
Early Signs of Molecular Defects in iPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Human iPSC-Derived NPCs
2.2. RNA Sequencing and Analysis
2.3. RNA Isolation, cDNA Synthesis, and qPCR
2.4. Immunofluorescence Staining
2.5. Artificial Synapse Formation Assays and Synapse Induction Analysis
3. Results
3.1. Generation and Characterization of p.A53T-αSyn NPCs
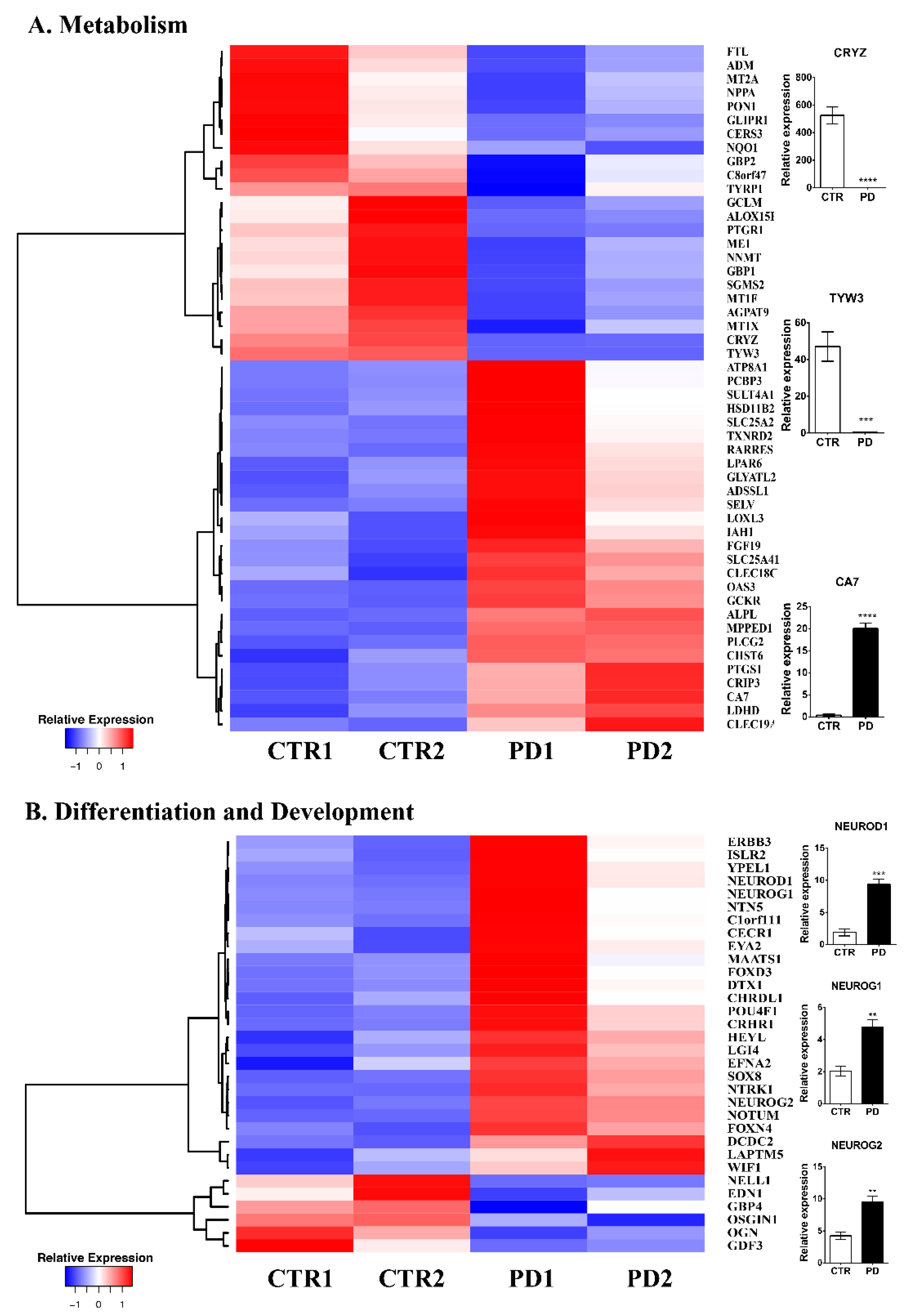
3.2. Comparative Gene Expression Profiling of p.A53T-αSyn NPCs
3.3. Artificial Synapse Formation in p.A53T-αSyn Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.; Debetto, P.; Negro, A.; Guidolin, D.; Skaper, S.D.; Giusti, P. Alpha-synuclein and Parkinson’s disease. FASEB J. 2004, 18, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Markopoulou, K.; Dickson, D.W.; McComb, R.D.; Wszolek, Z.K.; Katechalidou, L.; Avery, L.; Stansbury, M.S.; Chase, B.A. Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease. Acta Neuropathol. 2008, 116, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Klein, C. The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef]
- Dolt, K.S.; Hammachi, F.; Kunath, T. Modeling Parkinson’s disease with induced pluripotent stem cells harboring alpha-synuclein mutations. Brain Pathol. 2017, 27, 545–551. [Google Scholar] [CrossRef]
- Lin, L.; Göke, J.; Cukuroglu, E.; Dranias, M.R.; VanDongen, A.M.; Stanton, L.W. Molecular Features Underlying Neurodegeneration Identified through In Vitro Modeling of Genetically Diverse Parkinson’s Disease Patients. Cell Rep. 2016, 15, 2411–2426. [Google Scholar] [CrossRef]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Breza, M.; Koutsis, G.; Karadima, G.; Potagas, C.; Kartanou, C.; Papageorgiou, S.G.; Paraskevas, G.P.; Kapaki, E.; Stefanis, L.; Panas, M. The different faces of the p. A53T alpha-synuclein mutation: A screening of Greek patients with parkinsonism and/or dementia. Neurosci. Lett. 2018, 672, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.J.; Savas, J.N.; Kim, Y.; Tran, N.; Di Simone, A.; Hashimototorii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2014, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Dolatabadi, N.; Gao, R.; Wu, Y.; Scott, H.; Trudler, D.; Sultan, A.; Ambasudhan, R.; Nakamura, T.; Masliah, E.; et al. NitroSynapsin ameliorates hypersynchronous neural network activity in Alzheimer hiPSC models. Mol. Psychiatry 2020, 26, 5751–5765. [Google Scholar] [CrossRef]
- Tu, S.; Akhtar, M.W.; Escorihuela, R.M.; Amador-Arjona, A.; Swarup, V.; Parker, J.; Zaremba, J.D.; Holland, T.; Bansal, N.; Holohan, D.R.; et al. NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nat. Commun. 2017, 8, 1488. [Google Scholar] [CrossRef]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef]
- Son, M.Y.; Sim, H.; Son, Y.S.; Jung, K.B.; Lee, M.-O.; Oh, J.-H.; Chung, S.-K.; Jung, C.-R.; Kim, J. Distinctive genomic signature of neural and intestinal organoids from familial Parkinson’s disease patient-derived induced pluripotent stem cells. Neuropathol. Appl. Neurobiol. 2017, 43, 584–603. [Google Scholar] [CrossRef]
- Schwamborn, J.C. Is Parkinson’s Disease a Neurodevelopmental Disorder and Will Brain Organoids Help Us to Understand It? Stem Cells Dev. 2018, 27, 968–975. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Nussbaum Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Biederer, T.; Scheiffele, P. Mixed-culture assays for analyzing neuronal synapse formation. Nat. Protoc. 2007, 2, 670–676. [Google Scholar] [CrossRef]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Expression of α-synuclein is regulated in a neuronal cell type-dependent manner. Anat. Sci. Int. 2018, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Clémot, M.; Demarco, R.S.; Jones, D.L. Lipid Mediated Regulation of Adult Stem Cell Behavior. Front. Cell Dev. Biol. 2020, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.R.; Wang, Q.; Meechoovet, B.; Siniard, A.L.; Naymik, M.; De Both, M.; Huentelman, M.J.; Caselli, R.J.; Driver-Dunckley, E.; Dunckley, T. DNA Methylation and Expression Profiles of Whole Blood in Parkinson’s Disease. Front. Genet. 2021, 12, 640266. [Google Scholar] [CrossRef] [PubMed]
- Conway, O.J.; Carrasquillo, M.M.; Wang, X.; Bredenberg, J.M.; Reddy, J.S.; Strickland, S.L.; Younkin, C.S.; Burgess, J.D.; Allen, M.; Lincoln, S.J.; et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol. Neurodegener. 2018, 13, 53. [Google Scholar] [CrossRef]
- Geraldo, L.H.M.; de Sampaio Spohr, T.C.L.; do Amaral, R.F.; da Fonseca, A.C.C.; Garcia, C.; de Almeida Mendes, F.; Freitas, C.; Fabio dosSantos, M.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef]
- Okita, Y.; Rcom-H’Cheo-Gauthier, A.N.; Goulding, M.; Chung, R.; Faller, P.; Pountney, D.L. Metallothionein, Copper and Alpha-Synuclein in Alpha-Synucleinopathies. Front. Neurosci. 2017, 11, 114. [Google Scholar] [CrossRef]
- Bertling, E.; Blaesse, P.; Seja, P.; Kremneva, E.; Gateva, G.; Virtanen, A.M.; Summanen, M.; Spoljaric, I.; Uvarov, P.; Blaesse, M.; et al. Carbonic anhydrase seven bundles filamentous actin and regulates dendritic spine morphology and density. EMBO Rep. 2021, 22, e50145. [Google Scholar] [CrossRef]
- Wei, L.; Tian, Y.; Chen, Y.; Wei, Q.; Chen, F.; Cao, B.; Wu, Y.; Zhao, B.; Chen, X.; Xie, C. Identification of TYW3/CRYZ and FGD4 as susceptibility genes for amyotrophic lateral sclerosis. Neurol. Genet. 2019, 5, e375. [Google Scholar] [CrossRef]
- Victor, M.B.; Richner, M.; Olsen, H.E.; Lee, S.W.; Monteys, A.M.; Ma, C.; Huh, C.J.; Zhang, B.; Davidson, B.L.; Yang, X.W.; et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018, 21, 341–352. [Google Scholar] [CrossRef]
- Conforti, P.; Besusso, D.; Bocchi, V.D.; Faedo, A.; Cesana, E.; Rossetti, G.; Ranzani, V.; Svendsen, C.N.; Thompson, L.M.; Toselli, M.; et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl. Acad. Sci. USA 2018, 115, E762–E771. [Google Scholar] [CrossRef]
- Scopa, C.; Marrocco, F.; Latina, V.; Ruggeri, F.; Corvaglia, V.; la Regina, F.; Ammassari-Teule, M.; Middei, S.; Amadoro, G.; Meli, G.; et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Abeta oligomers. Cell Death Differ. 2020, 27, 934–948. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F.; Hodge, R.D.; Daza, R.A.; Englund, C. Transcription factors in glutamatergic neurogenesis: Conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci. Res. 2006, 55, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Aprea, J.; Nonaka-Kinoshita, M.; Calegari, F. Generation and characterization of Neurod1-CreER(T2) mouse lines for the study of embryonic and adult neurogenesis. Genesis 2014, 52, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Velkey, J.M.; O’Shea, K.S. Expression of Neurogenin 1 in mouse embryonic stem cells directs the differentiation of neuronal precursors and identifies unique patterns of down-stream gene expression. Dev. Dyn. 2013, 242, 230–253. [Google Scholar] [CrossRef]
- Lacomme, M.; Liaubet, L.; Pituello, F.; Bel-Vialar, S. NEUROG2 Drives Cell Cycle Exit of Neuronal Precursors by Specifically Repressing a Subset of Cyclins Acting at the G 1 and S Phases of the Cell Cycle. Mol. Cell. Biol. 2012, 32, 2596–2607. [Google Scholar] [CrossRef]
- Smith, R.S.; Walsh, C.A. Ion Channel Functions in Early Brain Development. Trends Neurosci. 2020, 43, 103–114. [Google Scholar] [CrossRef]
- Linta, L.; Boeckers, T.M.; Kleger, A.; Liebau, S. Calcium activated potassium channel expression during human iPS cell-derived neurogenesis. Ann. Anat. 2013, 195, 303–311. [Google Scholar] [CrossRef]
- Compagnucci, C.; Piermarini, E.; Sferra, A.; Borghi, R.; Niceforo, A.; Petrini, S.; Piemonte, F.; Bertini, E. Cytoskeletal dynamics during in vitro neurogenesis of induced pluripotent stem cells (iPSCs). Mol. Cell. Neurosci. 2016, 77, 113–124. [Google Scholar] [CrossRef]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef]
- Tang, N.H.; Jin, Y. Shaping neurodevelopment: Distinct contributions of cytoskeletal proteins. Curr. Opin. Neurobiol. 2018, 51, 111–118. [Google Scholar] [CrossRef]
- Han, W.; Šestan, N. Cortical Projection Neurons: Sprung from the Same Root. Neuron 2013, 80, 1103–1105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, C.; Zhang, W.; Rondard, P.; Pin, J.-P.; Liu, J. Complex GABAB receptor complexes: How to generate multiple functionally distinct units from a single receptor. Front. Pharmacol. 2014, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S. Neuronal development: Signalling synaptogenesis. Nat. Rev. Neurosci. 2016, 17, 372. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Satterfield, R.; Young, S.; Jonas, P. Triple Function of Synaptotagmin 7 Ensures Efficiency of High-Frequency Transmission at Central GABAergic Synapses. Cell Rep. 2017, 21, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Kandel, R.S.; Mishra, R.; Gautam, J.; Alaref, A.; Jahan, N. Diabetes Mellitus and Parkinson’s Disease: Shared Pathophysiological Links and Possible Therapeutic Implications. Cureus 2020, 12, e9853. [Google Scholar] [CrossRef]
- Horikawa, Y. Maturity-onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J. Diabetes Investig. 2018, 9, 704–712. [Google Scholar] [CrossRef]
- Garg, A.; Agarwal, A.K. Caveolin-1: A New Locus for Human Lipodystrophy. J. Clin. Endocrinol. Metab. 2008, 93, 1183–1185. [Google Scholar] [CrossRef]
- Ehrhart, F.; Coort, S.; Cirillo, E.; Smeets, E.; Evelo, C.; Curfs, L. New insights in Rett syndrome using pathway analysis for transcriptomics data. Wien. Med. Wochenschr. 2016, 166, 346–352. [Google Scholar] [CrossRef]
- Kurian, M.A. SLC6A3-Related Dopamine Transporter Deficiency Syndrome. In GeneReviews((R)); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Jiang, W.; Wei, M.; Liu, M.; Pan, Y.; Cao, D.; Yang, X.; Zhang, C. Identification of Protein Tyrosine Phosphatase Receptor Type O (PTPRO) as a Synaptic Adhesion Molecule that Promotes Synapse Formation. J. Neurosci. 2017, 37, 9828–9843. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Koros, C.; Yousaf, T.; Picillo, M.; Polychronis, S.; Simitsi, A.; Giordano, B.; Chappell, Z.; et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: A cross-sectional study. Lancet Neurol. 2019, 18, 748–759. [Google Scholar] [CrossRef]
- Brennand, K.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.-H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef] [PubMed]
- Cope, E.C.; Briones, B.A.; Brockett, A.T.; Martinez, S.; Vigneron, P.-A.; Opendak, M.; Wang, S.S.-H.; Gould, E. Immature Neurons and Radial Glia, but Not Astrocytes or Microglia, Are Altered in Adult Cntnap2 and Shank3 Mice, Models of Autism. Eneuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, L.-M.; Stock, R.; Haag, K.; Buckenmaier, S.; Eberle, M.-C.; Wildgruber, D.; Storchak, H.; Kriebel, M.; Weißgraeber, S.; Mathew, L.; et al. Comparative characterization of human induced pluripotent stem cells (hiPSC) derived from patients with schizophrenia and autism. Transl. Psychiatry 2019, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef]
- Xie, X.; Mahmood, S.R.; Gjorgjieva, T.; Percipalle, P. Emerging roles of cytoskeletal proteins in regulating gene expression and genome organization during differentiation. Nucleus 2020, 11, 53–65. [Google Scholar] [CrossRef]
- Boraas, L.C.; Guidry, J.B.; Pineda, E.T.; Ahsan, T. Cytoskeletal Expression and Remodeling in Pluripotent Stem Cells. PLoS ONE 2016, 11, e0145084. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akrioti, E.; Karamitros, T.; Gkaravelas, P.; Kouroupi, G.; Matsas, R.; Taoufik, E. Early Signs of Molecular Defects in iPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease. Biomolecules 2022, 12, 876. https://doi.org/10.3390/biom12070876
Akrioti E, Karamitros T, Gkaravelas P, Kouroupi G, Matsas R, Taoufik E. Early Signs of Molecular Defects in iPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease. Biomolecules. 2022; 12(7):876. https://doi.org/10.3390/biom12070876
Chicago/Turabian StyleAkrioti, Elissavet, Timokratis Karamitros, Panagiotis Gkaravelas, Georgia Kouroupi, Rebecca Matsas, and Era Taoufik. 2022. "Early Signs of Molecular Defects in iPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease" Biomolecules 12, no. 7: 876. https://doi.org/10.3390/biom12070876
APA StyleAkrioti, E., Karamitros, T., Gkaravelas, P., Kouroupi, G., Matsas, R., & Taoufik, E. (2022). Early Signs of Molecular Defects in iPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease. Biomolecules, 12(7), 876. https://doi.org/10.3390/biom12070876