rev. Biophys. 2022, 51, 377–408. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-Synuclein Promotes SNARE-Complex Assembly in Vivo and In Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein Assembles into Higher-Order Multimers upon Membrane Binding to Promote SNARE Complex Formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Definition of a Molecular Pathway Mediating α-Synuclein Neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein Occurs Physiologically as a Helically Folded Tetramer That Resists Aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A Soluble α-Synuclein Construct Forms a Dynamic Tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; von Saucken, V.E.; Bartels, T.; Selkoe, D. KTKEGV Repeat Motifs Are Key Mediators of Normal α-Synuclein Tetramerization: Their Mutation Causes Excess Monomers and Neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In Vivo Cross-Linking Reveals Principally Oligomeric Forms of α-Synuclein and β-Synuclein in Neurons and Non-Neural Cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-Causing α-Synuclein Missense Mutations Shift Native Tetramers to Monomers as a Mechanism for Disease Initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [PubMed]
- Nuber, S.; Rajsombath, M.; Minakaki, G.; Winkler, J.; Müller, C.P.; Ericsson, M.; Caldarone, B.; Dettmer, U.; Selkoe, D.J. Abrogating Native α-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease. Neuron 2018, 100, 75–90.e5. [Google Scholar] [CrossRef]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, H.; et al. GBA1 Deficiency Negatively Affects Physiological α-Synuclein Tetramers and Related Multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef]
- Imberdis, T.; Negri, J.; Ramalingam, N.; Terry-Kantor, E.; Ho, G.P.H.; Fanning, S.; Stirtz, G.; Kim, T.-E.; Levy, O.A.; Young-Pearse, T.L.; et al. Cell Models of Lipid-Rich α-Synuclein Aggregation Validate Known Modifiers of α-Synuclein Biology and Identify Stearoyl-CoA Desaturase. Proc. Natl. Acad. Sci. USA 2019, 116, 20760–20769. [Google Scholar] [CrossRef]
- Nuber, S.; Nam, A.Y.; Rajsombath, M.M.; Cirka, H.; Hronowski, X.; Wang, J.; Hodgetts, K.; Kalinichenko, L.S.; Müller, C.P.; Lambrecht, V.; et al. A Stearoyl-Coenzyme A Desaturase Inhibitor Prevents Multiple Parkinson Disease Phenotypes in α-Synuclein Mice. Ann. Neurol. 2021, 89, 74–90. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and Its Relevance to Parkinson Disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of Native Brain α-Synuclein. Nature 2013, 498, E4–E6; discussion E6–E7. [Google Scholar] [CrossRef]
- Bartels, T.; Selkoe, D.J. Bartels & Selkoe Reply. Nature 2013, 498, E6–E7. [Google Scholar] [CrossRef]
- Binolfi, A.; Theillet, F.-X.; Selenko, P. Bacterial In-Cell NMR of Human α-Synuclein: A Disordered Monomer by Nature? Biochem. Soc. Trans. 2012, 40, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Fares, M.-B.; Samuel, F.; Dikiy, I.; Tandon, A.; Eliezer, D.; Lashuel, H.A. Characterization of Semisynthetic and Naturally Nα-Acetylated α-Synuclein In Vitro and in Intact Cells: Implications for Aggregation and Cellular Properties of α-Synuclein. J. Biol. Chem. 2012, 287, 28243–28262. [Google Scholar] [CrossRef]
- Fauvet, B.; Mbefo, M.K.; Fares, M.-B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in Central Nervous System and from Erythrocytes, Mammalian Cells, and Escherichia Coli Exists Predominantly as Disordered Monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [PubMed]
- Waudby, C.A.; Camilloni, C.; Fitzpatrick, A.W.P.; Cabrita, L.D.; Dobson, C.M.; Vendruscolo, M.; Christodoulou, J. In-Cell NMR Characterization of the Secondary Structure Populations of a Disordered Conformation of α-Synuclein within E. Coli Cells. PLoS ONE 2013, 8, e72286. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a Protein That Purifies with the Scrapie Prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Leak, R.K.; Frosch, M.P.; Beach, T.G.; Halliday, G.M. Alpha-Synuclein: Prion or Prion-Like? Acta Neuropathol. 2019, 138, 509–514. [Google Scholar] [CrossRef]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-Ribose) Drives Pathologic α-Synuclein Neurodegeneration in Parkinson’s Disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy Bodies in Grafted Neurons in Subjects with Parkinson’s Disease Suggest Host-to-Graft Disease Propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy Body-like Pathology in Long-Term Embryonic Nigral Transplants in Parkinson’s Disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of Multiple System Atrophy Prions to Transgenic Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-Synuclein Prions Causing Multiple System Atrophy in Humans with Parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Lee, J.; Monteiro, O.; Woerman, A.L.; Lazar, A.A.; Condello, C.; Paras, N.A.; Prusiner, S.B. Different α-Synuclein Prion Strains Cause Dementia with Lewy Bodies and Multiple System Atrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2113489119. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Parkinson’s Disease Is Not Simply a Prion Disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef]
- Comellas, G.; Lemkau, L.R.; Zhou, D.H.; George, J.M.; Rienstra, C.M. Structural Intermediates during α-Synuclein Fibrillogenesis on Phospholipid Vesicles. J. Am. Chem. Soc. 2012, 134, 5090–5099. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef]
- Iljina, M.; Garcia, G.A.; Horrocks, M.H.; Tosatto, L.; Choi, M.L.; Ganzinger, K.A.; Abramov, A.Y.; Gandhi, S.; Wood, N.W.; Cremades, N.; et al. Kinetic Model of the Aggregation of Alpha-Synuclein Provides Insights into Prion-like Spreading. Proc. Natl. Acad. Sci. USA 2016, 113, E1206–E1215. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural Basis of Membrane Disruption and Cellular Toxicity by α-Synuclein Oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of Alpha-Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ Is a Key Factor in α-Synuclein-Induced Neurotoxicity. J. Cell. Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef]
- Farzadfard, A.; Pedersen, J.N.; Meisl, G.; Somavarapu, A.K.; Alam, P.; Goksøyr, L.; Nielsen, M.A.; Sander, A.F.; Knowles, T.P.J.; Pedersen, J.S.; et al. The C-Terminal Tail of α-Synuclein Protects against Aggregate Replication but Is Critical for Oligomerization. Commun. Biol. 2022, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Gracia, P.; Colom, A.; Camino, J.D.; Fernández-Higuero, J.Á.; Orozco, N.; Dulebo, A.; Saiz, L.; Cremades, N.; Vilar, J.M.G.; et al. All-or-None Amyloid Disassembly via Chaperone-Triggered Fibril Unzipping Favors Clearance of α-Synuclein Toxic Species. Proc. Natl. Acad. Sci. USA 2021, 118, e2105548118. [Google Scholar] [CrossRef]
- Singh, A.; Dawson, T.M.; Kulkarni, S. Neurodegenerative Disorders and Gut-Brain Interactions. J. Clin. Investig. 2021, 131, 143775. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-Released Oligomeric α-Synuclein Is an Endogenous Agonist of TLR2 for Paracrine Activation of Microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-Dependent TLR1/2 Signaling by Misfolded α-Synuclein, a Protein Linked to Neurodegenerative Disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Léna, C.; et al. α-Synuclein Assemblies Sequester Neuronal A3-Na+/K+-ATPase and Impair Na+ Gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.R.; Cha, S.-H.; Kang, S.-J.; Kim, J.-B.; Jou, I.; Park, S.M. Prion-like Propagation of α-Synuclein Is Regulated by the FcγRIIB-SHP-1/2 Signaling Pathway in Neurons. Cell. Rep. 2018, 22, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Urrea, L.; Segura-Feliu, M.; Masuda-Suzukake, M.; Hervera, A.; Pedraz, L.; García Aznar, J.M.; Vila, M.; Samitier, J.; Torrents, E.; Ferrer, I.; et al. Involvement of Cellular Prion Protein in α-Synuclein Transport in Neurons. Mol. Neurobiol. 2018, 55, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Wojcik, S.P.; Miranker, A.D.; Rhoades, E. Identification of N-Linked Glycans as Specific Mediators of Neuronal Uptake of Acetylated α-Synuclein. PLoS Biol. 2019, 17, e3000318. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, Y.; Lee, J.; Jarnik, M.; Wu, X.; Bonifacino, J.S.; Shen, J.; Ye, Y. A Myosin-7B-Dependent Endocytosis Pathway Mediates Cellular Entry of α-Synuclein Fibrils and Polycation-Bearing Cargos. Proc. Natl. Acad. Sci. USA 2020, 117, 10865–10875. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic Basis for Receptor-Mediated Pathological α-Synuclein Fibril Cell-to-Cell Transmission in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118. [Google Scholar] [CrossRef]
- Gu, H.; Yang, X.; Mao, X.; Xu, E.; Qi, C.; Wang, H.; Brahmachari, S.; York, B.; Sriparna, M.; Li, A.; et al. Lymphocyte Activation Gene 3 (Lag3) Contributes to α-Synucleinopathy in α-Synuclein Transgenic Mice. Front. Cell. Neurosci. 2021, 15, 656426. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Gu, H.; Kim, D.; Kimura, Y.; Wang, N.; Xu, E.; Wang, H.; Chen, C.; Zhang, S.; Jia, C.; et al. Aplp1 and the Aplp1-Lag3 Complex Facilitates Transmission of Pathologic α-Synuclein. Neuroscience 2021. Preprint. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal Cell-to-Cell Transmission of Alpha Synuclein Oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 Regulates Secretion of Exosomes and α-Synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef]
- Fussi, N.; Höllerhage, M.; Chakroun, T.; Nykänen, N.-P.; Rösler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Höglinger, G.U. Exosomal Secretion of α-Synuclein as Protective Mechanism after Upstream Blockage of Macroautophagy. Cell Death. Dis. 2018, 9, 757. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial Exosomes Facilitate α-Synuclein Transmission in Parkinson’s Disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Manzanza, N.; Sedlackova, L.; Kalaria, R.N. Alpha-Synuclein Post-Translational Modifications: Implications for Pathogenesis of Lewy Body Disorders. Front. Aging Neurosci. 2021, 13, 690293. [Google Scholar] [CrossRef]
- Gadhavi, J.; Patel, M.; Bhatia, D.; Gupta, S. Neurotoxic or Neuroprotective: Post-Translational Modifications of α-Synuclein at the Cross-Roads of Functions. Biochimie 2022, 192, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated Alpha-Synuclein Is Ubiquitinated in Alpha-Synucleinopathy Lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef]
- Kim, Y.M.; Jang, W.H.; Quezado, M.M.; Oh, Y.; Chung, K.C.; Junn, E.; Mouradian, M.M. Proteasome Inhibition Induces α-Synuclein SUMOylation and Aggregate Formation. J. Neurol. Sci. 2011, 307, 157–161. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of Alpha-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef]
- Chau, K.-Y.; Ching, H.L.; Schapira, A.H.V.; Cooper, J.M. Relationship between Alpha Synuclein Phosphorylation, Proteasomal Inhibition and Cell Death: Relevance to Parkinson’s Disease Pathogenesis. J. Neurochem. 2009, 110, 1005–1013. [Google Scholar] [CrossRef]
- Liu, Y.-Q.; Mao, Y.; Xu, E.; Jia, H.; Zhang, S.; Dawson, V.L.; Dawson, T.M.; Li, Y.-M.; Zheng, Z.; He, W.; et al. Nanozyme Scavenging ROS for Prevention of Pathologic α-Synuclein Transmission in Parkinson’s Disease. Nano Today 2021, 36, 101027. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The Process of Lewy Body Formation, Rather than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, Y.; Wang, W.; Wang, Y.; Liu, H.; Liu, F.; Chen, R.; Dawson, V.L.; Dawson, T.M.; Lu, F.; et al. Molecular Mediation of Prion-like α-Synuclein Fibrillation from Toxic PFFs to Nontoxic Species. ACS Appl. Bio. Mater. 2020, 3, 6096–6102. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular Milieu Imparts Distinct Pathological α-Synuclein Strains in α-Synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Asi, Y.T.; Simpson, J.E.; Heath, P.R.; Wharton, S.B.; Lees, A.J.; Revesz, T.; Houlden, H.; Holton, J.L. Alpha-Synuclein MRNA Expression in Oligodendrocytes in MSA. Glia 2014, 62, 964–970. [Google Scholar] [CrossRef]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-Synuclein Transfers from Neurons to Oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive Detection of Pathological Prion Protein by Cyclic Amplification of Protein Misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Atarashi, R.; Sano, K.; Satoh, K.; Nishida, N. Real-Time Quaking-Induced Conversion: A Highly Sensitive Assay for Prion Detection. Prion 2011, 5, 150–153. [Google Scholar] [CrossRef]
- Vilas, D. Moving Forward the in Vivo Diagnosis of the Synucleinopathies. Clin. Auton. Res. 2019, 29, 575–576. [Google Scholar] [CrossRef]
- Kang, U.J.; Boehme, A.K.; Fairfoul, G.; Shahnawaz, M.; Ma, T.C.; Hutten, S.J.; Green, A.; Soto, C. Comparative Study of Cerebrospinal Fluid α-Synuclein Seeding Aggregation Assays for Diagnosis of Parkinson’s Disease. Mov. Disord. 2019, 34, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Wang, X.; Vander Stel, K.; Chu, Y.; Kordower, J.; Ma, J. Detecting Alpha Synuclein Seeding Activity in Formaldehyde-Fixed MSA Patient Tissue by PMCA. Mol. Neurobiol. 2018, 55, 8728–8737. [Google Scholar] [CrossRef] [PubMed]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-Synuclein RT-QuIC in the CSF of Patients with Alpha-Synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, A.; Fairfoul, G.; Ayudhaya, A.C.N.; Serradell, M.; Gelpi, E.; Vilaseca, I.; Sanchez-Valle, R.; Gaig, C.; Santamaria, J.; Tolosa, E.; et al. Detection of α-Synuclein in CSF by RT-QuIC in Patients with Isolated Rapid-Eye-Movement Sleep Behaviour Disorder: A Longitudinal Observational Study. Lancet Neurol. 2021, 20, 203–212. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Campese, N.; Beatino, M.F.; Del Gamba, C.; Belli, E.; Giampietri, L.; Del Prete, E.; Galgani, A.; Vergallo, A.; Siciliano, G.; Ceravolo, R.; et al. Ultrasensitive Techniques and Protein Misfolding Amplification Assays for Biomarker-Guided Reconceptualization of Alzheimer’s and Other Neurodegenerative Diseases. Expert Rev. Neurother. 2021, 21, 949–967. [Google Scholar] [CrossRef]
- Fenyi, A.; Leclair-Visonneau, L.; Clairembault, T.; Coron, E.; Neunlist, M.; Melki, R.; Derkinderen, P.; Bousset, L. Detection of Alpha-Synuclein Aggregates in Gastrointestinal Biopsies by Protein Misfolding Cyclic Amplification. Neurobiol. Dis. 2019, 129, 38–43. [Google Scholar] [CrossRef]
- Wang, Z.; Becker, K.; Donadio, V.; Siedlak, S.; Yuan, J.; Rezaee, M.; Incensi, A.; Kuzkina, A.; Orrú, C.D.; Tatsuoka, C.; et al. Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol. 2020, 78, 30–40. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-Synuclein Strains in Parkinson’s Disease and Multiple System Atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Lipp, A.; Sandroni, P.; Ahlskog, J.E.; Fealey, R.D.; Kimpinski, K.; Iodice, V.; Gehrking, T.L.; Weigand, S.D.; Sletten, D.M.; Gehrking, J.A.; et al. Prospective Differentiation of Multiple System Atrophy from Parkinson Disease, with and without Autonomic Failure. Arch. Neurol. 2009, 66, 742–750. [Google Scholar] [CrossRef]
- Singer, W.; Schmeichel, A.M.; Shahnawaz, M.; Schmelzer, J.D.; Boeve, B.F.; Sletten, D.M.; Gehrking, T.L.; Gehrking, J.A.; Olson, A.D.; Savica, R.; et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain in Spinal Fluid Differentiate Multiple System Atrophy from Lewy Body Synucleinopathies. Ann. Neurol. 2020, 88, 503–512. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Rastegar, C.; Mao, X. α-Synuclein Conformational Plasticity: Physiologic States, Pathologic Strains, and Biotechnological Applications. Biomolecules 2022, 12, 994. https://doi.org/10.3390/biom12070994
Li A, Rastegar C, Mao X. α-Synuclein Conformational Plasticity: Physiologic States, Pathologic Strains, and Biotechnological Applications. Biomolecules. 2022; 12(7):994. https://doi.org/10.3390/biom12070994
Chicago/Turabian StyleLi, Amanda, Cyrus Rastegar, and Xiaobo Mao. 2022. "α-Synuclein Conformational Plasticity: Physiologic States, Pathologic Strains, and Biotechnological Applications" Biomolecules 12, no. 7: 994. https://doi.org/10.3390/biom12070994
APA StyleLi, A., Rastegar, C., & Mao, X. (2022). α-Synuclein Conformational Plasticity: Physiologic States, Pathologic Strains, and Biotechnological Applications. Biomolecules, 12(7), 994. https://doi.org/10.3390/biom12070994