
Pitfalls of Antiretroviral Therapy: Current Status and Long-Term CNS Toxicity


Abstract
:1. Introduction
2. HIV Infection within the CNS
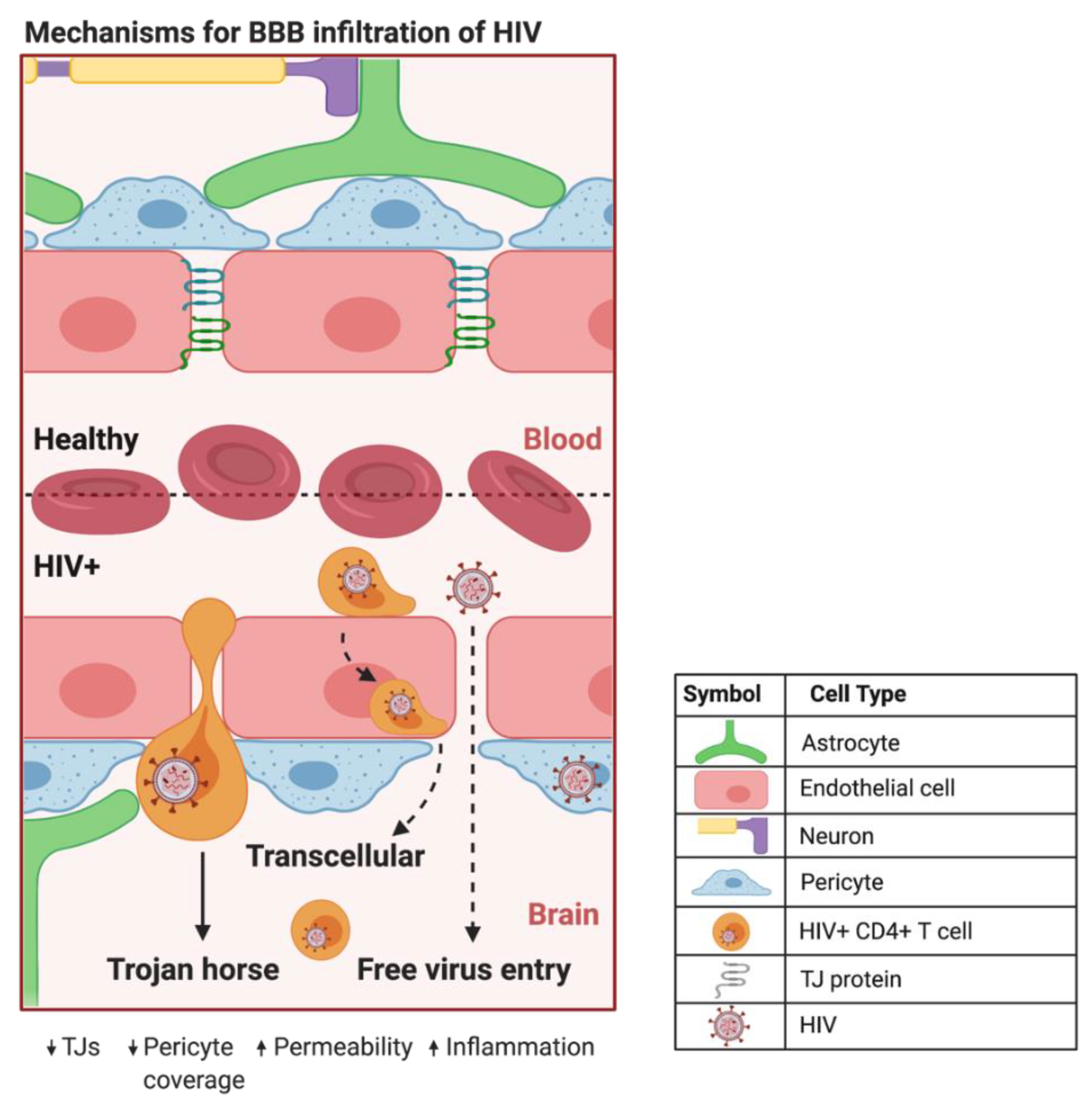
2.1. The Blood-Brain Barrier
2.2. A Trojan Horse Mechanism for HIV Infection of the CNS
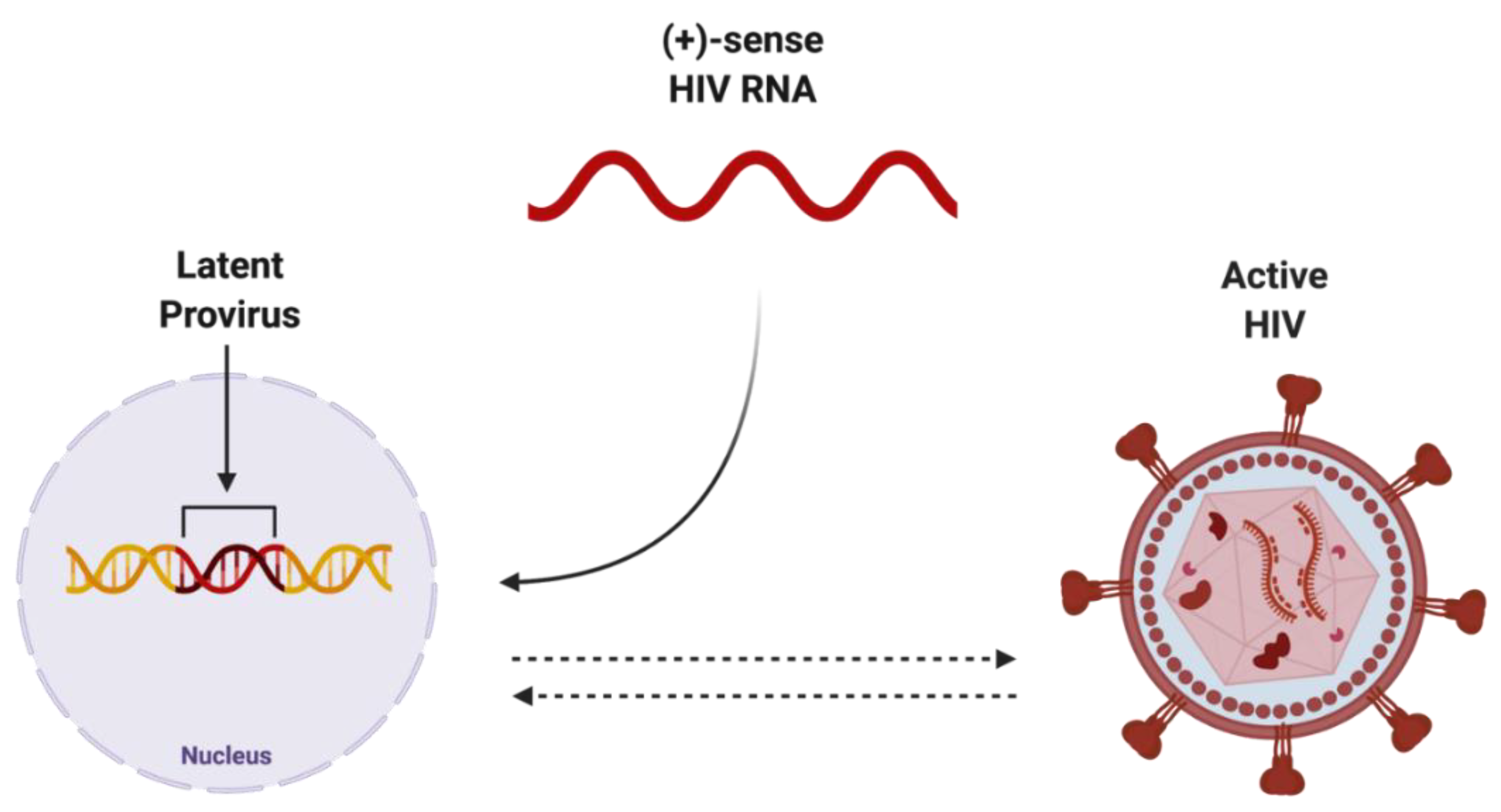
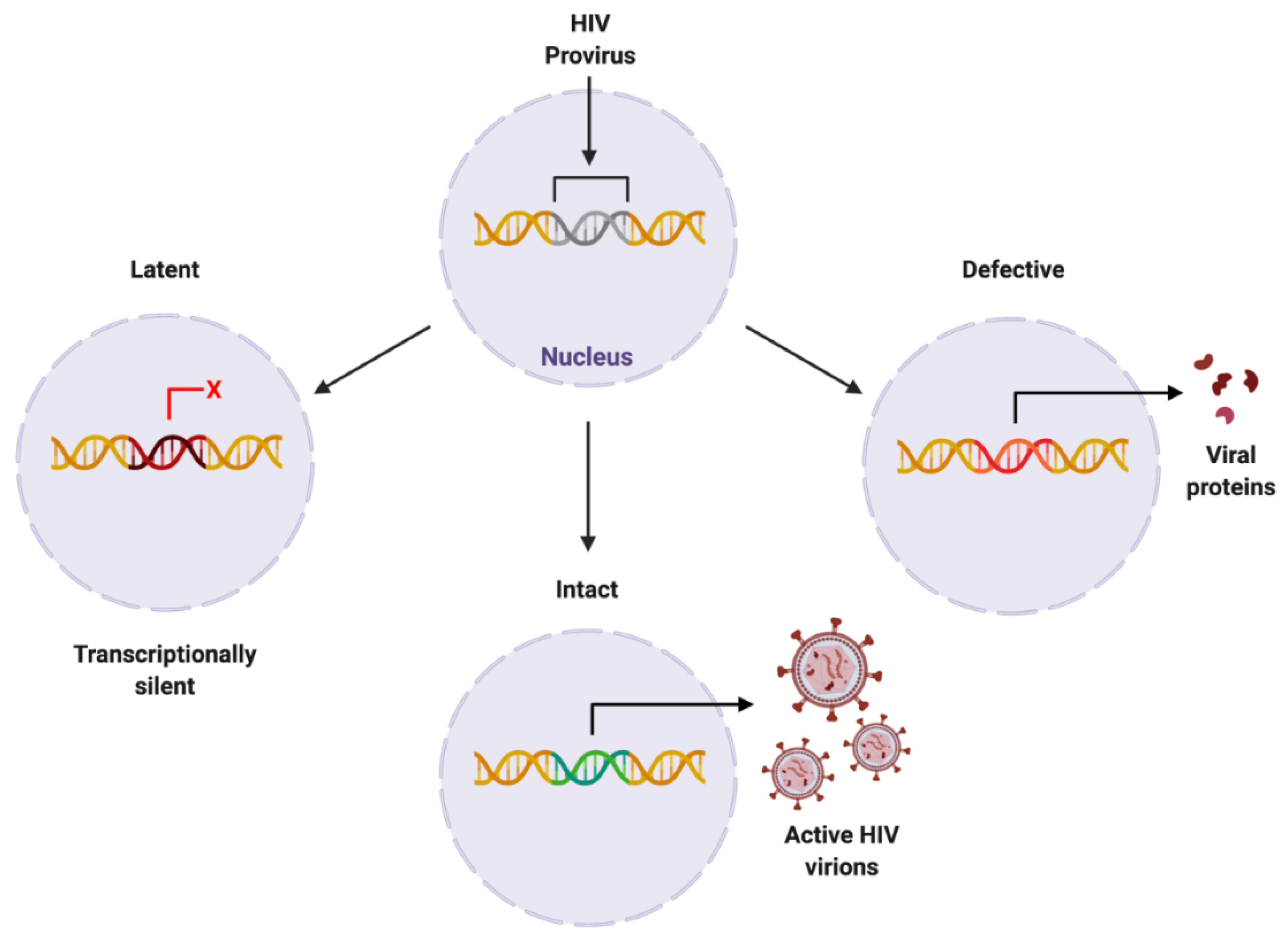
2.3. Elusive Latent Proviral Reservoirs within the CNS
3. Pitfalls of ART: Focus on the CNS
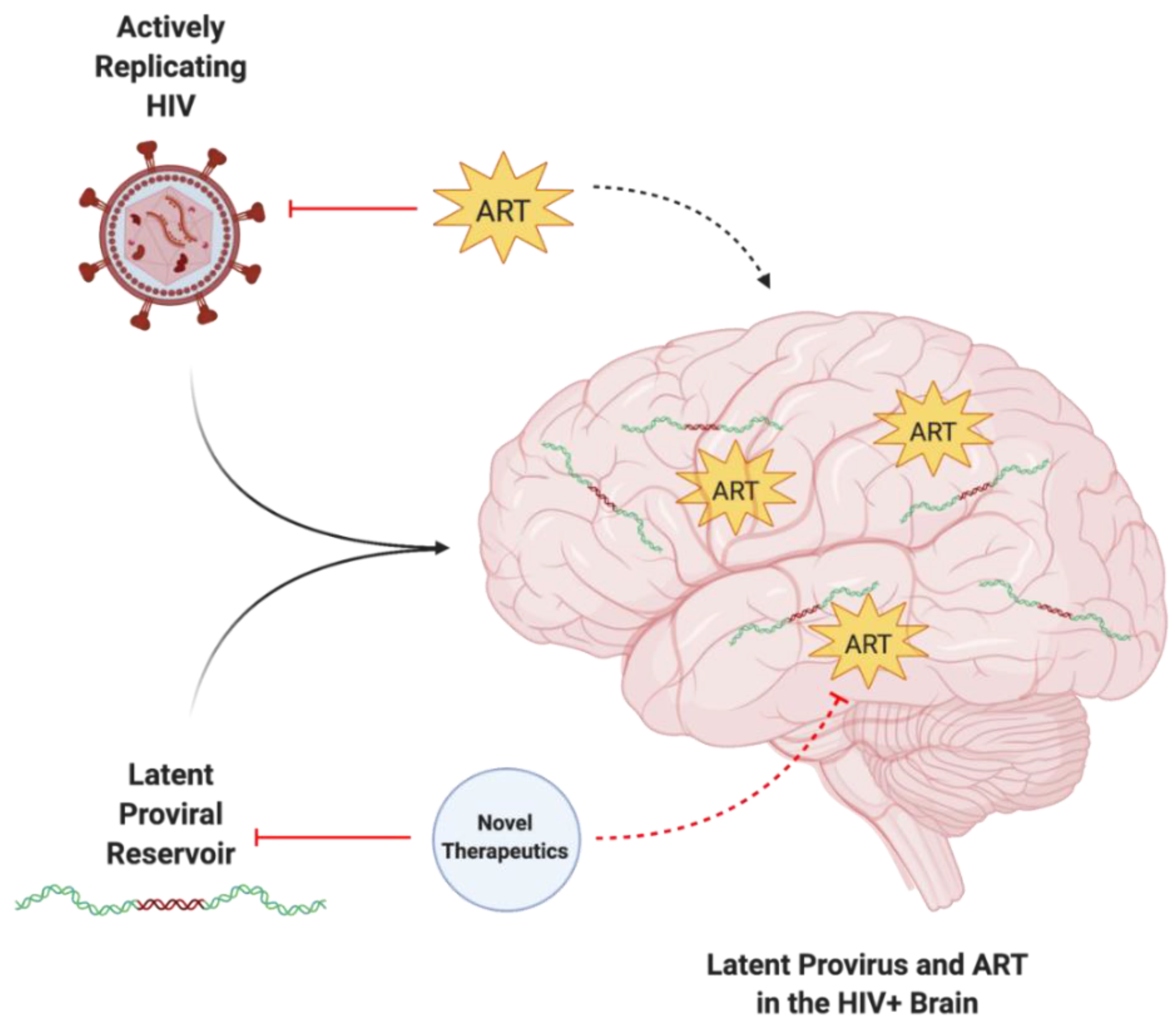
3.1. ART Does Not Protect against Latent HIV Infections
3.2. ART Has Limited BBB Penetrance
3.3. ART-Induced CNS Toxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Impact from ART | References |
|---|---|---|
| Astrocyte | ↓ Mitochondria function and metabolism ↓ MMPs ↑ Senescence ↑ ER stress | [3,13,77] |
| Endothelial Cell | ↓ Viability ↓ Mitochondria function ↓ Autophagosome formation ↓ TJPs ↑ ROS production ↑ ER stress ↑ Inflammatory cytokine production | [3,12,36,75,78,79] |
| Microglial Cell | ↓ Lysosomal function ↓ Autophagosomal function ↑ ROS production ↑ Expression of pro-inflammatory cytokines | [10] |
| Neuron | ↓ Axonal length ↓ Neurogenesis ↑ Neuronal death ↑ Oxidative stress ↑ ROS accumulation | [4,73,80,81] |
| Neural Progenitor Cell | ↓ Cell proliferation ↓ Mitochondrial function ↑ Senescence ↑ ROS production ↑ MMP production | [82,83,84] |
| Oligodendrocyte | ↓ Maturation ↑ ROS production ↑ Oxidative stress ↑ Lysosomal stress | [85,86] |
| Pericyte | ↓ Coverage | [87] |
4. Novel Strategies in Developing HIV Therapeutics
4.1. CNS Targeting of Latent HIV Provirus
4.2. Obstructing Infection by HIV
5. Concluding Comments
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. HIV Suveillance Report. 2018. Available online: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html (accessed on 19 September 2019).
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2015, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Velichkovska, M.; Toborek, M. Cerebral vascular toxicity of antiretroviral therapy. J. Neuroimmune Pharmacol. 2019, 16, 74–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akay, C.; Cooper, M.; Odeleye, A.; Jensen, B.K.; White, M.G.; Vassoler, F.; Gannon, P.J.; Mankowski, J.; Dorsey, J.L.; Buch, A.M.; et al. Antiretroviral drugs induce oxidative stress and neuronal damage in the Central Nervous System. J. NeuroVirology 2014, 20, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A.; Wang, C.; Patton, C.; Murray, M.; Punekar, Y.; de Ruiter, A.; Steinhart, C. A review of long-term toxicity of antiretroviral treatment regimens and implications for an aging population. Infect. Dis. Ther. 2018, 7, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.A.; Swinton, M.K.; Carson, A.; Soontornniyomkij, B.; Lindsay, C.; Han, M.M.; Frizzi, K.; Sambhwani, S.; Murphy, A.; Achim, C.L.; et al. Tenofovir disoproxil fumarate induces peripheral neuropathy and alters inflammation and mitochondrial biogenesis in the Brains of Mice. Sci. Rep. 2019, 9, 17158. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.; Landay, A.; Miyahara, S.; Vecchio, A.; Masters, M.C.; Brown, T.T.; Taiwo, B.O. Limited correlation between systemic biomarkers and neurocognitive performance before and during HIV treatment. J. NeuroVirology 2019, 26, 107–113. [Google Scholar] [CrossRef]
- Williams, D.W.; Li, Y.; Dastgheyb, R.; Fitzgerald, K.C.; Maki, P.M.; Spence, A.B.; Gustafson, D.R.; Milam, J.; Sharma, A.; Adimora, A.A.; et al. Associations between antiretroviral drugs on depressive symptomatology in homogenous subgroups of women with HIV. J. Neuroimmune Pharmacol. 2020, 16, 181–194. [Google Scholar] [CrossRef]
- Velichkovska, M.; Surnar, B.; Nair, M.; Dhar, S.; Toborek, M. Targeted mitochondrial COQ10 delivery attenuates antiretroviral-drug-induced senescence of neural progenitor cells. Mol. Pharm. 2018, 16, 724–736. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Guo, M.-L.; Buch, S. Antiretroviral-mediated microglial activation involves dysregulated autophagy and lysosomal dysfunction. Cells 2019, 8, 1168. [Google Scholar] [CrossRef] [Green Version]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The impact of HIV- and art-induced mitochondrial dysfunction in cellular senescence and aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef]
- Smith, R.L.; Tan, J.M.; Jonker, M.J.; Jongejan, A.; Buissink, T.; Veldhuijzen, S.; van Kampen, A.H.; Brul, S.; van der Spek, H. Beyond the polymerase-γ theory: Production of ROS as a mode of NRTI-induced mitochondrial toxicity. PLoS ONE 2017, 12, e0187424. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; D’Agostino, L.; Wilson, J.; Tuzer, F.; Torres, C. Astrocyte senescence and metabolic changes in response to HIV antiretroviral therapy drugs. Front. Aging Neurosci. 2017, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhang, J.; Geng, M.; Tang, S.-J.; Zhang, W.; Shu, J. Nucleoside reverse transcriptase inhibitors (nrtis) induce proinflammatory cytokines in the CNS via WNT5A signaling. Sci. Rep. 2017, 7, 4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Toborek, M.; Lee, Y.W.; Flora, G.; Pu, H.; András, I.E.; Wylegala, E.; Hennig, B.; Nath, A. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell. Mol. Neurobiol. 2005, 25, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nagai, N.; Umemura, K. A review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front. Cell. Neurosci. 2016, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Kurmann, L.; Okoniewski, M.; Dubey, R.K. Transcryptomic analysis of human brain -microvascular endothelial cell driven changes in -vascular pericytes. Cells 2021, 10, 1784. [Google Scholar] [CrossRef]
- Miller, D.S.; Bauer, B.; Hartz, A.M. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Xie, Y.; Chen, Y. Effect of Neuroinflammation on ABC Transporters: Possible Contribution to Refractory Epilepsy. CNS Neurol. Disord. Drug Targets 2018, 17, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, B.; Li, X.; Liu, G.; Liu, R.; Guo, J.; Xu, B.; Li, Y.; Fang, W. Significance and Mechanisms of P-glycoprotein in Central Nervous System Diseases. Curr. Cancer Drug Targets 2019, 20, 1141–1155. [Google Scholar] [CrossRef]
- Hayashi, K.; Pu, H.; Tian, J.; Andras, I.E.; Lee, Y.W.; Hennig, B.; Toborek, M. HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J. Neurochem. 2005, 93, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Hennig, B.; Toborek, M. Intact lipid rafts regulate HIV-1 Tat protein-induced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J. Cereb. Blood Flow Metab. 2010, 30, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, U.; Bulot, C.; Honer zu Bentrup, K.; Mondal, D. Specific increase in MDR1 mediated drug-efflux in human brain endothelial cells following co-exposure to HIV-1 and saquinavir. PLoS ONE 2013, 8, e75374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.N.; Patel, R.; Cummins, C.L.; Bendayan, R. Induction of P-glycoprotein by several antiretroviral drugs in human brain microvessel endothelial cells. Antimicrob. Agents Chemother. 2013, 57, 4481–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The paradox of HIV blood-brain barrier penetrance and antiretroviral drug delivery deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T cell development, localization, and function throughout life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, L.; Cho, H.J.; Toborek, M. Blood-brain barrier pericytes as a target for HIV-1 infection. Brain 2019, 142, 502–511. [Google Scholar] [CrossRef]
- Li, G.-H.; Henderson, L.; Nath, A. Astrocytes as an HIV reservoir: Mechanism of HIV infection. Curr. HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef]
- Joseph, S.B.; Arrildt, K.T.; Sturdevant, C.B.; Swanstrom, R. HIV-1 target cells in the CNS. J. NeuroVirology 2014, 21, 276–289. [Google Scholar] [CrossRef] [PubMed]
- András, I.E.; Toborek, M. HIV-1-induced alterations of Claudin-5 expression at the blood-brain barrier level. Methods Mol. Biol. 2011, 762, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Castro, V.; Toborek, M. Infection of human pericytes by HIV-1 disrupts the integrity of the blood-brain barrier. J. Cell. Mol. Med. 2012, 16, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, H.; Minato, N. Myeloid cells. Int. J. Biochem. Cell Biol. 2004, 36, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Dygert, L.; Toborek, M. Antiretroviral treatment with Efavirenz disrupts the blood-brain barrier integrity and increases stroke severity. Sci. Rep. 2016, 6, 39738. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Zhang, B.; Eum, S.Y.; Toborek, M. HIV-1 TAT triggers nuclear localization of ZO-1 via rho signaling and camp response element-binding protein activation. J. Neurosci. 2012, 32, 143–150. [Google Scholar] [CrossRef] [Green Version]
- András, I.E.; Pu, H.; Tian, J.; Deli, M.A.; Nath, A.; Hennig, B.; Toborek, M. Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J. Cereb. Blood Flow Metab. 2005, 25, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.W.; Eum, S.Y.; Nath, A.; Toborek, M. Estrogen-mediated protection against HIV Tat protein-induced inflammatory pathways in human vascular endothelial cells. Cardiovasc. Res. 2004, 63, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and art. FEBS J. 2018, 286, 1256–1270. [Google Scholar] [CrossRef] [Green Version]
- Henderson, L.J.; Reoma, L.B.; Kovacs, J.A.; Nath, A. Advances toward curing HIV-1 infection in tissue reservoirs. J. Virol. 2020, 94, e00375-19. [Google Scholar] [CrossRef]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial cells: The main HIV-1 reservoir in the brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249. [Google Scholar] [CrossRef] [Green Version]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef]
- Gu, C.J.; Borjabad, A.; Hadas, E.; Kelschenbach, J.; Kim, B.H.; Chao, W.; Arancio, O.; Suh, J.; Polsky, B.; McMillan, J.; et al. EcoHIV infection of mice establishes latent viral reservoirs in T cells and active viral reservoirs in macrophages that are sufficient for induction of neurocognitive impairment. PLoS Pathog. 2018, 14, e1007061. [Google Scholar] [CrossRef] [Green Version]
- Delery, E.C.; MacLean, A.G. Culture Model for Non-human Primate Choroid Plexus. Front. Cell. Neurosci. 2019, 13, 396. [Google Scholar] [CrossRef]
- Meeker, R.B.; Williams, K.; Killebrew, D.A.; Hudson, L.C. Cell trafficking through the choroid plexus. Cell Adhes. Migr. 2012, 6, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Burkala, E.J.; He, J.; West, J.T.; Wood, C.; Petito, C.K. Compartmentalization of HIV-1 in the central nervous system: Role of the choroid plexus. AIDS 2005, 19, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.V.; Dahlheimer, J.L.; Bardgett, M.E.; Snyder, A.Z.; Finch, R.A.; Sartorelli, A.C.; Piwnica-Worms, D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 3900–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, L.; Nair, M.; Toborek, M. Solving the blood-brain barrier challenge for the effective treatment of HIV replication in the central nervous system. Curr. Pharm. Des. 2016, 22, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J.; Kwon, K.J.; Farber, D.L.; Siliciano, R.F. The Latent Reservoir for HIV-1: How immunologic memory and clonal expansion contribute to HIV-1 persistence. J. Immunol. 2016, 197, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.-H.; Lichterfeld, M. Recent progress in understanding HIV reservoirs. Curr. Opin. HIV AIDS 2018, 13, 137–142. [Google Scholar] [CrossRef]
- Darcis, G.; Maes, N.; Pasternak, A.O.; Sauvage, A.-S.; Frippiat, F.; Meuris, C.; Uurlings, F.; Lecomte, M.; Léonard, P.; Elmoussaoui, M.; et al. Detectability of HIV residual viremia despite therapy is highly associated with treatment with a protease inhibitor-based combination antiretroviral therapy. Antimicrob. Agents Chemother. 2020, 64, e01902–e01919. [Google Scholar] [CrossRef]
- Sörstedt, E.; Nilsson, S.; Blaxhult, A.; Gisslén, M.; Flamholc, L.; Sönnerborg, A.; Yilmaz, A. Viral blips during suppressive antiretroviral treatment are associated with high baseline HIV-1 RNA levels. BMC Infect. Dis. 2016, 16, 305. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, N.; von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; Kok, Y.L.; et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive art. Nat. Commun. 2019, 10, 3193. [Google Scholar] [CrossRef] [Green Version]
- Avalos, C.R.; Abreu, C.M.; Queen, S.E.; Li, M.; Price, S.; Shirk, E.N.; Engle, E.L.; Forsyth, E.; Bullock, B.T.; Mac Gabhann, F.; et al. Brain macrophages in simian immunodeficiency virus-infected, antiretroviral-suppressed macaques: A functional latent reservoir. MBio 2017, 8, e01186-17. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, L.; Méroth, F.; Tournebize, M.; Leda, A.R.; Sun, E.; Toborek, M. Targeting the HIV-infected brain to improve ischemic stroke outcome. Nat. Commun. 2019, 10, 2009. [Google Scholar] [CrossRef] [Green Version]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouri, S.; Kebodeaux, C.; Wilson, A.; Smith, D. A review of cardiovascular and Renal Function Monitoring: A consideration of older adults with HIV. HIV/AIDS Res. Palliat. Care 2013, 263, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, J.E.; Currier, J.S. Metabolic disease in HIV infection. Lancet Infect. Dis. 2013, 13, 964–975. [Google Scholar] [CrossRef]
- Bhatia, N.S.; Chow, F.C. Neurologic complications in treated HIV-1 infection. Curr. Neurol. Neurosci. Rep. 2016, 16. [Google Scholar] [CrossRef]
- Martin-Iguacel, R.; Negredo, E.; Peck, R.; Friis-Møller, N. Hypertension is a key feature of the metabolic syndrome in subjects aging with HIV. Curr. Hypertens. Rep. 2016, 18, 46. [Google Scholar] [CrossRef] [Green Version]
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2016, 187, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Nduka, C.U.; Stranges, S.; Sarki, A.M.; Kimani, P.K.; Uthman, O.A. Evidence of increased blood pressure and hypertension risk among people living with HIV on antiretroviral therapy: A systematic review with meta-analysis. J. Hum. Hypertens. 2015, 30, 355–362. [Google Scholar] [CrossRef]
- d’Ettorre, G.; Ceccarelli, G.; Pavone, P.; Vittozzi, P.; De Girolamo, G.; Schietroma, I.; Serafino, S.; Giustini, N.; Vullo, V. What happens to cardiovascular system behind the undetectable level of HIV viremia? AIDS Res. Ther. 2016, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Peltenburg, N.C.; Schoeman, J.C.; Hou, J.; Mora, F.; Harms, A.C.; Lowe, S.H.; Bierau, J.; Bakker, J.A.; Verbon, A.; Hankemeier, T.; et al. Persistent metabolic changes in HIV-infected patients during the first year of combination antiretroviral therapy. Sci. Rep. 2018, 8, 16947. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.; Liner, J.; Meeker, R.B. Antiretroviral neurotoxicity. J. NeuroVirology 2012, 18, 388–399. [Google Scholar] [CrossRef]
- Wang, P.; Tian, X.; Tang, J.; Duan, X.; Wang, J.; Cao, H.; Qiu, X.; Wang, W.; Mai, M.; Yang, Q.; et al. Artemisinin protects endothelial function and vasodilation from oxidative damage via activation of PI3K/AKT/Enos pathway. Exp. Gerontol. 2021, 147, 111270. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, I.; Trunfio, M.; Guastamacchia, G.; Bonora, S.; Calcagno, A. A review of the potential mechanisms of neuronal toxicity associated with antiretroviral drugs. J. Neuro.Virol. 2020, 26, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; D’Agostino, L.; Tuzer, F.; Torres, C. HIV antiretroviral therapy drugs induce premature senescence and altered physiology in HUVECS. Mech. Ageing Dev. 2018, 175, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Peyravian, N.; Dikici, E.; Deo, S.; Toborek, M.; Daunert, S. Opioid antagonists as potential therapeutics for ischemic stroke. Prog. Neurobiol. 2019, 182, 101679. [Google Scholar] [CrossRef]
- Voirin, A.-C.; Perek, N.; Roche, F. Inflammatory stress induced by a combination of cytokines (IL-6, IL-17, TNF-α) leads to a loss of integrity on bend.3 endothelial cells in vitro BBB model. Brain Res. 2020, 1730, 146647. [Google Scholar] [CrossRef]
- Nooka, S.; Ghorpade, A. HIV-1-associated inflammation and antiretroviral therapy regulate astrocyte endoplasmic reticulum stress responses. Cell Death Discov. 2017, 3, 17061. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, L.; Toborek, M. Dysregulation of endoplasmic reticulum stress and autophagic responses by the Antiretroviral Drug Efavirenz. Mol. Pharmacol. 2015, 88, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Dugas, T.R. Endothelial mitochondrial senescence accelerates cardiovascular disease in antiretroviral-receiving HIV patients. Toxicol. Lett. 2019, 317, 13–23. [Google Scholar] [CrossRef]
- Guha, D.; Mukerji, S.S.; Chettimada, S.; Misra, V.; Lorenz, D.R.; Morgello, S.; Gabuzda, D. Cerebrospinal fluid extracellular vesicles and neurofilament light protein as biomarkers of central nervous system injury in HIV-infected patients on antiretroviral therapy. AIDS 2019, 33, 615–625. [Google Scholar] [CrossRef]
- Blas-Garcia, A.; Polo, M.; Alegre, F.; Funes, H.A.; Martinez, E.; Apostolova, N.; Esplugues, J.V. Lack of mitochondrial toxicity of darunavir, raltegravir and rilpivirine in neurons and hepatocytes: A comparison with Efavirenz. J. Antimicrob. Chemother. 2014, 69, 2995–3000. [Google Scholar] [CrossRef] [Green Version]
- Tan, I.L.; McArthur, J.C. HIV-associated neurological disorders. CNS Drugs 2012, 26, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Grimmig, B.; Izzo, J.; Brown, L.A.; Hudson, C.; Smith, A.J.; Tan, J.; Bickford, P.C.; Giunta, B. HIV non-nucleoside reverse transcriptase inhibitor Efavirenz reduces neural stem cell proliferation in vitro and in vivo. Cell Transplant. 2016, 25, 1967–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Wang, Y.; Qin, Z.; Qiu, L.; Zhang, M.; Huang, Y.; Zheng, J.C. Combined medication of antiretroviral drugs tenofovir disoproxil fumarate, emtricitabine, and raltegravir reduces neural progenitor cell proliferation in vivo and in vitro. J. Neuroimmune Pharmacol. 2017, 12, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Festa, L.; Roth, L.M.; KJensen, B.; Geiger, J.D.; Jordan-Sciutto, K.L.; Grinspan, J.B. Protease inhibitors, saquinavir and darunavir, inhibit oligodendrocyte maturation: Implications for lysosomal stress. J. Neuroimmune Pharmacol. 2019, 16, 169–180. [Google Scholar] [CrossRef]
- Shah, A.; Gangwani, M.R.; Chaudhari, N.S.; Glazyrin, A.; Bhat, H.K.; Kumar, A. Neurotoxicity in the post-HAART era: Caution for the antiretroviral therapeutics. Neurotox. Res. 2016, 30, 677–697. [Google Scholar] [CrossRef]
- Persidsky, Y.; Hill, J.; Zhang, M.; Dykstra, H.; Winfield, M.; Reichenbach, N.L.; Potula, R.; Mukherjee, A.; Ramirez, S.H.; Rom, S. Dysfunction of brain pericytes in chronic neuroinflammation. J. Cereb. Blood Flow Metab. 2015, 36, 794–807. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shim, E.; Crespo-Mejias, Y.; Nguyen, P.G.; Gibbons, A.; Liu, D.; Shide, E.; Poirier, M.C. Cardiomyocytes are protected from antiretroviral nucleoside analog-induced mitochondrial toxicity by overexpression of pgc-1α. Cardiovasc. Toxicol. 2014, 15, 224–231. [Google Scholar] [CrossRef]
- Smith, R.L.; de Boer, R.; Brul, S.; Budovskaya, Y.; van Spek, H. Premature and accelerated aging: HIV or Haart? Front. Genet. 2013, 3, 328. [Google Scholar] [CrossRef] [Green Version]
- Bañó, M.; Morén, C.; Barroso, S.; Juárez, D.L.; Guitart-Mampel, M.; González-Casacuberta, I.; Canto-Santos, J.; Lozano, E.; León, A.; Pedrol, E.; et al. Mitochondrial toxicogenomics for antiretroviral management: HIV Post-exposure prophylaxis in uninfected patients. Front. Genet. 2020, 11, 497. [Google Scholar] [CrossRef]
- Hukezalie, K.R.; Thumati, N.R.; Côté, H.C.; Wong, J.M. In vitro and ex vivo inhibition of human telomerase by Anti-HIV nucleoside reverse transcriptase inhibitors (nrtis) but not by Non-NRTIs. PLoS ONE 2012, 7, e47505. [Google Scholar] [CrossRef] [Green Version]
- Leeansyah, E.; Cameron, P.U.; Solomon, A.; Tennakoon, S.; Velayudham, P.; Gouillou, M.; Spelman, T.; Hearps, A.; Fairley, C.; Smit, D.V.; et al. Inhibition of telomerase activity by human immunodeficiency virus (HIV) nucleos(t)ide reverse transcriptase inhibitors: A potential factor contributing to HIV-associated accelerated aging. J. Infect. Dis. 2013, 207, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Bollmann, F.M. Telomerase inhibition may contribute to accelerated mitochondrial aging induced by anti-retroviral HIV treatment. Med. Hypotheses 2013, 81, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Oeck, S.; West, A.P.; Mangalhara, K.C.; Sainz, A.G.; Newman, L.E.; Zhang, X.-O.; Wu, L.; Yan, Q.; Bosenberg, M.; et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 2019, 1, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Yarosz, E.L.; Chang, C.-H. The role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw. 2018, 18. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The role of mitochondrial Ros in the aging brain. FEBS Lett. 2017, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, C.; LI, Z.; Kim, G.; Jeevanandam, V.; Uriel, N. Molecular mechanism of the association between Atrial Fibrillation and heart failure includes energy metabolic dysregulation due to mitochondrial dysfunction. J. Card. Fail. 2019, 25, 911–920. [Google Scholar] [CrossRef]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative stress-mediated blood-brain barrier (BBB) disruption in neurological diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Anasooya Shaji, C.; Robinson, B.D.; Yeager, A.; Beeram, M.R.; Davis, M.L.; Isbell, C.L.; Huang, J.H.; Tharakan, B. The tri-phasic role of hydrogen peroxide in blood-brain barrier endothelial cells. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Sulhan, S.; Lyon, K.A.; Shapiro, L.A.; Huang, J.H. Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J. Neurosci. Res. 2018, 98, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Diwanji, N.; Bergmann, A. Basement membrane damage by ros- and JNK-mediated MMP2 activation drives macrophage recruitment to overgrown tissue. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.-R.; Kim, J.Y.; Hyun, H.-W.; Kim, J.-E. Endothelial NOS activation induces the blood-brain barrier disruption via ER stress following status epilepticus. Brain Res. 2015, 1622, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Castro, V.; Bertrand, L.; Luethen, M.; Dabrowski, S.; Lombardi, J.; Morgan, L.; Sharova, N.; Stevenson, M.; Blasig, I.E.; Toborek, M. Occludin controls HIV transcription in brain pericytes via regulation of SIRT-1 activation. FASEB J. 2015, 30, 1234–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, V.; Skowronska, M.; Lombardi, J.; He, J.; Seth, N.; Velichkovska, M.; Toborek, M. Occludin regulates glucose uptake and ATP production in pericytes by influencing AMP-activated protein kinase activity. J. Cereb. Blood Flow Metab. 2017, 38, 317–332. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Alegre, F.; Funes, H.A.; Victor, V.M.; Barrachina, M.D.; Blas-Garcia, A.; Esplugues, J.V. ER stress in human hepatic cells treated with Efavirenz: Mitochondria again. J. Hepatol. 2013, 59, 780–789. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Kawabata, T.; Yoshimori, T. Autophagosome Biogenesis and human health. Cell Discov. 2020, 6, 33. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. The J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.A.; Masliah, E.; Vinters, H.V.; Beizai, P.; Moore, D.J.; Achim, C.L. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS 2005, 19, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Swinton, M.K.; Soontornniyomkij, B.; Carson, A.; Achim, C.L. Beta amyloid levels in cerebrospinal fluid of HIV-infected people vary by exposure to antiretroviral therapy. AIDS 2020, 34, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Achim, C.L.; Adame, A.; Dumaop, W.; Everall, I.P.; Masliah, E. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J. Neuroimmune Pharmacol. 2009, 4, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Choi, M.-S.; Inn, K.-S.; Kim, B.-J. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a hsp90-dependent manner. Sci. Rep. 2016, 6, 28896. [Google Scholar] [CrossRef] [Green Version]
- Gavegnano, C.; Detorio, M.; Montero, C.; Bosque, A.; Planelles, V.; Schinazi, R.F. Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus ReactivationIn Vitro. Antimicrob. Agents Chemother. 2014, 58, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using JAK inhibitors. PLoS Pathog. 2017, 13, e1006740. [Google Scholar] [CrossRef] [Green Version]
- Lichterfeld, M. Reactivation of latent HIV moves shock-and-kill treatments forward. Nature 2020, 578, 42–43. [Google Scholar] [CrossRef]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.; Thompson, G.R.; et al. Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-KB signaling in combination with JQ1 induced P-tefb activation. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, E.K.; Spicer, L.; Smith, S.A.; Lee, D.; Fast, R.; Paganini, S.; Lawson, B.O.; Nega, M.; Easley, K.; Schmitz, J.E.; et al. Cd8 + lymphocytes are required for maintaining viral suppression in siv-infected macaques treated with short-term antiretroviral therapy. Immunity 2016, 45, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, R.; Conway, J.M.; Margolis, D.M.; Perelson, A.S. Determinants of the efficacy of HIV latency-reversing agents and implications for drug and treatment design. JCI Insight 2018, 3, e123052. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-and-lock strategies to cure HIV infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In vivo suppression of HIV rebound by Didehydro-cortistatin a, a “block-and-lock” strategy for HIV-1 treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR complex controls HIV latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of signaling pathways that block reversal of HIV-1 latency. Antimicrob. Agents Chemother. 2019, 63, e01744-18. [Google Scholar] [CrossRef] [Green Version]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef]
- Kaushik, A.; Yndart, A.; Atluri, V.; Tiwari, S.; Tomitaka, A.; Gupta, P.; Jayant, R.D.; Alvarez-Carbonell, D.; Khalili, K.; Nair, M. Magnetically guided non-invasive CRISPR-Cas9/grna delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci. Rep. 2019, 9, 3928. [Google Scholar] [CrossRef] [Green Version]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential Laser Art and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halper-Stromberg, A.; Lu, C.-L.; Klein, F.; Horwitz, J.A.; Bournazos, S.; Nogueira, L.; Eisenreich, T.R.; Liu, C.; Gazumyan, A.; Schaefer, U.; et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell 2014, 158, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. Talen gene editing takes aim on HIV. Hum. Genet. 2016, 135, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Rathore, A.; Iketani, S.; Wang, P.; Jia, M.; Sahi, V.; Ho, D.D. CRISPR-based gene knockout screens reveal deubiquitinases involved in HIV-1 latency in two Jurkat cell models. Sci. Rep. 2020, 10, 5350. [Google Scholar] [CrossRef] [Green Version]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.-W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.-C.; Hütter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef]
- Gupta, R.K.; Peppa, D.; Hill, A.L.; Gálvez, C.; Salgado, M.; Pace, M.; McCoy, L.E.; Griffith, S.A.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: A case report. Lancet HIV 2020, 7, e340–e347. [Google Scholar] [CrossRef] [Green Version]
- Skundric, D.S.; Tse, H.Y.; Montgomery, P.C. Functional phenotypes of CCR5 on CD4+ T cells of relevance to its genetic and epigenetic associations with HIV infection. Cell. Mol. Immunol. 2020, 17, 680–681. [Google Scholar] [CrossRef]
- Peterson, C.W.; Kiem, H.-P. Lessons from London and Berlin: Designing A scalable gene therapy approach for HIV cure. Cell Stem Cell 2019, 24, 685–687. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef]
- Berg, C.; Daugvilaite, V.; Steen, A.; Jørgensen, A.S.; Våbenø, J.; Rosenkilde, M.M. Inhibition of HIV fusion by small molecule agonists through efficacy-engineering of CXCR4. ACS Chem. Biol. 2018, 13, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-S.; Yin, J.; Leng, F.; Teng, R.-F.; Xu, C.; Xia, X.-Y.; Pan, X.-M. HIV coreceptor tropism determination and mutational pattern identification. Sci. Rep. 2016, 6, 21280. [Google Scholar] [CrossRef] [PubMed]
- ElZohary, L.; Weglicki, W.B.; Chmielinska, J.J.; Kramer, J.H.; Mak, I.T. MG-supplementation attenuated lipogenic and oxidative/nitrosative gene expression caused by combination antiretroviral therapy (CART) in HIV-1-transgenic rats. PLoS ONE 2019, 14, e0210107. [Google Scholar] [CrossRef] [Green Version]
- Coulibaly, F.S.; Ezoulin, M.J.; Purohit, S.S.; Ayon, N.J.; Oyler, N.A.; Youan, B.-B.C. Layer-by-layer engineered microbicide drug delivery system targeting HIV-1 gp120: Physicochemical and biological properties. Mol. Pharm. 2017, 14, 3512–3527. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, F.S.; Thomas, D.N.; Youan, B.-B.C. Anti-HIV lectins and current delivery strategies. AIMS Mol. Sci. 2018, 5, 96–116. [Google Scholar] [CrossRef]
- Iannazzo, D.; Pistone, A.; Ferro, S.; De Luca, L.; Monforte, A.M.; Romeo, R.; Buemi, M.R.; Pannecouque, C. Graphene quantum dots based systems as HIV inhibitors. Bioconjugate Chem. 2018, 29, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Surnar, B.; Shah, A.S.; Park, M.; Kalathil, A.A.; Kamran, M.Z.; Ramirez Jaime, R.; Toborek, M.; Nair, M.; Kolishetti, N.; Dhar, S. Brain-Accumulating Nanoparticles for Assisting Astrocytes to Reduce Human Immunodeficiency Virus and Drug Abuse-Induced Neuroinflammation and Oxidative Stress. ACS Nano 2021, 15, 15741–15753. [Google Scholar] [CrossRef]


| ARVd Class | Function | Drug Examples | CPE Score | Side Effects | Adult Dosage Schedule |
|---|---|---|---|---|---|
| Nucleoside reverse transcriptase inhibitor (NRTI) | Inhibits reverse transcriptase, blocking production of viral DNA | Lamivudine (3TC) | 2 | Nausea, dizziness, lactic acidosis, pancreatitis, IRIS | 300 mg once or 150 mg twice daily |
| Zidovudine (ZDV) | 4 | Nausea, dizziness, lactic acidosis, liver problems, myopathy, severe anemia, neutropenia, IRIS, lipoatrophy | 250–300 mg twice daily | ||
| Emtricitabine (FTC) | 3 | Nausea, dizziness, lactic acidosis, IRIS, possible HBV flare up | 200 mg daily | ||
| Tenofovir (TFV) | 1 | Nausea, dizziness, lactic acidosis, kidney problems including kidney failure | 300 mg daily | ||
| Non-nucleoside reverse transcriptase inhibitors (NNRTI) | Binds to and blocks HIV reverse transcriptase, blocking production of viral DNA | Efavirenz (EFV) | 3 | Nausea, dizziness, mental health problems, liver problems, severe rash, nervous system issues, seizures, IRIS, lipodystrophy, hyperlipidemia | 600 mg daily with a NRTI or PI |
| Nevirapine (NVP) | 4 | Nausea, dizziness, severe liver problems, skin rash, IRIS, lipodystrophy syndrome | 200 mg twice daily | ||
| Fusion inhibitors (FI) | Inhibits viral binding or fusion of HIV to host target cells preventing the entry of HIV | Albuvirtide * (ABT) | ND | Nausea, headache, diarrhea, rashes, hyperlipidemia | ND |
| Enfuvirtide (T20) | 1 | Allergic reaction, nausea, headache, pneumonia, neuralgia, IRIS | 90 mg twice daily | ||
| Protease inhibitors (PI) | Blocks proteases required for proteolytic cleavage of precursors necessary viral replication | Atazanavir (ATV) | 2 | Nausea, dizziness, heart arrhythmia, severe rash, liver problems, life-threatening drug interaction, chronic kidney disease, kidney stones, gallbladder problems, IRIS, lipodystrophy, increased bleeding in hemophiliacs, diabetes, and hyperglycemia | 300 mg with 100 mg RTV daily |
| Darunavir (DRV) | 3 | Nausea, dizziness, liver problems sever skin reactions, diabetes, hyperglycemia, lipodystrophy, IRIS | 600–800 mg with 100 mg RTV daily | ||
| Ritonavir (RTV) ⊥ | 1 | Nausea, dizziness, pancreatitis, heart arrhythmia, severe allergic reactions, liver problems, hyperlipidemia, hyperglycemia, IRIS, lipodystrophy, increased bleeding in hemophiliacs, gastrointestinal problems | 600 mg twice daily | ||
| Integrase strand transfer inhibitors (INSTI) | Prevents the integration of HIV DNA into host DNA | Dolutegravir (DTG) | ND | Nausea, dizziness, allergic reactions, liver problems, IRIS, sleep problems | 50 mg once or twice daily |
| Raltegravir (RAL) | 3 | Nausea, dizziness, severe skin reactions, allergic reactions, liver problems, IRIS | 1200 mg daily or 400–800 mg twice daily | ||
| Chemokine coreceptor antagonists | Blocks coreceptors (CCR5/CXCR4) preventing the entry of HIV | Maraviroc (MVC)—CCR5 | 3 | Nausea, dizziness, liver problems, heart problems (including heart attack), skin reactions, allergic reactions, postural hypotension, IRIS, possible increased risk of other infections or cancer | 150 mg, 300 mg, or 600 mg twice daily depending on concomitant medications |
| Leronlimab * (PA14) | ND | Diarrhea, headache, swollen lymph nodes, hypertension, local injection site reactions | ND | ||
| CD4 attachment inhibitors/post-attachment inhibitors | Binds to host CD4 receptor blocking HIV attachment and entry | Ibalizumab-uiyk (IBA) | ND | Nausea, dizziness, IRIS, diarrhea, rashes | Loading dose of 2000 mg and maintenance doses of 800 mg every two weeks |
| UB-421 * (mAb dB4) | ND | Rash, hives, increased eosinophil count, elevated liver enzyme levels | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudd, H.; Toborek, M. Pitfalls of Antiretroviral Therapy: Current Status and Long-Term CNS Toxicity. Biomolecules 2022, 12, 894. https://doi.org/10.3390/biom12070894
Rudd H, Toborek M. Pitfalls of Antiretroviral Therapy: Current Status and Long-Term CNS Toxicity. Biomolecules. 2022; 12(7):894. https://doi.org/10.3390/biom12070894
Chicago/Turabian StyleRudd, Harrison, and Michal Toborek. 2022. "Pitfalls of Antiretroviral Therapy: Current Status and Long-Term CNS Toxicity" Biomolecules 12, no. 7: 894. https://doi.org/10.3390/biom12070894
APA StyleRudd, H., & Toborek, M. (2022). Pitfalls of Antiretroviral Therapy: Current Status and Long-Term CNS Toxicity. Biomolecules, 12(7), 894. https://doi.org/10.3390/biom12070894