
scEpiLock: A Weakly Supervised Learning Framework for cis-Regulatory Element Localization and Variant Impact Quantification for Single-Cell Epigenetic Data

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Processing
2.1.1. PBMC Data from 10× Genomics
2.1.2. Brain scATAC-seq Data
2.1.3. ENCODE Bulk ATAC-Seq Data
2.1.4. Genome Features as Model Inputs
2.1.5. Negative Regions
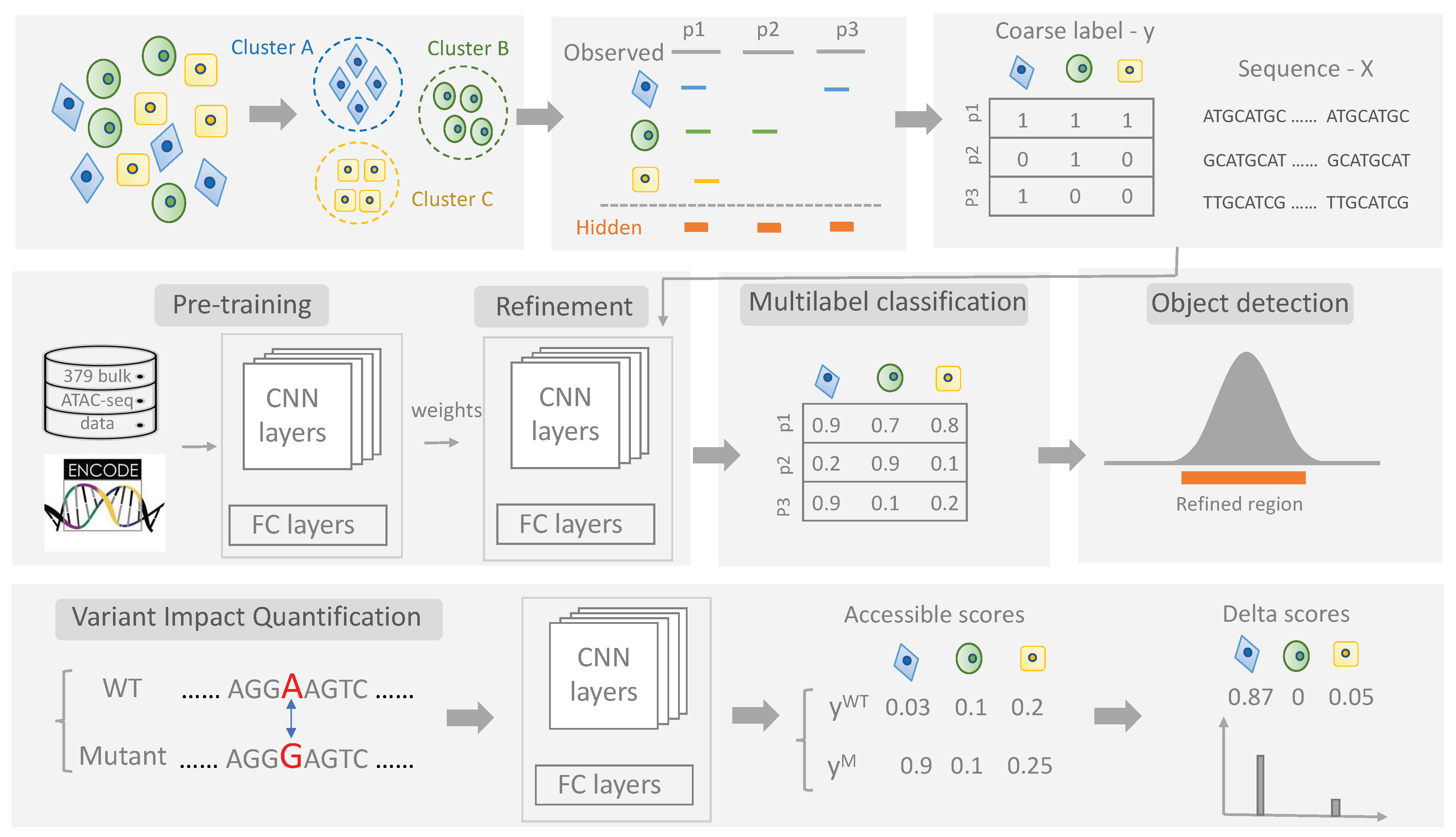
2.2. scEpiLock Module 1—Multi-Label Classifier Module
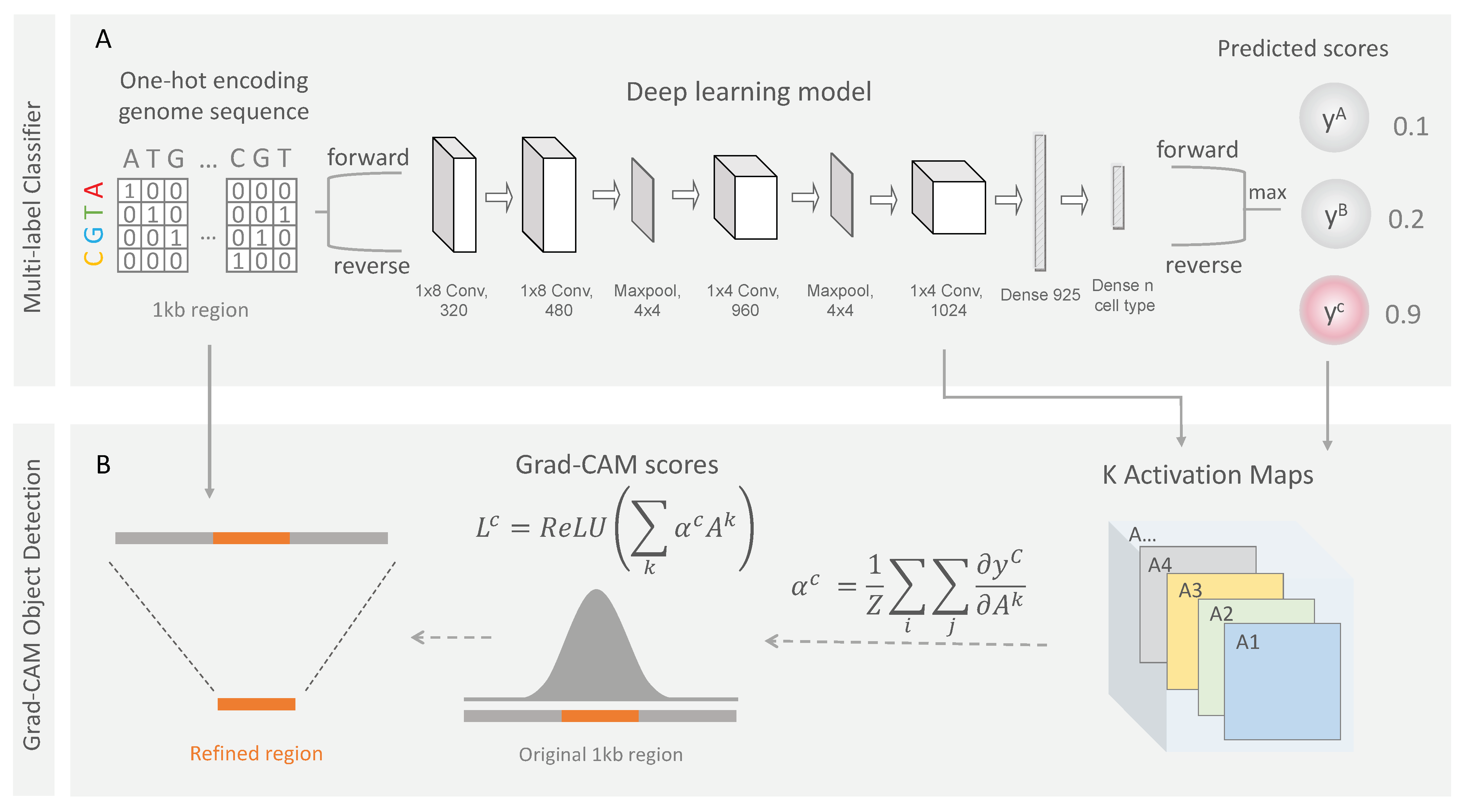
2.2.1. Neural Network Architecture
2.2.2. Multi-Label Classifier Performance Evaluations
2.2.3. Performance Benchmarking with Other Methods
2.3. scEpiLock Module 2—Object Detection for CRE Boundary Refinement
2.3.1. Grad-CAM
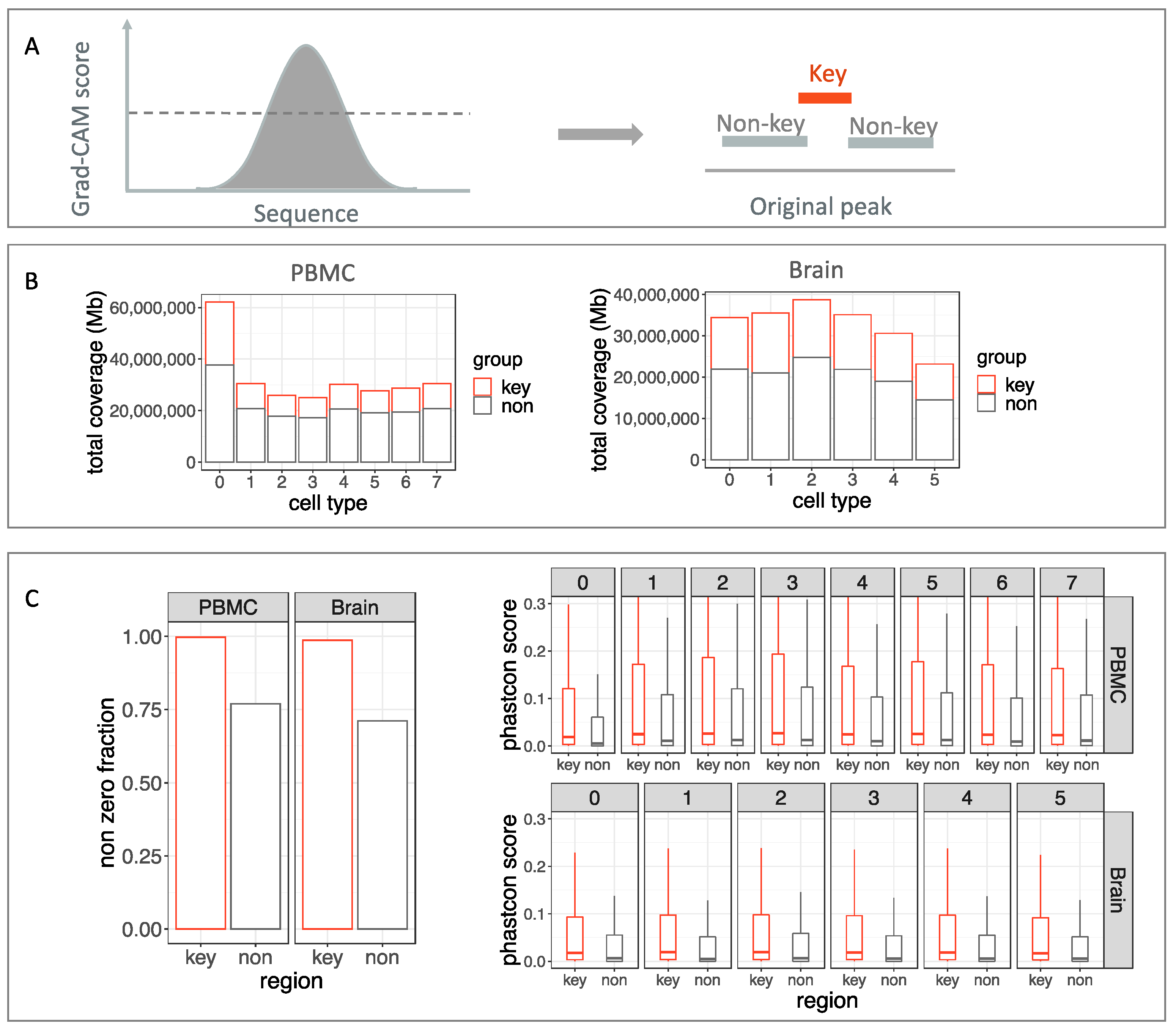
2.3.2. Conservation Scores for Refined Regions
2.4. scEpiLock Module 3—Variant Impact Quantification
2.4.1. GWAS Data Used and SNP Extraction
2.4.2. Sequencing Tracks
3. Results
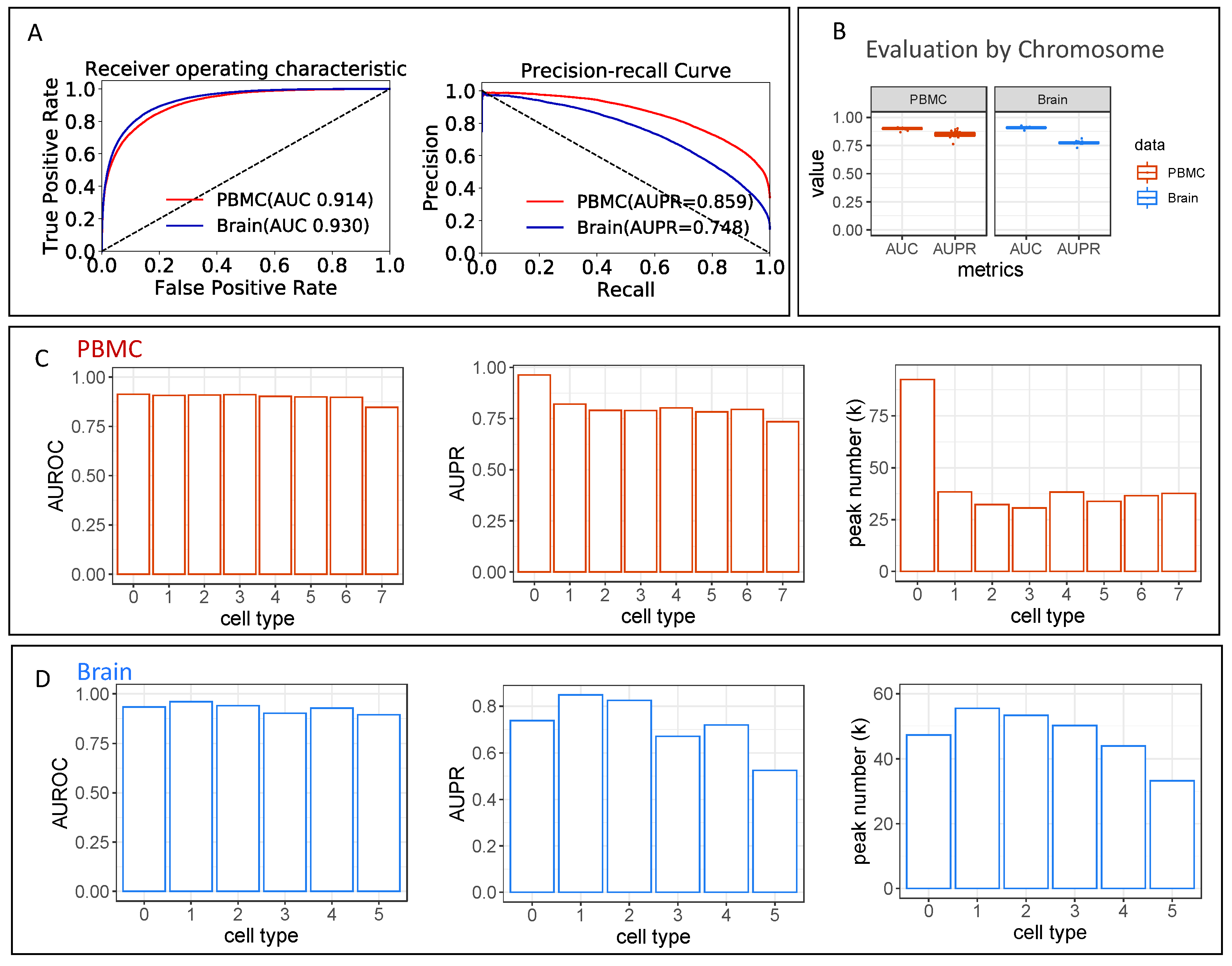
3.1. The Multi-Label Classifier in scEpiLock Precisely Predicts Accessible Peaks
3.2. Object Detection Module Precisely Refines Peak Boundry
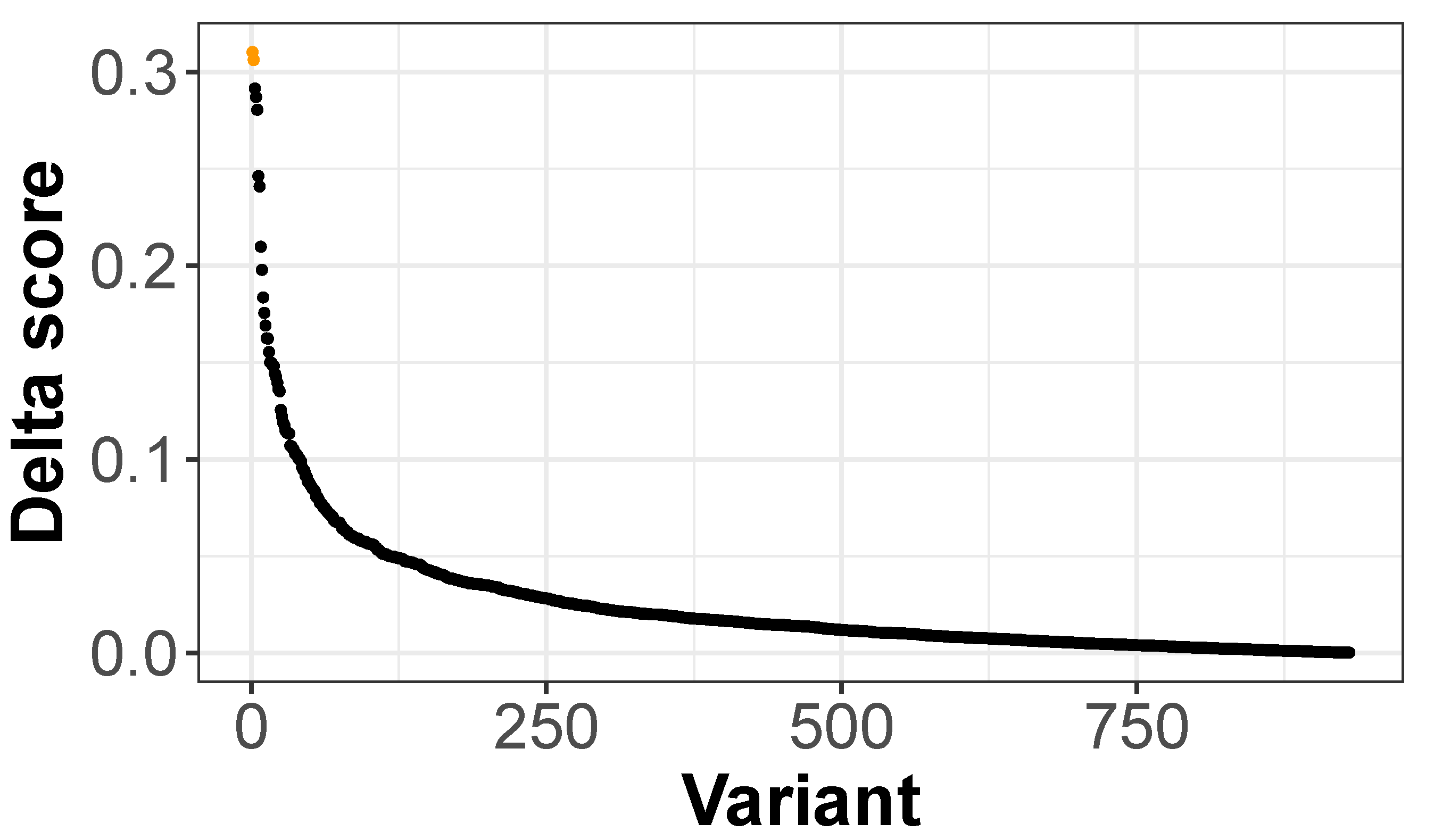
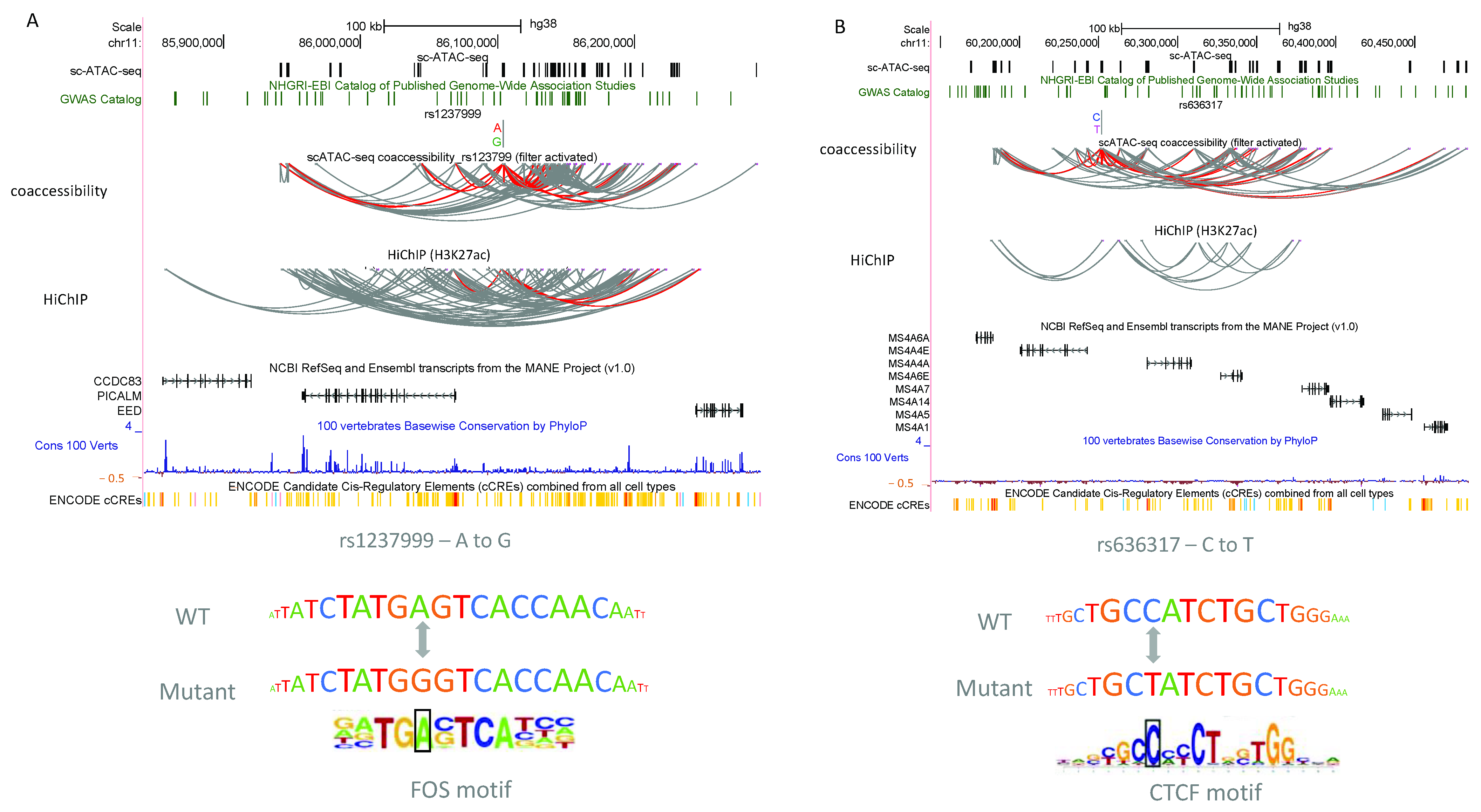
3.3. scEpiLock Predicts Functional Disease-Associated SNPs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casamassimi, A.; Ciccodicola, A. Transcriptional Regulation: Molecules, Involved Mechanisms, and Misregulation. Int. J. Mol. Sci. 2019, 20, 1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-Cell Chromatin Accessibility Reveals Principles of Regulatory Variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex Single-Cell Profiling of Chromatin Accessibility by Combinatorial Cellular Indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of Computational Methods for the Analysis of Single-Cell ATAC-Seq Data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Preissl, S.; Li, Y.; Hou, X.; Lucero, J.; Wang, X.; Motamedi, A.; Shiau, A.K.; Zhou, X.; Xie, F.; et al. Comprehensive Analysis of Single Cell ATAC-Seq Data with SnapATAC. Nat. Commun. 2021, 12, 1337. [Google Scholar] [CrossRef] [PubMed]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR Is a Scalable Software Package for Integrative Single-Cell Chromatin Accessibility Analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef]
- Baker, S.M.; Rogerson, C.; Hayes, A.; Sharrocks, A.D.; Rattray, M. Classifying Cells with Scasat, a Single-Cell ATAC-Seq Analysis Tool. Nucleic Acids Res. 2019, 47, e10. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.; Zhang, S. Network Diffusion for Scalable Embedding of Massive Single-Cell ATAC-Seq Data. Sci. Bull. 2021, 66, 2271–2276. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for Occurrences of a given Motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Machanick, P. Inferring Direct DNA Binding from ChIP-Seq. Nucleic Acids Res. 2012, 40, e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts Cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 2018, 71, 858–871.e8. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.; Zhang, S. Joint Reconstruction of Cis -Regulatory Interaction Networks across Multiple Tissues Using Single-Cell Chromatin Accessibility Data. Brief. Bioinform. 2021, 22, bbaa120. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.J.; Hannenhalli, S.; Plotkin, J.B. Why Transcription Factor Binding Sites Are Ten Nucleotides Long. Genetics 2012, 192, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, Z.; Lou, S.; Bedford, J.; Mu, X.J.; Yip, K.Y.; Khurana, E.; Gerstein, M. FunSeq2: A Framework for Prioritizing Noncoding Regulatory Variants in Cancer. Genome Biol. 2014, 15, 480. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, G.R.S.; Dunham, I.; Zeggini, E.; Flicek, P. Functional Annotation of Noncoding Sequence Variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef]
- Cao, Z.; Huang, Y.; Duan, R.; Jin, P.; Qin, Z.S.; Zhang, S. Disease Category-Specific Annotation of Variants Using an Ensemble Learning Framework. Brief. Bioinform. 2022, 23, bbab438. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting Effects of Noncoding Variants with Deep Learning–Based Sequence Model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Quang, D.; Xie, X. DanQ: A Hybrid Convolutional and Recurrent Deep Neural Network for Quantifying the Function of DNA Sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pu, Y.; Tang, J.; Zou, Q.; Guo, F. DeepATT: A Hybrid Category Attention Neural Network for Identifying Functional Effects of DNA Sequences. Brief. Bioinform. 2021, 22, bbaa159. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. In Proceedings of the 2017 IEEE International Conference on Computer Vision (ICCV), Venice, Italy, 22 October 2017; pp. 618–626. [Google Scholar]
- Chen, Z.; Zhang, J.; Liu, J.; Dai, Y.; Lee, D.; Min, M.R.; Xu, M.; Gerstein, M. DECODE: A Deep-Learning Framework for Condensing Enhancers and Refining Boundaries with Large-Scale Functional Assays. Bioinformatics 2021, 37, i280–i288. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Lamkin, M.; Zhao, H.; Wu, C.; Su, H.; Gymrek, M. Deep Neural Networks Identify Sequence Context Features Predictive of Transcription Factor Binding. Nat. Mach. Intell. 2021, 3, 172–180. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-Cell Epigenomic Analyses Implicate Candidate Causal Variants at Inherited Risk Loci for Alzheimer’s and Parkinson’s Diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA Elements (ENCODE): Data Portal Update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. arXiv 2019, arXiv:1912.01703. [Google Scholar]
- Breiman, L. Random Forest. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. arXiv 2018, arXiv:1201.0490 [cs]. [Google Scholar]
- Cooper, G.M.; Brown, C.D. Qualifying the Relationship between Sequence Conservation and Molecular Function. Genome Res. 2008, 18, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Asthana, S.; Roytberg, M.; Stamatoyannopoulos, J.; Sunyaev, S. Analysis of Sequence Conservation at Nucleotide Resolution. PLoS Comput. Biol. 2007, 3, e254. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. A Space-Time Process Model for the Evolution of DNA Sequences. Genetics 1995, 139, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily Conserved Elements in Vertebrate, Insect, Worm, and Yeast Genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac Separates Active from Poised Enhancers and Predicts Developmental State. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Lupski, J.R. Non-Coding Genetic Variants in Human Disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef] [Green Version]
- Grubert, F.; Srivas, R.; Spacek, D.V.; Kasowski, M.; Ruiz-Velasco, M.; Sinnott-Armstrong, N.; Greenside, P.; Narasimha, A.; Liu, Q.; Geller, B.; et al. Landscape of Cohesin-Mediated Chromatin Loops in the Human Genome. Nature 2020, 583, 737–743. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Yu, J.-T. The Role of PICALM in Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 399–413. [Google Scholar] [CrossRef]
- Ma, J.; Yu, J.-T.; Tan, L. MS4A Cluster in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1240–1248. [Google Scholar] [CrossRef]
- Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.M.; Mee, E.W.; Faull, R.L.M.; Dragunow, M. The Transcription Factor PU.1 is Critical for Viability and Function of Human Brain Microglia: Critical Role of PU.1 in Human Microglia. Glia 2013, 61, 929–942. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Smith, A.M.; Smyth, L.C.; Jansson, D.; Scotter, E.L.; Swanson, M.E.V.; Aalderink, M.; Coppieters, N.; Narayan, P.; Handley, R.; et al. PU.1 Regulates Alzheimer’s Disease-Associated Genes in Primary Human Microglia. Mol. Neurodegener. 2018, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Andrews, R.; Holmans, P.; Hill, M.; Taylor, P.R. Modest Changes in Spi1 Dosage Reveal the Potential for Altered Microglial Function as Seen in Alzheimer’s Disease. Sci. Rep. 2021, 11, 14935. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhay, A.; Sarkar, A.; Howlader, P.; Balasubramanian, V.N. Grad-CAM++: Generalized Gradient-Based Visual Explanations for Deep Convolutional Networks. In Proceedings of the 2018 IEEE Winter Conference on Applications of Computer Vision (WACV), Lake Tahoe, NV, USA, 12–15 March 2018; pp. 839–847. [Google Scholar]
- Srinivas, S.; Fleuret, F. Full-Gradient Representation for Neural Network Visualization. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 3 December 2019. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Model | AUROC | AUPR | F1 Score |
|---|---|---|---|---|
| PBMC | scEpiLock | 0.914 | 0.854 | 0.752 |
| DeepSEA | 0.906 | 0.841 | 0.742 | |
| DanQ | 0.905 | 0.841 | 0.753 | |
| RF | 0.79 | 0.681 | 0.451 | |
| Brain | scEpiLock | 0.93 | 0.745 | 0.669 |
| DeepSEA | 0.92 | 0.72 | 0.634 | |
| DanQ | 0.92 | 0.717 | 0.642 | |
| RF | 0.576 | 0.249 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Srinivasan, S.S.; Zhang, R.; Kessenbrock, K.; Zhang, J. scEpiLock: A Weakly Supervised Learning Framework for cis-Regulatory Element Localization and Variant Impact Quantification for Single-Cell Epigenetic Data. Biomolecules 2022, 12, 874. https://doi.org/10.3390/biom12070874
Gong Y, Srinivasan SS, Zhang R, Kessenbrock K, Zhang J. scEpiLock: A Weakly Supervised Learning Framework for cis-Regulatory Element Localization and Variant Impact Quantification for Single-Cell Epigenetic Data. Biomolecules. 2022; 12(7):874. https://doi.org/10.3390/biom12070874
Chicago/Turabian StyleGong, Yanwen, Shushrruth Sai Srinivasan, Ruiyi Zhang, Kai Kessenbrock, and Jing Zhang. 2022. "scEpiLock: A Weakly Supervised Learning Framework for cis-Regulatory Element Localization and Variant Impact Quantification for Single-Cell Epigenetic Data" Biomolecules 12, no. 7: 874. https://doi.org/10.3390/biom12070874
APA StyleGong, Y., Srinivasan, S. S., Zhang, R., Kessenbrock, K., & Zhang, J. (2022). scEpiLock: A Weakly Supervised Learning Framework for cis-Regulatory Element Localization and Variant Impact Quantification for Single-Cell Epigenetic Data. Biomolecules, 12(7), 874. https://doi.org/10.3390/biom12070874