
Structural and Functional Basis of JAMM Deubiquitinating Enzymes in Disease


Abstract
1. Introduction
2. Structural Characteristic of JAMMs
3. Catalytic Mechanism of JAMMs
4. Structural and Functional Basis of JAMMs
4.1. Functional Basis of AMSH in Receptor Endocytosis
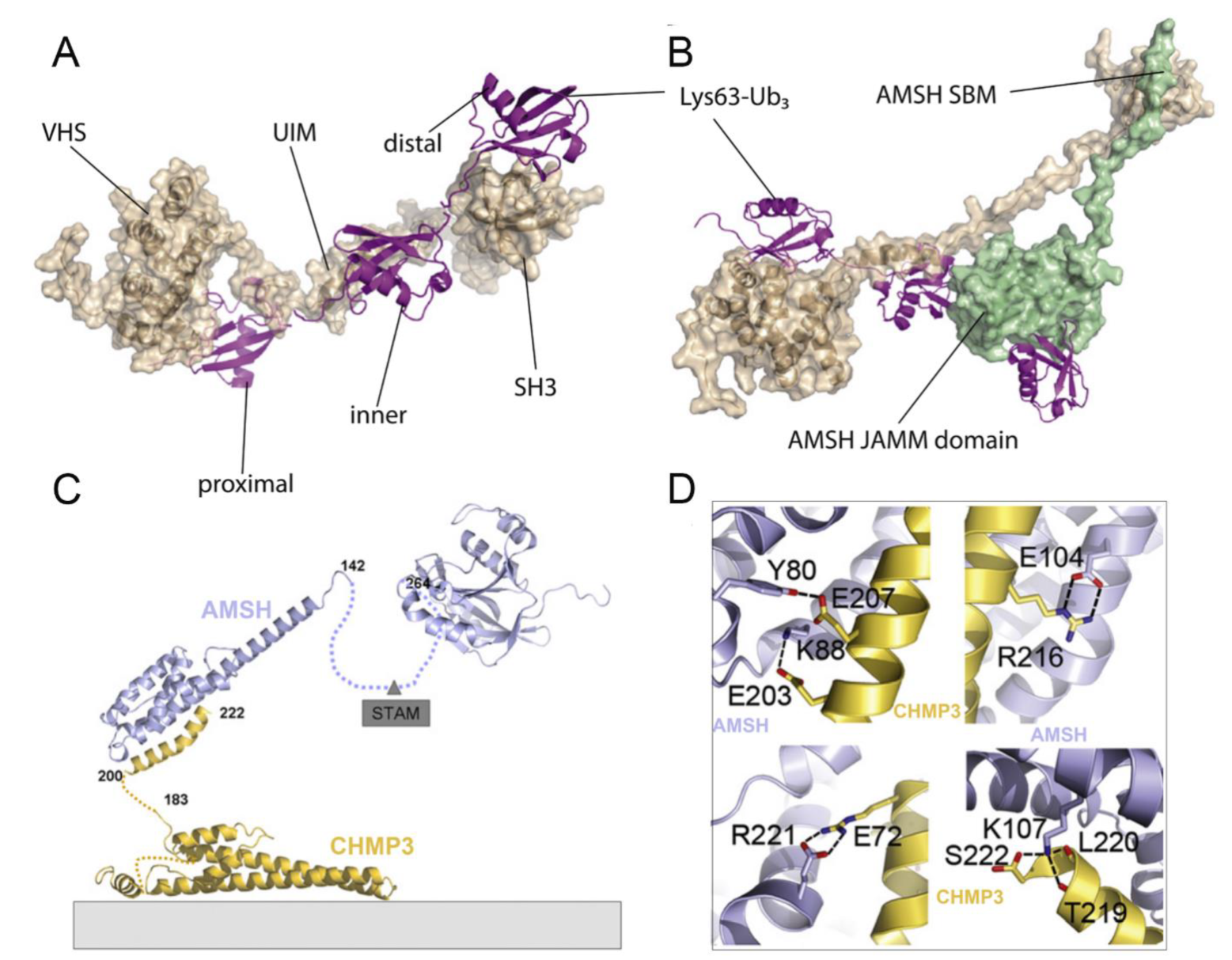
4.2. Structural Basis of AMSH
4.3. Comparison of AMSH and AMSH-LP
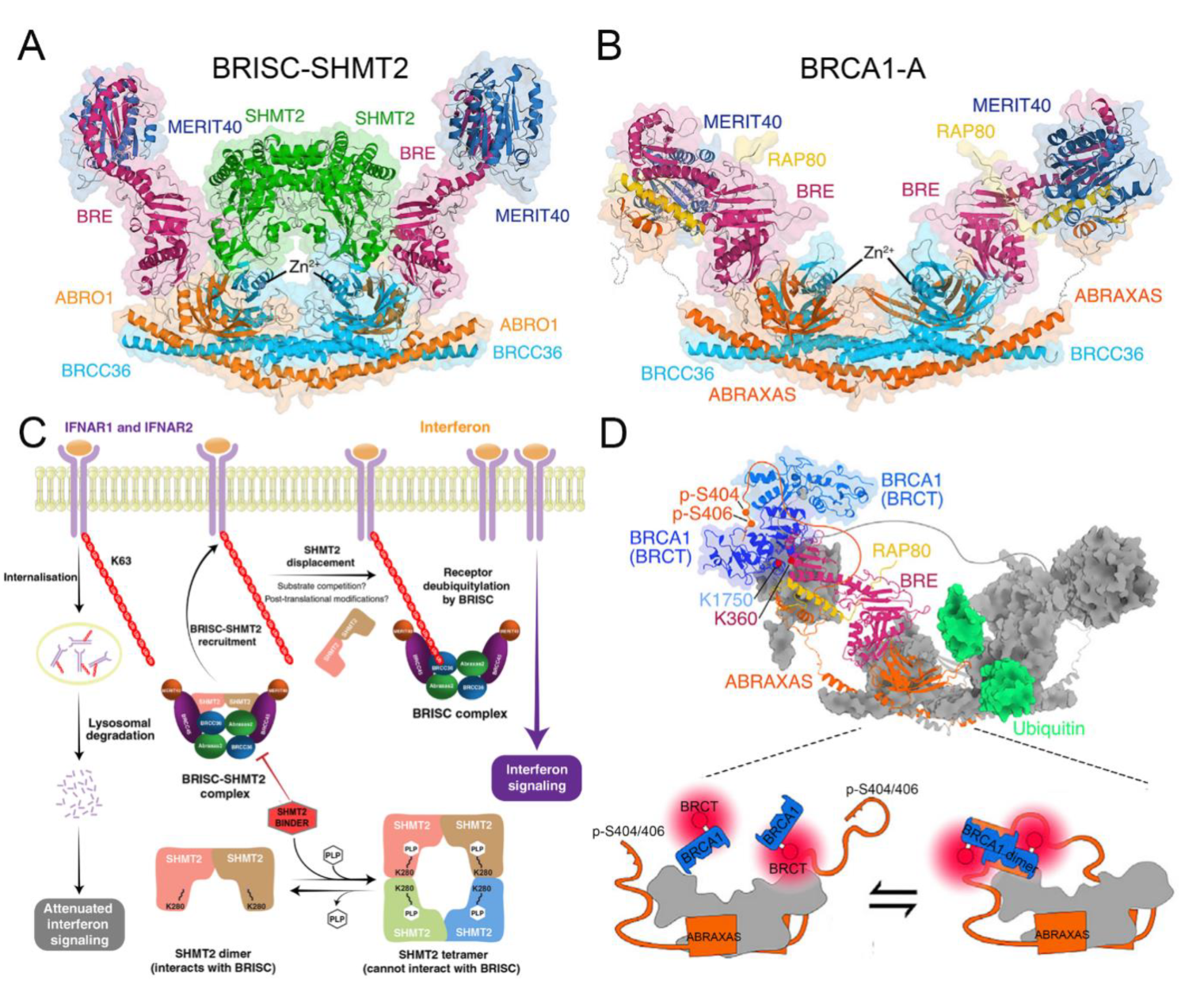
4.4. Functional Basis of BRISC in Inflammation, Immune Response, Mitosis, and Hematopoiesis
4.5. Structural Basis of BRISC
4.6. Functional Basis of BRCA1-A in DNA Damage Repair
4.7. Structural Basis of BRCA1-A
4.8. Functional Basis of Rpn11 in Proteasome-Dependent Versatility
4.9. Structural Basis of Rpn11
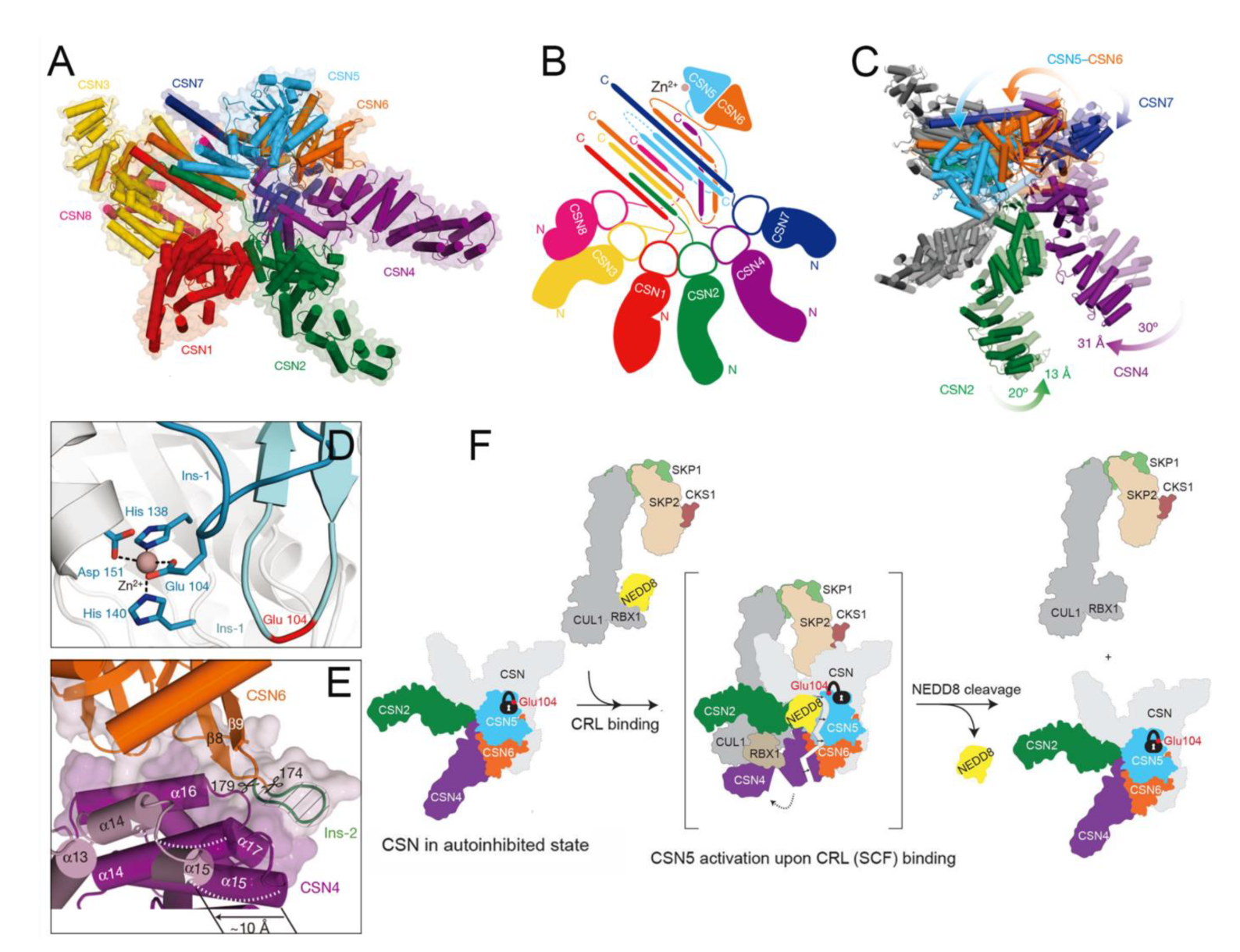
4.10. Functional Basis of CSN5 in Regulating the Cullin-RING E3 Ubiquitin Ligases
4.11. Structural Basis of CSN5
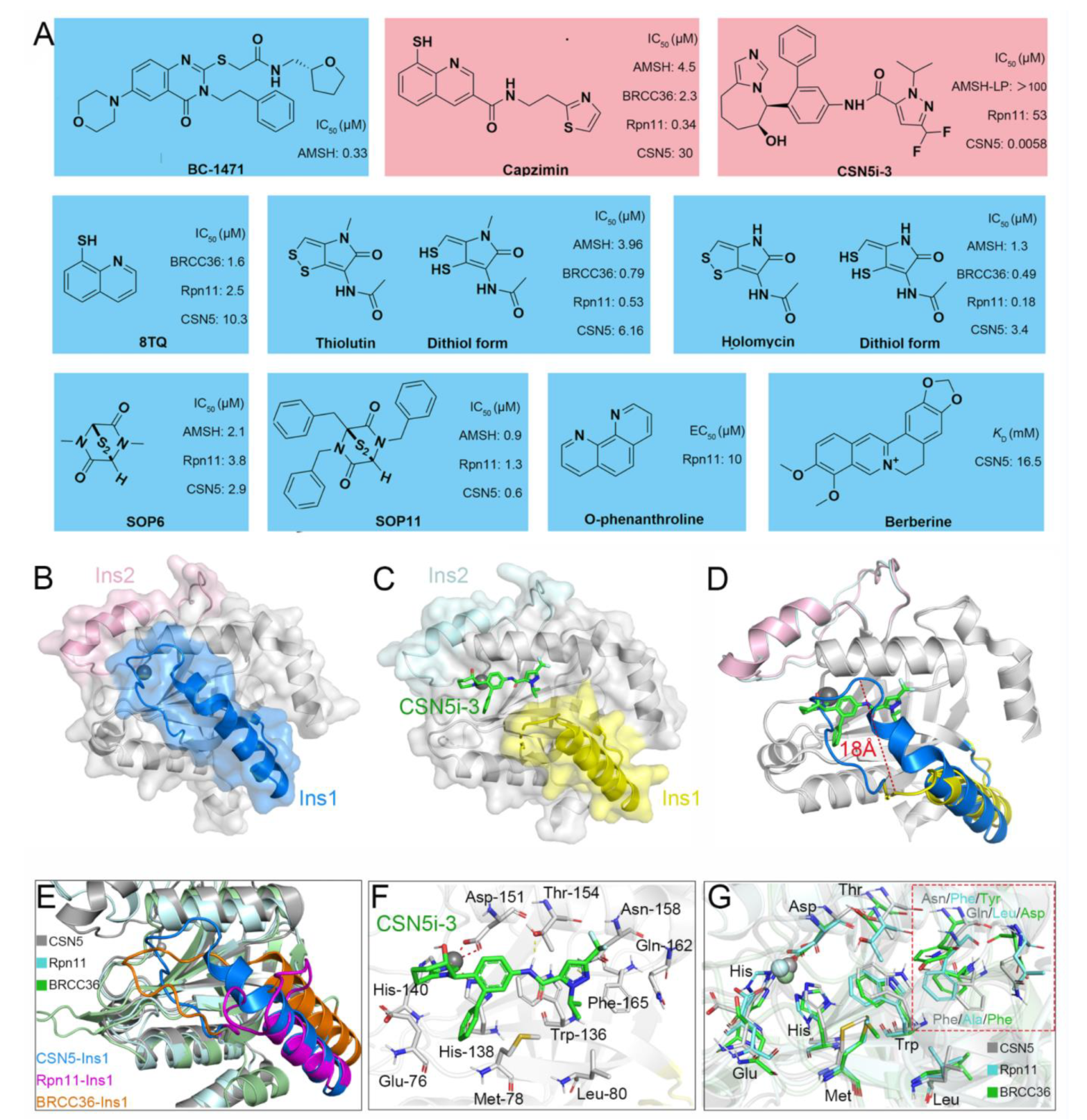
5. Currently Reported Inhibitors Targeting JAMMs
6. Challenges and Future Prospects
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DUB | Deubiquitinating enzyme |
| JAMM | JAB1/MPN/Mov34 metalloenzymes |
| AMSH | Associated molecule with the SH3 domain of STAM |
| AMSH-LP | AMSH-like protein |
| BRCA1 | Breast cancer susceptibility protein 1 |
| BRCC36 | BRCA1-BRCA2-containing complex subunit 36 |
| Rpn11 | Regulatory particle non-ATPase 11 |
| CSN | COP9 signalosome |
| SUMO | Small ubiquitin-like modifier |
| NEDD8 | Neuronal precursor cell-expressed developmentally downregulated protein 8 |
| USP | Ubiquitin-specific peptidase |
| OTU | Ovarian tumor protease |
| MINDYs | Motif interacting with ubiquitin-containing novel DUB family |
| UCH | Ubiquitin C-terminal hydroxylase |
| MJD | Machado-Josephin domain protease |
| ZUP1 | Zinc finger-containing ubiquitin peptidase 1 |
| ZnF | Zinc finger domain |
| UIM | Ubiquitin-interacting motif |
| UBA | Ubiquitin-associated domain |
| eIF3h | Eukaryotic translation initiation factor 3 subunit H |
| MYSM1 | Myb-like, SWIRM, and MPN domains 1 protein |
| MPN | Mpr1/Pad1 N-terminal |
| SH3 | Src-homology domain 3 |
| STAM | Signal transducing adapter molecule |
| ESCRT | Endosomal sorting complexes required for transport |
| EGFR | Epidermal growth factor receptor |
| GPCR | G protein-coupled receptor |
| NALP | NACHT, LRR and PYD domains-containing protein |
| SBM | SH3 binding motif |
| CHMP | Charged multivesicular body proteins |
| MIM | MIT-interacting motif |
| MIT | Microtubule interacting and transport |
| UIM | Ubiquitin-interacting motif |
| VHS | Vps27/Hrs/STAM |
| ASC | Apoptosis-associated speck-like protein containing a caspase recruitment domain |
| SHMT2 | Serine hydroxymethyltransferase 2 |
| JAK2 | Janus kinase 2 |
| PLP | Pyridoxal-5′-phosphate |
| DSB | DNA double-strand breaks |
| HR | Homologous recombination |
| NHEJ | Non-homologous end joining |
| CP | Core particle |
| RPs | Regulatory particles |
| Mitf | Microphthalmia-associated transcription factor |
| CRLs | Cullin-RING E3 ubiquitin ligases |
| PD-L1 | Programmed death-ligand 1 |
| MYC | Myelocytomatosis oncogene |
References
- Imai, Y.; Soda, M.; Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000, 275, 35661–35664. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Pagano, M.; Tam, S.W.; Theodoras, A.M.; Beer-Romero, P.; Del Sal, G.; Chau, V.; Yew, P.R.; Draetta, G.F.; Rolfe, M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A.; Varshavsky, A. Basic medical research award. The ubiquitin system. Nat. Med. 2000, 6, 1073–1081. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Hospenthal, M.K.; Freund, S.M.; Komander, D. Assembly, analysis and architecture of atypical ubiquitin chains. Nat. Struct. Mol. Biol. 2013, 20, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Blaser, G.; Horrocks, M.H.; Ruedas-Rama, M.J.; Ibrahim, S.; Zhukov, A.A.; Orte, A.; Klenerman, D.; Jackson, S.E.; Komander, D. Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature 2012, 492, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Erpapazoglou, Z.; Walker, O.; Haguenauer-Tsapis, R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells 2014, 3, 1027–1088. [Google Scholar] [CrossRef]
- Clague, M.J.; Urbé, S. Endocytosis: The DUB version. Trends Cell Biol. 2006, 16, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Schauer, N.J.; Magin, R.S.; Liu, X.; Doherty, L.M.; Buhrlage, S.J. Advances in discovering deubiquitinating enzyme (DUB) inhibitors. J. Med. Chem. 2020, 63, 2731–2750. [Google Scholar] [CrossRef] [PubMed]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and characterization of ZUFSP/ZUP1, a distinct deubiquitinase class important for genome stability. Mol. Cell 2018, 70, 150–164.e156. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Kumar, P.; Kapuganti, S.K.; Uversky, V.N.; Giri, R. Unstructured biology of proteins from ubiquitin-proteasome system: Roles in cancer and neurodegenerative diseases. Biomolecules 2020, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Cockram, P.E.; Kist, M.; Prakash, S.; Chen, S.H.; Wertz, I.E.; Vucic, D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ. 2021, 28, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Ronau, J.A.; Beckmann, J.F.; Hochstrasser, M. Substrate specificity of the ubiquitin and Ubl proteases. Cell Res. 2016, 26, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Patterson-Fortin, J.; Shao, G.; Bretscher, H.; Messick, T.E.; Greenberg, R.A. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J. Biol. Chem. 2010, 285, 30971–30981. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshikawa, A.; Yamagata, A.; Mimura, H.; Yamashita, M.; Ookata, K.; Nureki, O.; Iwai, K.; Komada, M.; Fukai, S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 2008, 455, 358–362. [Google Scholar] [CrossRef]
- McCullough, J.; Clague, M.J.; Urbé, S. AMSH is an endosome-associated ubiquitin isopeptidase. J. Cel. Biol. 2004, 166, 487–492. [Google Scholar] [CrossRef]
- Marchione, R.; Leibovitch, S.A.; Lenormand, J.L. The translational factor eIF3f: The ambivalent eIF3 subunit. Cell Mol. Life Sci. 2013, 70, 3603–3616. [Google Scholar] [CrossRef]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R.; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef]
- Birol, M.; Echalier, A. Structure and function of MPN (Mpr1/PadN-terminal) domain-containing proteins. Curr. Protein Pept. Sci. 2014, 15, 504–517. [Google Scholar] [CrossRef]
- Ambroggio, X.I.; Rees, D.C.; Deshaies, R.J. JAMM: A metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004, 2, 0113–0119. [Google Scholar] [CrossRef] [PubMed]
- Dambacher, C.M.; Worden, E.J.; Herzik, M.A.; Martin, A.; Lander, G.C. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. eLife 2016, 5, e13027. [Google Scholar] [CrossRef] [PubMed]
- Galej, W.P.; Nguyen, T.H.D.; Newman, A.J.; Nagai, K. Structural studies of the spliceosome: Zooming into the heart of the machine. Curr. Opin. Struct. Biol. 2014, 25, 57–66. [Google Scholar] [CrossRef]
- Lingaraju, G.M.; Bunker, R.D.; Cavadini, S.; Hess, D.; Hassiepen, U.; Renatus, M.; Fischer, E.S.; Thomä, N.H. Crystal structure of the human COP9 signalosome. Nature 2014, 512, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.K.; Ronau, J.A.; Davies, C.W.; Guenette, R.G.; Strieter, E.R.; Paul, L.N.; Das, C. Insights into the mechanism of deubiquitination by JAMM deubiquitinases from cocrystal structures of the enzyme with the substrate and product. Biochemistry 2014, 53, 3199–3217. [Google Scholar] [CrossRef]
- De Poot, S.A.H.; Tian, G.; Finley, D. Meddling with fate: The proteasomal deubiquitinating enzymes. J. Mol. Biol. 2017, 429, 3525–3545. [Google Scholar] [CrossRef]
- Tran, H.J.T.T.; Allen, M.D.; Löwe, J.; Bycroft, M. Structure of the Jab1/MPN domain and its implications for proteasome function. Biochemistry 2003, 42, 11460–11465. [Google Scholar] [CrossRef]
- Worden, E.J.; Padovani, C.; Martin, A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat. Struct. Mol. Biol. 2014, 21, 220–227. [Google Scholar] [CrossRef]
- Worden, E.J.; Dong, K.C.; Martin, A. An AAA motor-driven mechanical switch in Rpn11 controls deubiquitination at the 26S proteasome. Mol. Cell 2017, 67, 799–811. [Google Scholar] [CrossRef]
- Pathare, G.R.; Nagy, I.; Śledź, P.; Anderson, D.J.; Zhou, H.-J.; Pardon, E.; Steyaert, J.; Förster, F.; Bracher, A.; Baumeister, W. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc. Natl. Acad. Sci. USA 2014, 111, 2984–2989. [Google Scholar] [CrossRef]
- Echalier, A.; Pan, Y.; Birol, M.; Tavernier, N.; Pintard, L.; Hoh, F.; Ebel, C.; Galophe, N.; Claret, F.X.; Dumas, C. Insights into the regulation of the human COP9 signalosome catalytic subunit, CSN5/Jab1. Proc. Natl. Acad. Sci. USA 2013, 110, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef]
- Sanches, M.; Alves, B.S.C.; Zanchin, N.I.T.; Guimarães, B.G. The crystal structure of the human MovMPN domain reveals a metal-free dimer. J. Mol. Biol. 2007, 370, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; Quesada, V.; Rodríguez, D.; Freije, J.M.P.; López-Otín, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388. [Google Scholar] [CrossRef] [PubMed]
- Maytal-Kivity, V.; Reis, N.; Hofmann, K.; Glickman, M.H. MPN+, a putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC Biochem. 2002, 3, 28. [Google Scholar] [CrossRef]
- Liu, Y.; Shah, S.V.; Xiang, X.; Wang, J.; Deng, Z.-B.; Liu, C.; Zhang, L.; Wu, J.; Edmonds, T.; Jambor, C.; et al. COP9-associated CSN5 regulates exosomal protein deubiquitination and sorting. Am. J. Pathol. 2009, 174, 1415–1425. [Google Scholar] [CrossRef]
- Iadevaia, V.; Caldarola, S.; Tino, E.; Amaldi, F.; Loreni, F. All translation elongation factors and the e, f, and h subunits of translation initiation factor 3 are encoded by 5′-terminal oligopyrimidine (TOP) mRNAs. RNA 2008, 14, 1730–1736. [Google Scholar] [CrossRef]
- Dong, Y.; Hakimi, M.-A.; Chen, X.; Kumaraswamy, E.; Cooch, N.S.; Godwin, A.K.; Shiekhattar, R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell 2003, 12, 1087–1099. [Google Scholar] [CrossRef]
- Zeqiraj, E.; Tian, L.; Piggott, C.A.; Pillon, M.C.; Duffy, N.M.; Ceccarelli, D.F.; Keszei, A.F.A.; Lorenzen, K.; Kurinov, I.; Orlicky, S.; et al. Higher-order assembly of BRCC36-KIAA0157 is required for DUB activity and biological function. Mol. Cell 2015, 59, 970–983. [Google Scholar] [CrossRef]
- Nakamura, M.; Tanaka, N.; Kitamura, N.; Komada, M. Clathrin anchors deubiquitinating enzymes, AMSH and AMSH-like protein, on early endosomes. Genes Cells 2006, 11, 593–606. [Google Scholar] [CrossRef]
- Holden, H.M.; Tronrud, D.E.; Monzingo, A.F.; Weaver, L.H.; Matthews, B.W. Slow- and fast-binding inhibitors of thermolysin display different modes of binding: Crystallographic analysis of extended phosphonamidate transition-state analogues. Biochemistry 1987, 26, 8542–8553. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Berlin, I.; Nash, P.D. Regulation of endocytic sorting by ESCRT-DUB-mediated deubiquitination. Cell Biochem. Biophys. 2011, 60, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Kyuuma, M.; Kikuchi, K.; Kojima, K.; Sugawara, Y.; Sato, M.; Mano, N.; Goto, J.; Takeshita, T.; Yamamoto, A.; Sugamura, K.; et al. AMSH, an ESCRT-III associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell Struct. Funct. 2007, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Agromayor, M.; Martin-Serrano, J. Interaction of AMSH with ESCRT-III and deubiquitination of endosomal cargo. J. Biol. Chem. 2006, 281, 23083–23091. [Google Scholar] [CrossRef]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTs. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef]
- Henne, W.M.; Stenmark, H.; Emr, S.D. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 2013, 5, 1288–1302. [Google Scholar] [CrossRef]
- Hasdemir, B.; Murphy, J.E.; Cottrell, G.S.; Bunnett, N.W. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor. J. Biol. Chem. 2009, 284, 28453–28466. [Google Scholar] [CrossRef]
- Hislop, J.N.; Henry, A.G.; Marchese, A.; von Zastrow, M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J. Biol. Chem. 2009, 284, 19361–19370. [Google Scholar] [CrossRef]
- Meijer, I.M.J.; van Rotterdam, W.; van Zoelen, E.J.J.; van Leeuwen, J.E.M. Recycling of EGFR and ErbB2 is associated with impaired Hrs tyrosine phosphorylation and decreased deubiquitination by AMSH. Cell. Signal. 2012, 24, 1981–1988. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Catarino, S.; Marques, C.; Ferreira, J.V.; Martins-Marques, T.; Pereira, P.; Girão, H. AMSH-mediated deubiquitination of Cx43 regulates internalization and degradation of gap junctions. FASEB J. 2014, 28, 4629–4641. [Google Scholar] [CrossRef]
- Sierra, M.I.; Wright, M.H.; Nash, P.D. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR. J. Biol. Chem. 2010, 285, 13990–14004. [Google Scholar] [CrossRef] [PubMed]
- Bednash, J.S.; Weathington, N.; Londino, J.; Rojas, M.; Gulick, D.L.; Fort, R.; Han, S.; McKelvey, A.C.; Chen, B.B.; Mallampalli, R.K. Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat. Commun. 2017, 8, 15203. [Google Scholar] [CrossRef] [PubMed]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Zheng, H.; Gupta, V.; Patterson-Fortin, J.; Bhattacharya, S.; Katlinski, K.; Wu, J.; Varghese, B.; Carbone, C.J.; Aressy, B.; Fuchs, S.Y.; et al. A BRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep. 2013, 5, 180–193. [Google Scholar] [CrossRef]
- Xu, M.; Moresco, J.J.; Chang, M.; Mukim, A.; Smith, D.; Diedrich, J.K.; Yates, J.R.; Jones, K.A. SHMT2 and the BRCC36/BRISC deubiquitinase regulate HIV-1Tat K63-ubiquitylation and destruction by autophagy. PLoS Pathog. 2018, 14, e1007071. [Google Scholar] [CrossRef]
- Yan, K.; Li, L.; Wang, X.; Hong, R.; Zhang, Y.; Yang, H.; Lin, M.; Zhang, S.; He, Q.; Zheng, D.; et al. The deubiquitinating enzyme complex BRISC is required for proper mitotic spindle assembly in mammalian cells. J. Cell Biol. 2015, 210, 209–224. [Google Scholar] [CrossRef]
- Donaghy, R.; Han, X.; Rozenova, K.; Lv, K.; Jiang, Q.; Doepner, M.; Greenberg, R.A.; Tong, W. The BRISC deubiquitinating enzyme complex limits hematopoietic stem cell expansion by regulating JAKK63-ubiquitination. Blood 2019, 133, 1560–1571. [Google Scholar] [CrossRef]
- Shao, G.; Lilli, D.R.; Patterson-Fortin, J.; Coleman, K.A.; Morrissey, D.E.; Greenberg, R.A. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl. Acad. Sci. USA 2009, 106, 3166–3171. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Ribeiro, P. The 19S proteasomal subunit POH1 contributes to the regulation of c-Jun ubiquitination, stability, and subcellular localization. J. Biol. Chem. 2006, 281, 16099–16107. [Google Scholar] [CrossRef]
- Wang, B.; Ma, A.; Zhang, L.; Jin, W.-L.; Qian, Y.; Xu, G.; Qiu, B.; Yang, Z.; Liu, Y.; Xia, Q.; et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat. Commun. 2015, 6, 8704. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Buus, R.; Clague, M.J.; Urbé, S. Regulation of ErbB2 receptor status by the proteasomal DUB POH1. PLoS ONE 2009, 4, e5544. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.R.; Densham, R.M.; Jia, J.; Garvin, A.J.; Stone, H.R.; Shah, V.; Weekes, D.; Festy, F.; Beesley, J.; Morris, J.R. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012, 31, 3918–3934. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.; Sohn, C.; Kaiser, B.; Jensen, E.D.; Mansky, K.C. The 19S proteasomal lid subunit POH1 enhances the transcriptional activation by Mitf in osteoclasts. J. Cell. Biochem. 2010, 109, 967–974. [Google Scholar] [CrossRef]
- Cope, G.A.; Deshaies, R.J. COP9 signalosome: A multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell 2003, 114, 663–671. [Google Scholar] [CrossRef]
- Polo, S. Signaling-mediated control of ubiquitin ligases in endocytosis. BMC. Biol. 2012, 10, 25. [Google Scholar] [CrossRef]
- McCullough, J.; Row, P.E.; Lorenzo, O.; Doherty, M.; Beynon, R.; Clague, M.J.; Urbé, S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr. Biol. 2006, 16, 160–165. [Google Scholar] [CrossRef]
- Davies, C.W.; Paul, L.N.; Das, C. Mechanism of recruitment and activation of the endosome-associated deubiquitinase AMSH. Biochemistry 2013, 52, 7818–7829. [Google Scholar] [CrossRef]
- Hologne, M.; Cantrelle, F.-X.; Riviere, G.; Guillière, F.; Trivelli, X.; Walker, O. NMR reveals the interplay among the AMSH SH3 binding motif, STAM2, and Lys63-linked diubiquitin. J. Mol. Biol. 2016, 428, 4544–4558. [Google Scholar] [CrossRef]
- Azmi, I.F.; Davies, B.A.; Xiao, J.; Babst, M.; Xu, Z.; Katzmann, D.J. ESCRT-III family members stimulate Vps4ATPase activity directly or via Vta1. Dev. Cell 2008, 14, 50–61. [Google Scholar] [CrossRef]
- Tsang, H.T.H.; Connell, J.W.; Brown, S.E.; Thompson, A.; Reid, E.; Sanderson, C.M. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics 2006, 88, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Solomons, J.; Sabin, C.; Poudevigne, E.; Usami, Y.; Hulsik, D.L.; Macheboeuf, P.; Hartlieb, B.; Göttlinger, H.; Weissenhorn, W. Structural basis for ESCRT-III CHMP3 recruitment of AMSH. Structure 2011, 19, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Ishii, N.; Asao, H.; Sugamura, K. Identification of AMSH-LP containing a Jab1/MPN domain metalloenzyme motif. Biochem. Biophys. Res. Commun. 2003, 306, 637–643. [Google Scholar] [CrossRef]
- Davies, C.W.; Paul, L.N.; Kim, M.-I.; Das, C. Structural and thermodynamic comparison of the catalytic domain of AMSH and AMSH-LP: Nearly identical fold but different stability. J. Mol. Biol. 2011, 413, 416–429. [Google Scholar] [CrossRef]
- Rabl, J. BRCA1-A and BRISC: Multifunctional molecular machines for ubiquitin signaling. Biomolecules 2020, 10, 1503. [Google Scholar] [CrossRef]
- Rabl, J.; Bunker, R.D.; Schenk, A.D.; Cavadini, S.; Gill, M.E.; Abdulrahman, W.; Andrés-Pons, A.; Luijsterburg, M.S.; Ibrahim, A.F.M.; Branigan, E.; et al. Structural basis of BRCC36 function in DNA repair and immune regulation. Mol. Cell 2019, 75, 483–497.e489. [Google Scholar] [CrossRef]
- Walden, M.; Tian, L.; Ross, R.L.; Sykora, U.M.; Byrne, D.P.; Hesketh, E.L.; Masandi, S.K.; Cassel, J.; George, R.; Ault, J.R.; et al. Metabolic control of BRISC-SHMT2 assembly regulates immune signalling. Nature 2019, 570, 194–199. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Singh, M.; Kumari, B.; Yadav, U.C.S. Regulation of oxidized LDL-induced inflammatory process through NLRP3 inflammasome activation by the deubiquitinating enzyme BRCC36. Inflamm. Res. 2019, 68, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carvalho, L.P.; Bhattacharya, S.; Carbone, C.J.; Kumar, K.G.S.; Leu, N.A.; Yau, P.M.; Donald, R.G.K.; Weiss, M.J.; Baker, D.P.; et al. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol. Cell. Biol. 2009, 29, 6401–6412. [Google Scholar] [CrossRef]
- Mbonye, U.; Karn, J. The molecular basis for human immunodeficiency virus latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Giardina, G.; Brunotti, P.; Fiascarelli, A.; Cicalini, A.; Costa, M.G.S.; Buckle, A.M.; di Salvo, M.L.; Giorgi, A.; Marani, M.; Paone, A.; et al. How pyridoxal 5′-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015, 282, 1225–1241. [Google Scholar] [CrossRef] [PubMed]
- Szebenyi, D.M.; Liu, X.; Kriksunov, I.A.; Stover, P.J.; Thiel, D.J. Structure of a murine cytoplasmic serine hydroxymethyltransferase quinonoid ternary complex: Evidence for asymmetric obligate dimers. Biochemistry 2000, 39, 13313–13323. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Legube, G. Organizing DNA repair in the nucleus: DSBs hit the road. Curr. Opin. Cell Biol. 2017, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Mohammadian Gol, T.; Rodemann, H.P.; Dittmann, K. Depletion of Akt1 and Akt2 impairs the repair of radiation-induced DNA double strand breaks via homologous recombination. Int. J. Mol. Sci. 2019, 20, 6316. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Bian, C.; Wu, R.; Cho, K.; Yu, X. Loss of BRCA1-A complex function in RAP80 null tumor cells. PLoS ONE 2012, 7, e40406. [Google Scholar] [CrossRef]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Soo Lee, N.; Kim, Y.; Kim, H. Factors forming the BRCA1-A complex orchestrate BRCA1 recruitment to the sites of DNA damage. Acta Biochim. Biophys. Sin. 2016, 48, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Pfeiffer, A. Real estate in the DNA damage response: Ubiquitin and SUMO ligases home in on DNA double-strand breaks. Front. Genet. 2016, 7, 58. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef]
- Smeenk, G.; van Attikum, H. The chromatin response to DNA breaks: Leaving a mark on genome integrity. Annu. Rev. Biochem. 2013, 82, 55–80. [Google Scholar] [CrossRef]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef]
- Hu, X.; Paul, A.; Wang, B. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J. Biol. Chem. 2012, 287, 25510–25519. [Google Scholar] [CrossRef] [PubMed]
- Kyrieleis, O.J.P.; McIntosh, P.B.; Webb, S.R.; Calder, L.J.; Lloyd, J.; Patel, N.A.; Martin, S.R.; Robinson, C.V.; Rosenthal, P.B.; Smerdon, S.J. Three-dimensional architecture of the human BRCA1-A histone deubiquitinase core complex. Cell Rep. 2016, 17, 3099–3106. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Katsuki, Y.; Freire, R.; Shibata, A.; Jeggo, P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013, 41, 10298–10311. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Kastan, M.B. Repair versus checkpoint functions of BRCA1 are differentially regulated by site of chromatin binding. Cancer Res. 2015, 75, 2699–2707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar] [CrossRef]
- Solyom, S.; Aressy, B.; Pylkäs, K.; Patterson-Fortin, J.; Hartikainen, J.M.; Kallioniemi, A.; Kauppila, S.; Nikkilä, J.; Kosma, V.-M.; Mannermaa, A.; et al. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci. Transl. Med. 2012, 4, 122ra123. [Google Scholar] [CrossRef]
- Guzzo, C.M.; Berndsen, C.E.; Zhu, J.; Gupta, V.; Datta, A.; Greenberg, R.A.; Wolberger, C.; Matunis, M.J. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci. Signal. 2012, 5, ra88. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Wu, Q.; Paul, A.; Su, D.; Mehmood, S.; Foo, T.K.; Ochi, T.; Bunting, E.L.; Xia, B.; Robinson, C.V.; Wang, B.; et al. Structure of BRCA1-BRCT/Abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol.Cell 2016, 61, 434–448. [Google Scholar] [CrossRef]
- Vikrant; Sawant, U.U.; Varma, A.K. Role of MERIT40 in stabilization of BRCA1 complex: A protein-protein interaction study. Biochem. Biophys. Res. Commun. 2014, 446, 1139–1144. [Google Scholar] [CrossRef]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Glickman, M.H.; Adir, N. The proteasome and the delicate balance between destruction and rescue. PLoS Biol. 2004, 2, E13. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.M.; Aranda-Orgilles, B.; Strikoudis, A.; Apostolou, E.; Loizou, E.; Moran-Crusio, K.; Farnsworth, C.L.; Koller, A.A.; Dasgupta, R.; Silva, J.C.; et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem. Cell 2012, 11, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.-P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828. [Google Scholar] [CrossRef]
- Shin, J.Y.; Muniyappan, S.; Tran, N.-N.; Park, H.; Lee, S.B.; Lee, B.-H. Deubiquitination reactions on the proteasome for proteasome versatility. Int. J. Mol. Sci. 2020, 21, 5312. [Google Scholar] [CrossRef]
- Luo, G.; Hu, N.; Xia, X.; Zhou, J.; Ye, C. RPN11 deubiquitinase promotes proliferation and migration of breast cancer cells. Mol. Med. Rep. 2017, 16, 331–338. [Google Scholar] [CrossRef]
- De la Peña, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-engaged 26 proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef]
- Ding, Z.; Xu, C.; Sahu, I.; Wang, Y.; Fu, Z.; Huang, M.; Wong, C.C.L.; Glickman, M.H.; Cong, Y. Structural snapshots of 26S proteasome reveal tetraubiquitin-induced conformations. Mol. Cell 2019, 73, 1150–1161.e1156. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Sahu, I.; Glickman, M.H. Proteasome in action: Substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021, 49, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.; Hartmann-Petersen, R.; Seeger, M.; Bech-Otschir, D.; Wallace, M.; Gordon, C. Uch2/Uch37 is the major deubiquitinating enzyme associated with the 26S proteasome in fission yeast. J. Mol. Biol. 2004, 344, 697–706. [Google Scholar] [CrossRef]
- Park, K.C.; Woo, S.K.; Yoo, Y.J.; Wyndham, A.M.; Baker, R.T.; Chung, C.H. Purification and characterization of UBP6, a new ubiquitin-specific protease in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 1997, 347, 78–84. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Lander, G.C.; Martin, A. Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol. 2013, 20, 781–788. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Bio. 2005, 6, 519–529. [Google Scholar] [CrossRef]
- Wei, N.; Deng, X.W. The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 2003, 19, 261–286. [Google Scholar] [CrossRef]
- Wolf, D.A.; Zhou, C.; Wee, S. The COP9 signalosome: An assembly and maintenance platform for cullin ubiquitin ligases? Nat. Cell Biol. 2003, 5, 1029–1033. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Schulman, B.A.; Harper, J.W. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013, 14, 1050–1061. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Lyapina, S.; Cope, G.; Shevchenko, A.; Serino, G.; Tsuge, T.; Zhou, C.; Wolf, D.A.; Wei, N.; Shevchenko, A.; Deshaies, R.J. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science 2001, 292, 1382–1385. [Google Scholar] [CrossRef]
- Shackleford, T.J.; Claret, F.X. JAB1/CSN5: A new player in cell cycle control and cancer. Cell Div. 2010, 5, 26. [Google Scholar] [CrossRef]
- Pan, Y.; Yang, H.; Claret, F.X. Emerging roles of Jab1/CSN5 in DNA damage response, DNA repair, and cancer. Cancer Biol. Ther. 2014, 15, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, K.; Yoneda-Kato, N.; Fukumoto, A.; Yamanaka, S.; Kato, J.-Y. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J. Biol. Chem. 2004, 279, 43013–43018. [Google Scholar] [CrossRef]
- Lykke-Andersen, K.; Schaefer, L.; Menon, S.; Deng, X.-W.; Miller, J.B.; Wei, N. Disruption of the COP9 signalosome Csn2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol. Cell Biol. 2003, 23, 6790–6797. [Google Scholar] [CrossRef]
- Yan, J.; Walz, K.; Nakamura, H.; Carattini-Rivera, S.; Zhao, Q.; Vogel, H.; Wei, N.; Justice, M.J.; Bradley, A.; Lupski, J.R. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol. Cell Biol. 2003, 23, 6798–6808. [Google Scholar] [CrossRef]
- Peng, Z.; Shen, Y.; Feng, S.; Wang, X.; Chitteti, B.N.; Vierstra, R.D.; Deng, X.W. Evidence for a physical association of the COP9 signalosome, the proteasome, and specific SCF E3 ligases in vivo. Curr. Biol. 2003, 13, R504–R505. [Google Scholar] [CrossRef]
- Wan, M.; Tang, Y.; Tytler, E.M.; Lu, C.; Jin, B.; Vickers, S.M.; Yang, L.; Shi, X.; Cao, X. Smad4 protein stability is regulated by ubiquitin ligase SCF beta-TrCP1. J. Biol. Chem. 2004, 279, 14484–14487. [Google Scholar] [CrossRef]
- Kim, B.-C.; Lee, H.-J.; Park, S.H.; Lee, S.R.; Karpova, T.S.; McNally, J.G.; Felici, A.; Lee, D.K.; Kim, S.-J. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor beta signaling by binding to Smad7 and promoting its degradation. Mol. Cell Biol. 2004, 24, 2251–2262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cope, G.A.; Deshaies, R.J. Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 2006, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Wang, B.; Ma, Y.; Chen, P. CSN5/Jab1 facilitates non-small cell lung cancer cell growth through stabilizing survivin. Biochem. Biophys. Res. Commun. 2018, 500, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef]
- Adler, A.S.; Lin, M.; Horlings, H.; Nuyten, D.S.A.; van de Vijver, M.J.; Chang, H.Y. Genetic regulators of large-scale transcriptional signatures in cancer. Nat. Genet. 2006, 38, 421–430. [Google Scholar] [CrossRef]
- Wei, N.; Serino, G.; Deng, X.-W. The COP9 signalosome: More than a protease. Trends Biochem. Sci. 2008, 33, 592–600. [Google Scholar] [CrossRef]
- Bemis, L.; Chan, D.A.; Finkielstein, C.V.; Qi, L.; Sutphin, P.D.; Chen, X.; Stenmark, K.; Giaccia, A.J.; Zundel, W. Distinct aerobic and hypoxic mechanisms of HIF-alpha regulation by CSN5. Genes Dev. 2004, 18, 739–744. [Google Scholar] [CrossRef]
- Wee, S.; Geyer, R.K.; Toda, T.; Wolf, D.A. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat. Cell Biol. 2005, 7, 387–391. [Google Scholar] [CrossRef]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.-i.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar] [CrossRef]
- Holmberg, C.; Fleck, O.; Hansen, H.A.; Liu, C.; Slaaby, R.; Carr, A.M.; Nielsen, O. Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 2005, 19, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.-L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Powell, K.A.; Mundt, K.; Wu, L.; Carr, A.M.; Caspari, T. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 2003, 17, 1130–1140. [Google Scholar] [CrossRef]
- Higa, L.A.A.; Mihaylov, I.S.; Banks, D.P.; Zheng, J.; Zhang, H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 2003, 5, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Y.; Douglas, L.; Zhou, P. UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J. Biol. Chem. 2001, 276, 48175–48182. [Google Scholar] [CrossRef]
- Chen, L.C.; Manjeshwar, S.; Lu, Y.; Moore, D.; Ljung, B.M.; Kuo, W.L.; Dairkee, S.H.; Wernick, M.; Collins, C.; Smith, H.S. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res. 1998, 58, 3677–3683. [Google Scholar]
- Hori, T.; Osaka, F.; Chiba, T.; Miyamoto, C.; Okabayashi, K.; Shimbara, N.; Kato, S.; Tanaka, K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 1999, 18, 6829–6834. [Google Scholar] [CrossRef]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.J.; et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef]
- Enchev, R.I.; Scott, D.C.; da Fonseca, P.C.A.; Schreiber, A.; Monda, J.K.; Schulman, B.A.; Peter, M.; Morris, E.P. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2012, 2, 616–627. [Google Scholar] [CrossRef]
- Faull, S.V.; Lau, A.M.C.; Martens, C.; Ahdash, Z.; Hansen, K.; Yebenes, H.; Schmidt, C.; Beuron, F.; Cronin, N.B.; Morris, E.P.; et al. Structural basis of Cullin RING E3 ligase regulation by the COP9 signalosome. Nat. Commun. 2019, 10, 3814. [Google Scholar] [CrossRef]
- Ren, G.-M.; Li, J.; Zhang, X.-C.; Wang, Y.; Xiao, Y.; Zhang, X.-Y.; Liu, X.; Zhang, W.; Ma, W.-B.; Zhang, J.; et al. Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3-associated inflammatory diseases. Sci. Immunol. 2021, 6, eabe2933. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Da Silva Sil Dos Santos, B.; Wang, F.; Ma, Y.; Perez, C.; Yang, Y.; Peng, J.; Cohen, S.M.; Chou, T.-F.; et al. Epidithiodiketopiperazines inhibit protein degradation by targeting proteasome deubiquitinase Rpn11. Cell Chem. Biol. 2018, 25, 1350–1358.e1359. [Google Scholar] [CrossRef]
- Song, Y.; Li, S.; Ray, A.; Das, D.S.; Qi, J.; Samur, M.K.; Tai, Y.T.; Munshi, N.; Carrasco, R.D.; Chauhan, D.; et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene 2017, 36, 5631–5638. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yakushi, T.; Parlati, F.; Mackinnon, A.L.; Perez, C.; Ma, Y.; Carter, K.P.; Colayco, S.; Magnuson, G.; Brown, B.; et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat. Chem. Biol. 2017, 13, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Zhang, N.; Yin, M.; Dong, J.; Zeng, Q.; Mao, G.; Song, D.; Liu, L.; Deng, H. Berberine diminishes cancer cell PD-L1 expression and facilitates antitumor immunity inhibiting the deubiquitination activity of CSN5. Acta Pharm. Sin. B 2020, 10, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef]
- Mikhael, J.R.; Dingli, D.; Roy, V.; Reeder, C.B.; Buadi, F.K.; Hayman, S.R.; Dispenzieri, A.; Fonseca, R.; Sher, T.; Kyle, R.A.; et al. Management of newly diagnosed symptomatic multiple myeloma: Updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin. Proc. 2013, 88, 360–376. [Google Scholar] [CrossRef]
- Hadjiaggelidou, C.; Katodritou, E. Regulatory T-cells and multiple myeloma: Implications in tumor immune biology and treatment. J. Clin. Med. 2021, 10, 4588. [Google Scholar] [CrossRef]
- Van der Linden, W.A.; Willems, L.I.; Shabaneh, T.B.; Li, N.; Ruben, M.; Florea, B.I.; van der Marel, G.A.; Kaiser, M.; Kisselev, A.F.; Overkleeft, H.S. Discovery of a potent and highly β1 specific proteasome inhibitor from a focused library of urea-containing peptide vinyl sulfones and peptide epoxyketones. Org. Biomol. Chem. 2012, 10, 181–194. [Google Scholar] [CrossRef]
- Desvergne, A.; Genin, E.; Maréchal, X.; Gallastegui, N.; Dufau, L.; Richy, N.; Groll, M.; Vidal, J.; Reboud-Ravaux, M. Dimerized linear mimics of a natural cyclopeptide (TMC-95A) are potent noncovalent inhibitors of the eukaryotic 20S proteasome. J. Med. Chem. 2013, 56, 3367–3378. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Unno, Y.; List, A.; Mizuno, A.; Tanaka, M.; Sasaki, T.; Arisawa, M.; Asai, A.; Groll, M.; Shuto, S. Potent proteasome inhibitors derived from the unnatural cis-cyclopropane isomer of Belactosin A: Synthesis, biological activity, and mode of action. J. Med. Chem. 2013, 56, 3689–3700. [Google Scholar] [CrossRef]
- Ozcan, S.; Kazi, A.; Marsilio, F.; Fang, B.; Guida, W.C.; Koomen, J.; Lawrence, H.R.; Sebti, S.M. Oxadiazole-isopropylamides as potent and noncovalent proteasome inhibitors. J. Med. Chem. 2013, 56, 3783–3805. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Richardson, P.G.; Moreau, P.; Anderson, K.C. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat. Rev. Clin. Oncol. 2015, 12, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Li, J.; Parlati, F.; Rouffet, M.; Ma, Y.; Mackinnon, A.L.; Chou, T.-F.; Deshaies, R.J.; Cohen, S.M. Discovery of an inhibitor of the proteasome subunit Rpn11. J. Med. Chem. 2017, 60, 1343–1361. [Google Scholar] [CrossRef] [PubMed]
- Lauinger, L.; Li, J.; Shostak, A.; Cemel, I.A.; Ha, N.; Zhang, Y.; Merkl, P.E.; Obermeyer, S.; Stankovic-Valentin, N.; Schafmeier, T.; et al. Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat. Chem. Biol. 2017, 13, 709–714. [Google Scholar] [CrossRef]
- Li, B.; Wever, W.J.; Walsh, C.T.; Bowers, A.A. Dithiolopyrrolones: Biosynthesis, synthesis, and activity of a unique class of disulfide-containing antibiotics. Nat. Prod. Rep. 2014, 31, 905–923. [Google Scholar] [CrossRef]
- Lee, M.-H.; Zhao, R.; Phan, L.; Yeung, S.-C.J. Roles of COP9 signalosome in cancer. Cell Cycle 2011, 10, 3057–3066. [Google Scholar] [CrossRef]
- Lee, Y.H.; Judge, A.D.; Seo, D.; Kitade, M.; Gómez-Quiroz, L.E.; Ishikawa, T.; Andersen, J.B.; Kim, B.K.; Marquardt, J.U.; Raggi, C.; et al. Molecular targeting of CSN5 in human hepatocellular carcinoma: A mechanism of therapeutic response. Oncogene 2011, 30, 4175–4184. [Google Scholar] [CrossRef]
- Altmann, E.; Erbel, P.; Renatus, M.; Schaefer, M.; Schlierf, A.; Druet, A.; Kieffer, L.; Sorge, M.; Pfister, K.; Hassiepen, U.; et al. Azaindoles as zinc-binding small-molecule inhibitors of the JAMM protease CSN5. Angew. Chem. 2017, 56, 1294–1297. [Google Scholar] [CrossRef]
- Ni, W.-J.; Ding, H.-H.; Tang, L.-Q. Berberine as a promising anti-diabetic nephropathy drug: An analysis of its effects and mechanisms. Eur. J. Pharmacol. 2015, 760, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, G.; Yang, J.; Pan, X.; Yang, Z.; Zang, L. Berberine inhibits human hepatoma cell invasion without cytotoxicity in healthy hepatocytes. PLoS ONE 2011, 6, e21416. [Google Scholar] [CrossRef] [PubMed]
- Chidambara Murthy, K.N.; Jayaprakasha, G.K.; Patil, B.S. The natural alkaloid berberine targets multiple pathways to induce cell death in cultured human colon cancer cells. Eur. J. Pharmacol. 2012, 688, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Rose, I.A. Ubiquitin-aldehyde: A general inhibitor of ubiquitin-recycling processes. Proc. Natl. Acad. Sci. USA 1987, 84, 1829–1833. [Google Scholar] [CrossRef]
- Ndubaku, C.; Tsui, V. Inhibiting the deubiquitinating enzymes (DUBs). J. Med. Chem. 2015, 58, 1581–1595. [Google Scholar] [CrossRef]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.-K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 26979. [Google Scholar] [CrossRef]
- Kumar, V.; Naumann, M.; Stein, M. Computational studies on the inhibitor selectivity of human JAMM deubiquitinylases Rpn11 and CSN5. Front. Chem. 2018, 6, 480. [Google Scholar] [CrossRef]
- Fiore, A.; Liang, Y.; Lin, Y.H.; Tung, J.; Wang, H.; Langlais, D.; Nijnik, A. Deubiquitinase MYSM1 in the hematopoietic system and beyond: A current review. Int. J. Mol. Sci. 2020, 21, 3007. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Functional Complex | Target Protein | Linkage Type | Regulation Effects | Ref. |
|---|---|---|---|---|---|
| AMSH | N/A | EGFR | K63- | Promote the recycling of EGFR | [59] |
| AMSH | N/A | Cx43 | K63- | Protect gap junctions from degradation to regulate the intercellular communication | [60] |
| AMSH | N/A | NALP7 | K63- | Lead to the inflammasome-dependent IL-1β cleavage and release | [62] |
| AMSH | N/A | CXCR4 | Mono- | Regulate the stability and trafficking of CXCR4 | [61] |
| AMSH | N/A | PAR2 | Mono- | Regulate the trafficking and down-regulation of PAR2 | [57] |
| AMSH | N/A | DOR | Mono- | Regulate the downregulation of the DOR | [58] |
| BRCC36 | BRISC | NLRP3 | K63- | Activate NLRP3 and promote inflammasome assembly | [64] |
| BRCC36 | BRISC | IFNAR1/2 | K63- | Promote the cellular response to Type I interferons | [65] |
| BRCC36 | BRISC | HIV-1 Tat | K63- | Rescue Tat from destruction to potentiate the effectiveness of antiviral regimens | [66] |
| BRCC36 | BRISC | NuMA | K63- | Promote the assembly of functional bipolar spindle during mitosis | [67] |
| BRCC36 | BRISC | JAK2 | K63- | Limit hematopoietic stem cell expansion | [68] |
| BRCC36 | BRCA1-A | H2A/H2AX | K63- | Suppress hyperactive HR repair | [69] |
| Rpn11 | 26S proteasome | c-Jun | K48- | Maintain a stable intracellular concentration of c-Jun | [70] |
| Rpn11 | 26S proteasome | E2F1 | K63- | Stabilize E2F1 protein to promote tumorigenesis | [71] |
| Rpn11 | 26S proteasome | ErbB2 | ND | Regulate ErbB2 ubiquitylation and stability in cancer cells | [72] |
| Rpn11 | 26S proteasome | H2A/H2AX | K63- | Promote the correct coordination of the cellular response to DSB | [73] |
| Rpn11 | 26S proteasome | Mitf | ND | Allow more stable Mitf expression in osteoclast differentiation process | [74] |
| CSN5 | Cop9 signalosome | CRLs | NEDD8 | Maintain the proper activity of CRLs in myriad cellular processes | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Wu, S.; Wei, W.; Chen, Z.; Wu, Y.; Gong, K. Structural and Functional Basis of JAMM Deubiquitinating Enzymes in Disease. Biomolecules 2022, 12, 910. https://doi.org/10.3390/biom12070910
Pan X, Wu S, Wei W, Chen Z, Wu Y, Gong K. Structural and Functional Basis of JAMM Deubiquitinating Enzymes in Disease. Biomolecules. 2022; 12(7):910. https://doi.org/10.3390/biom12070910
Chicago/Turabian StylePan, Xin, Sihua Wu, Wenping Wei, Zixuan Chen, Yong Wu, and Kaizheng Gong. 2022. "Structural and Functional Basis of JAMM Deubiquitinating Enzymes in Disease" Biomolecules 12, no. 7: 910. https://doi.org/10.3390/biom12070910
APA StylePan, X., Wu, S., Wei, W., Chen, Z., Wu, Y., & Gong, K. (2022). Structural and Functional Basis of JAMM Deubiquitinating Enzymes in Disease. Biomolecules, 12(7), 910. https://doi.org/10.3390/biom12070910