
Development of a Biotechnology Platform for the Fast-Growing Cyanobacterium Synechococcus sp. PCC 11901

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Culture Conditions
2.2. Multicultivator MC-1000 Growth Conditions
2.3. Bioinformatics Analysis and Generation of a Metabolic and Electron Transport Map for PCC 11901
2.4. Oxygen Electrode Measurements
2.5. Development of Vitamin B12-Independent PCC 11901 Strains
2.6. Cloning of the metE Upstream Region and Generation of B12-Independent Strains by Targeted Mutagenesis
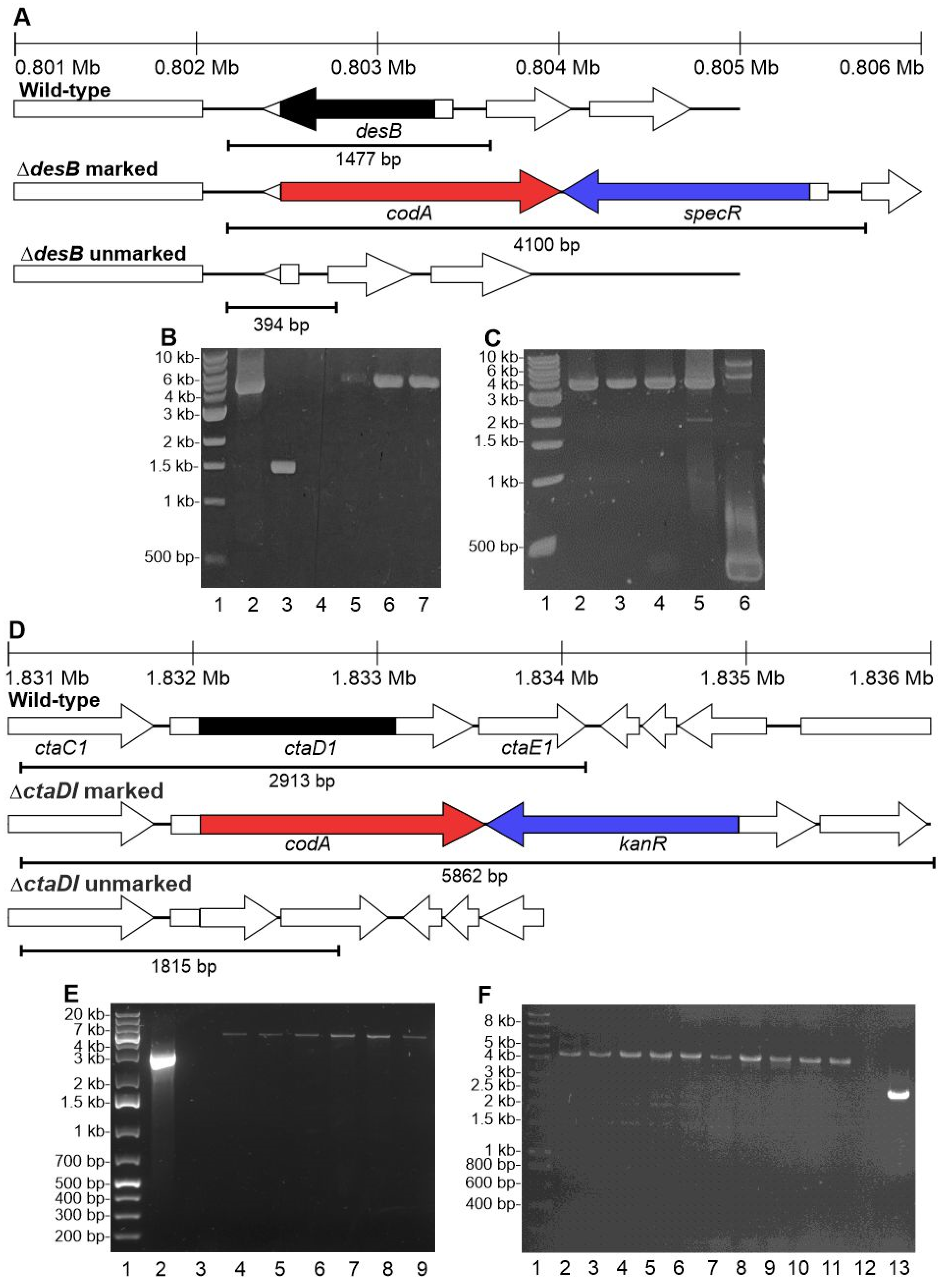
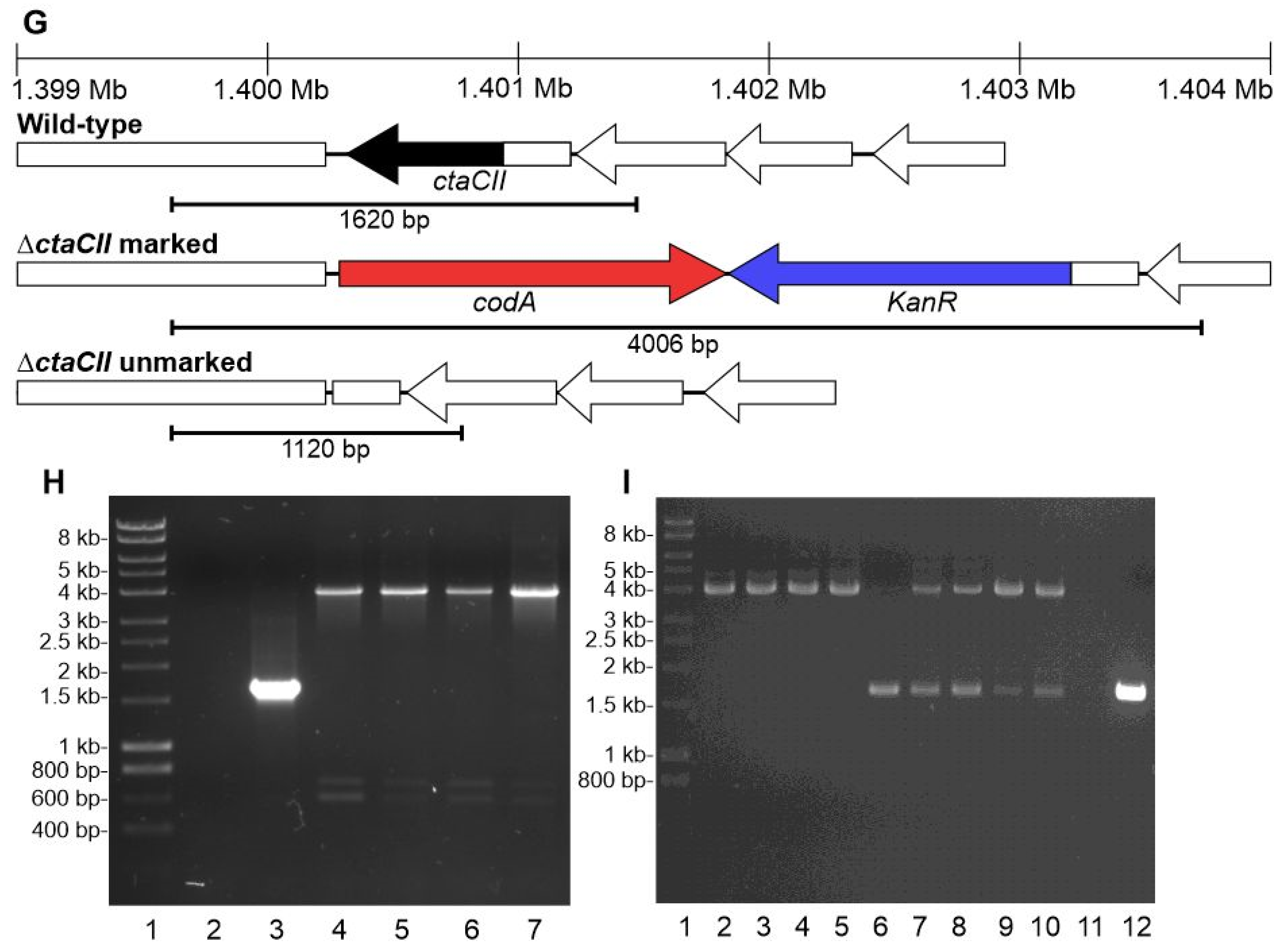
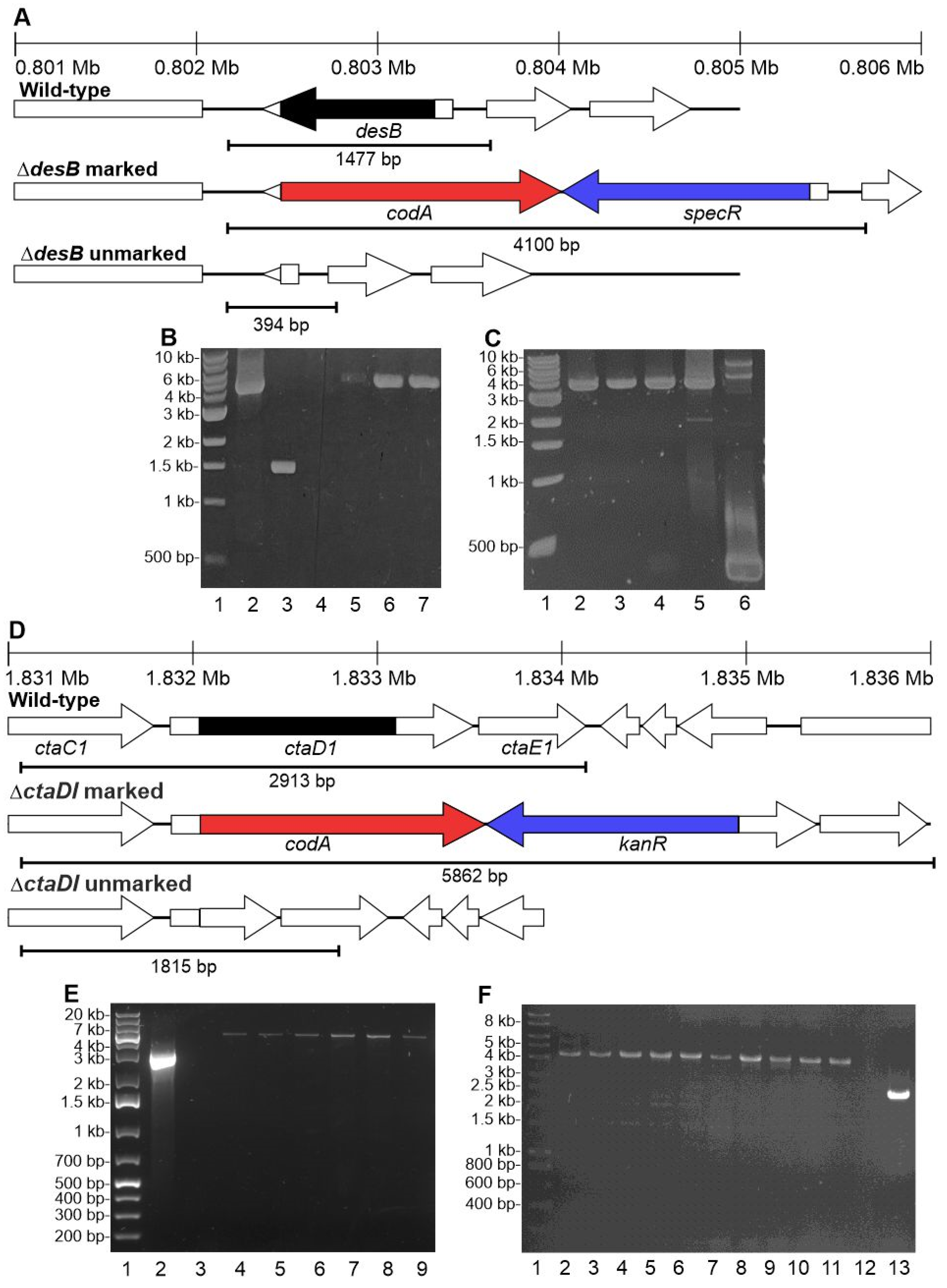
2.7. Generation of PCC 11901 Unmarked Deletion Mutants
3. Results
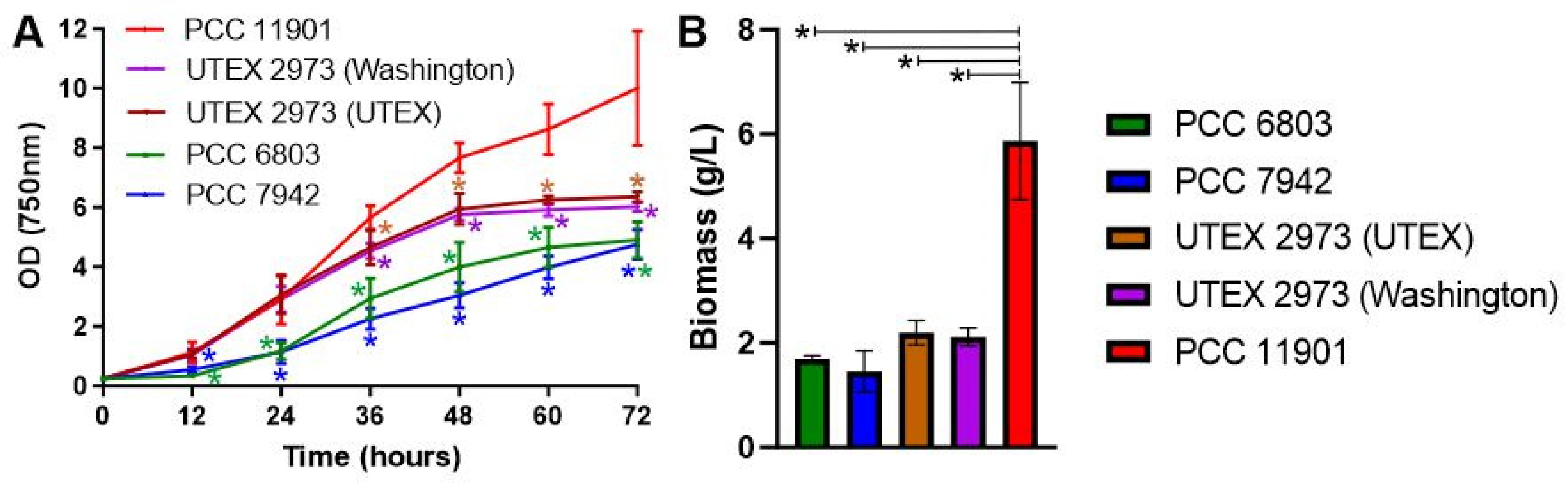
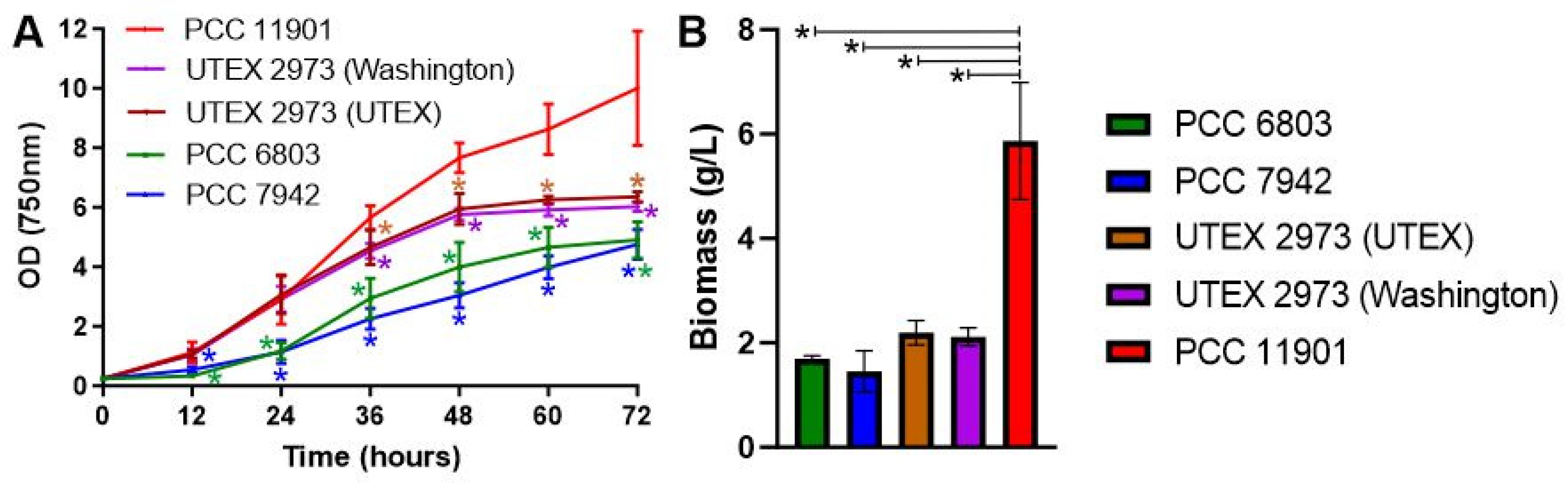
3.1. Synechococcus sp. PCC 11901 Is the Fastest Growing Species under High Light at 38 °C
3.2. Metabolic Pathways Are Highly Conserved between Synechococcus sp. PCC 11901 and Synechocystis sp. PCC 6803
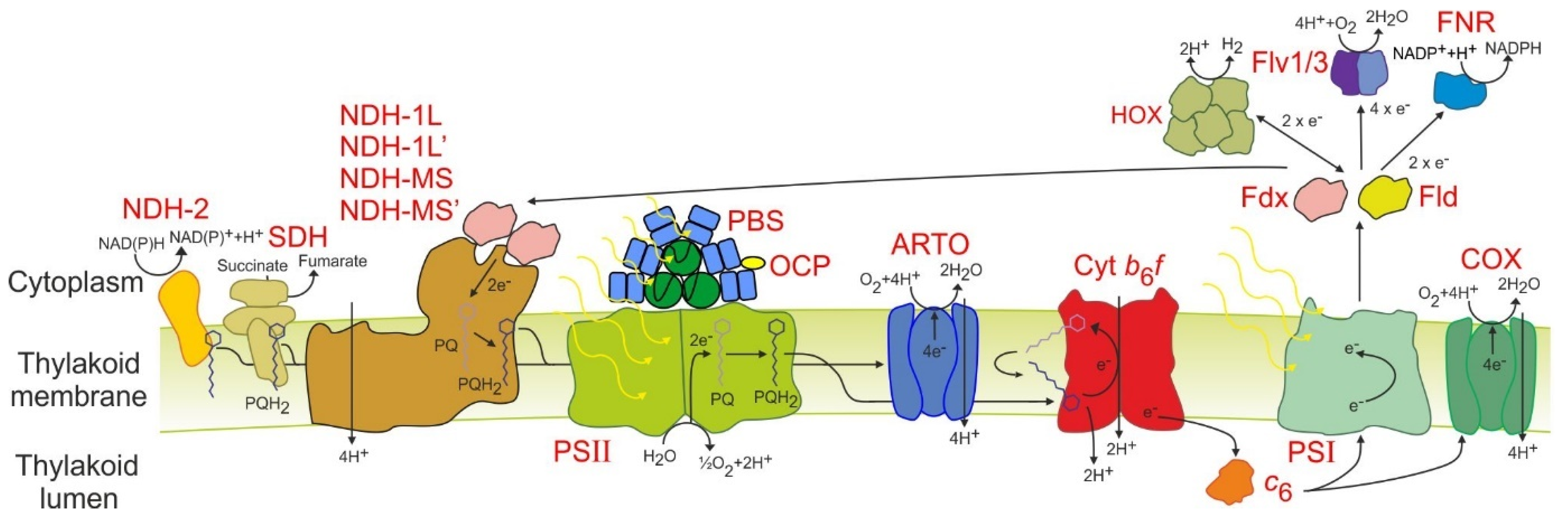
3.3. Electron Transport and Light Harvesting Is Streamlined in PCC 11901 Compared to PCC 6803
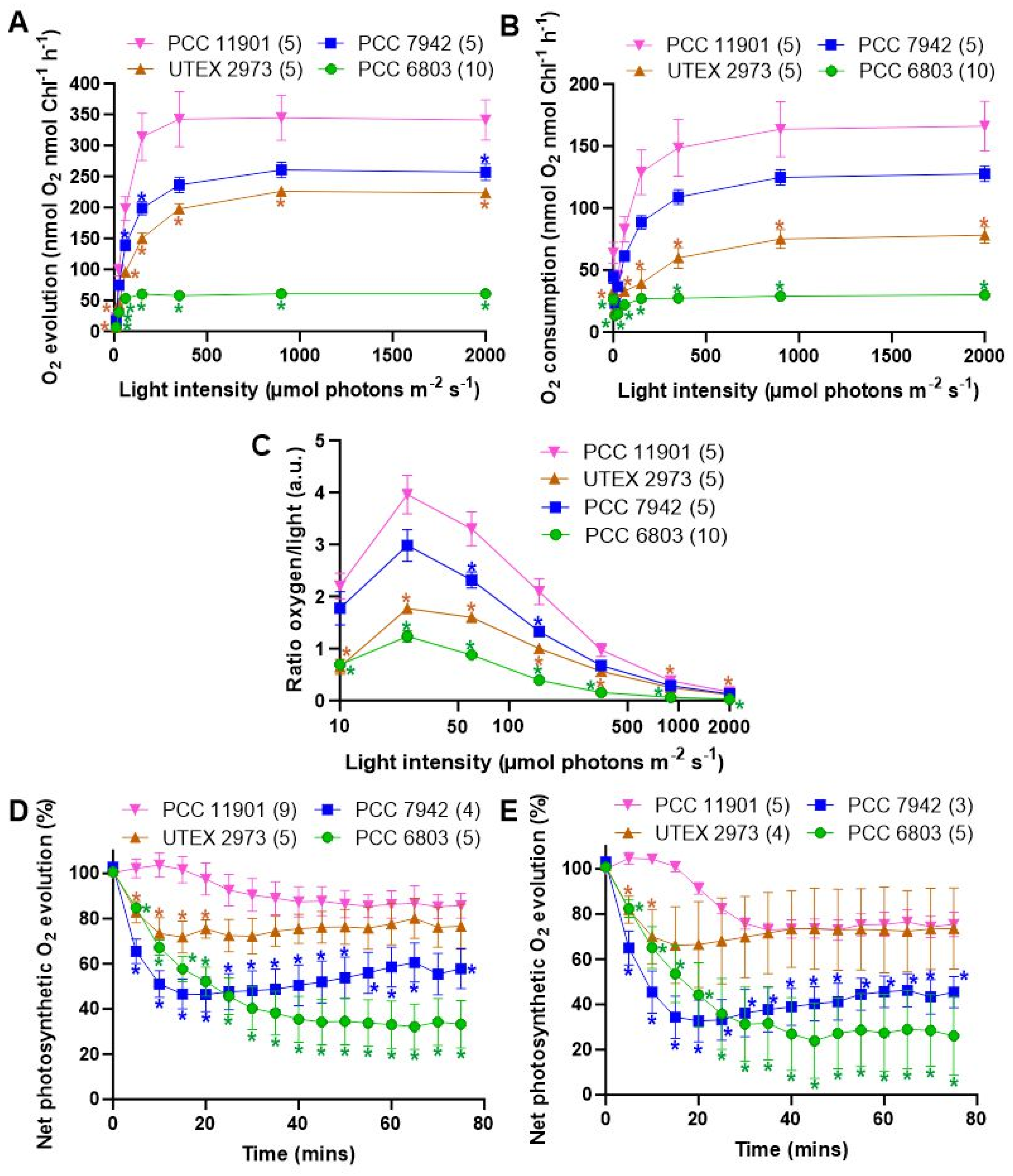
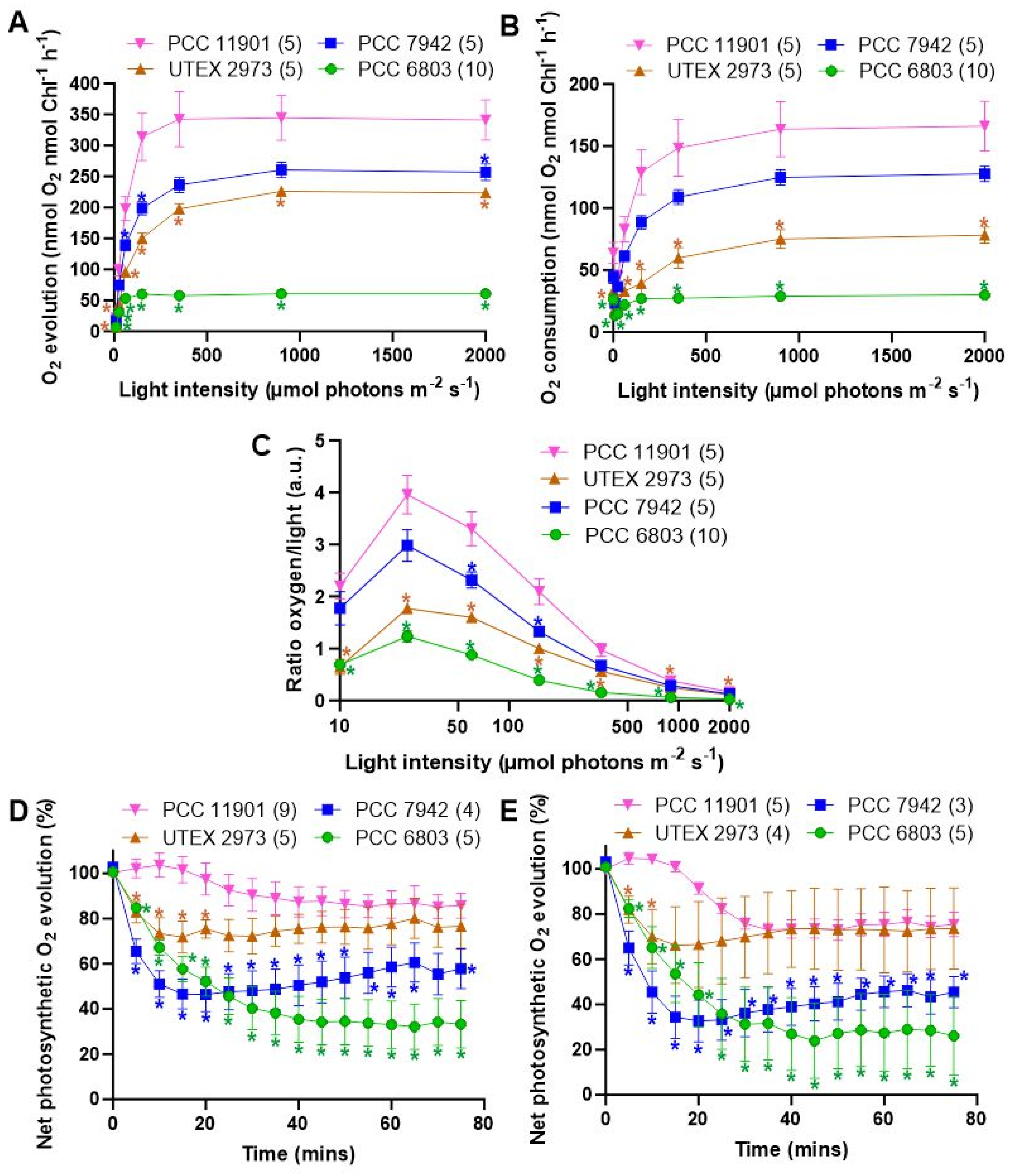
3.4. PCC 11901 Demonstrates Higher Photosynthetic and Respiratory Rates, Lower Photoinhibition and Superior Light Use Compared to Other Model Cyanobacteria
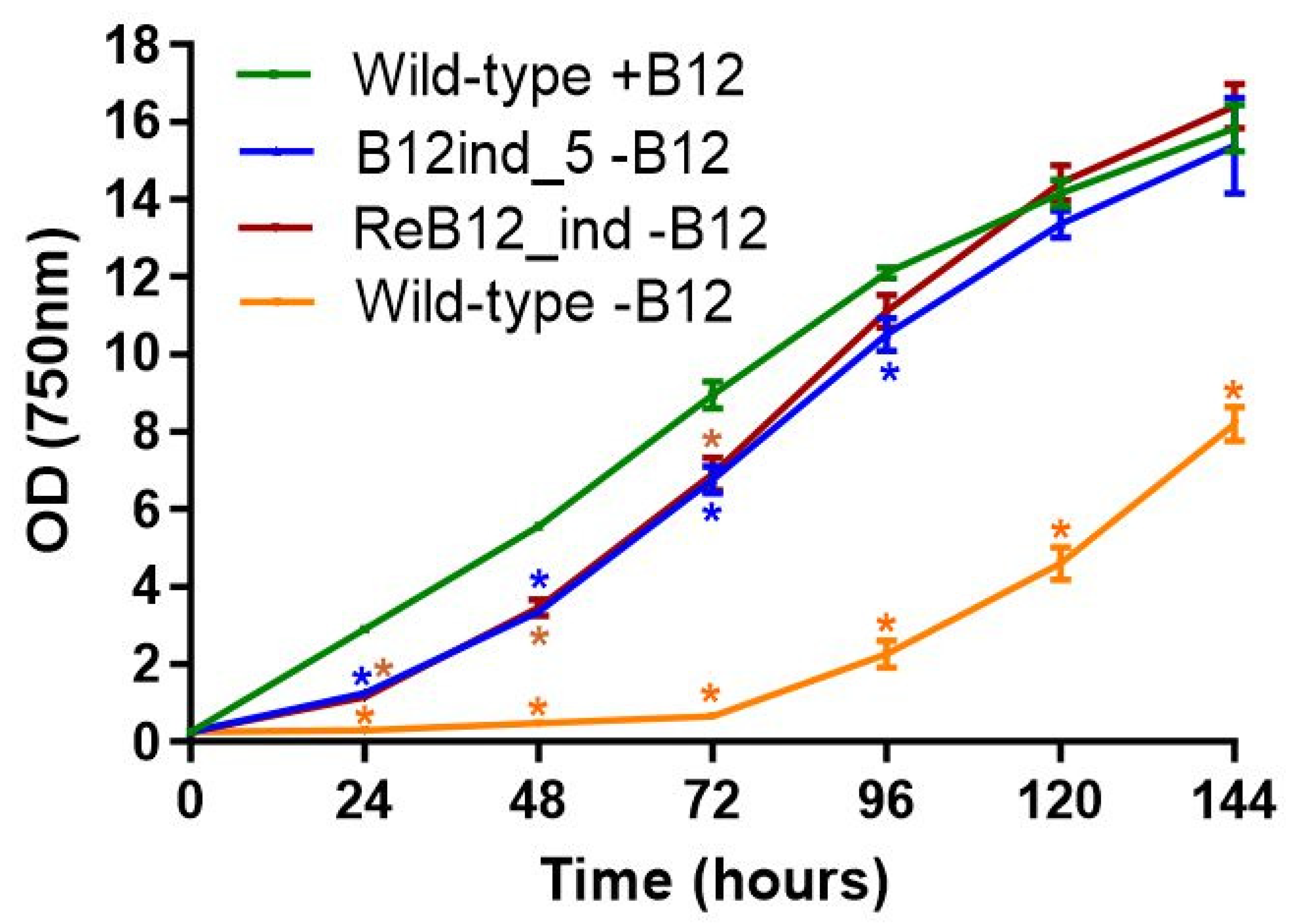
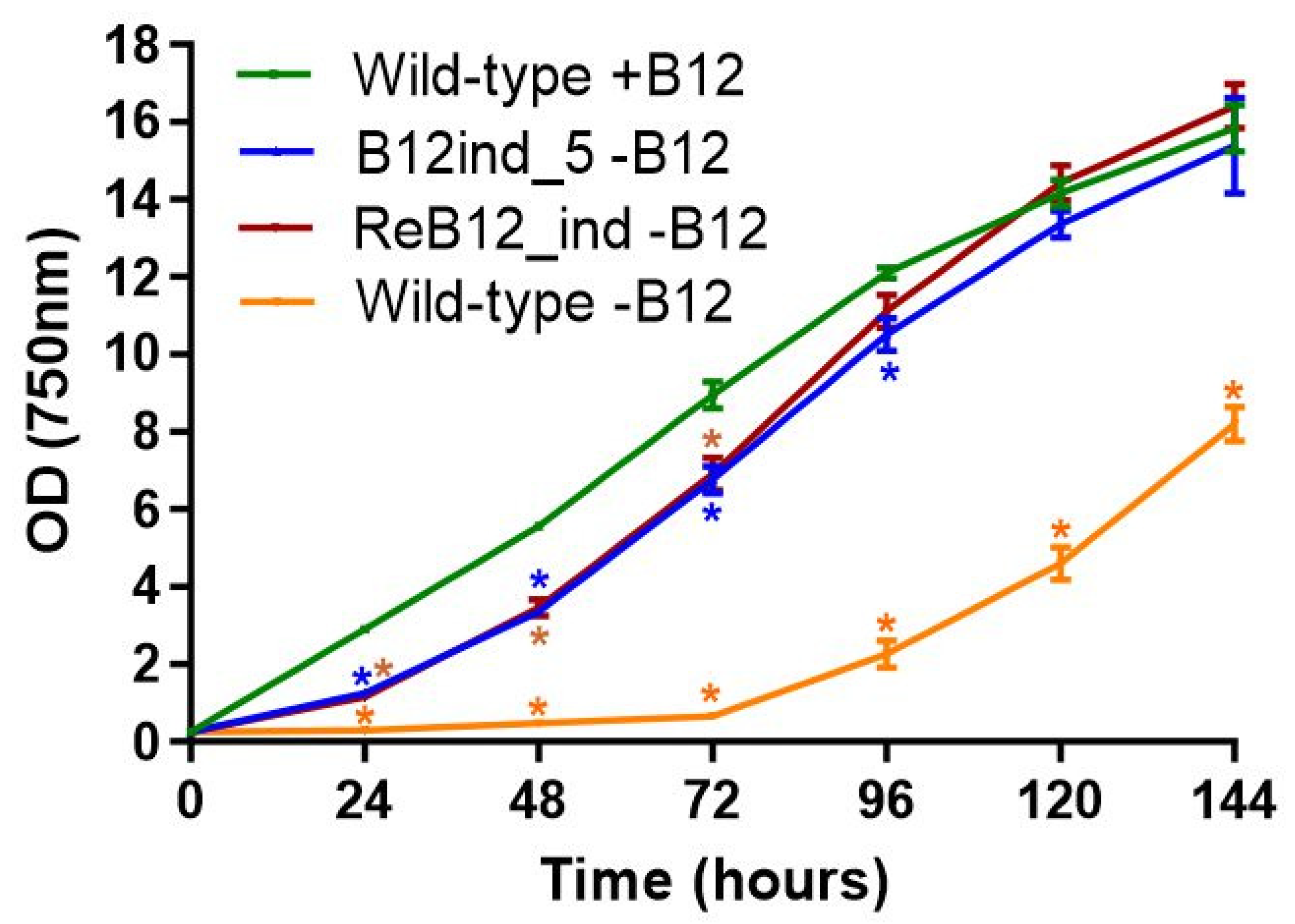
3.5. Generation of a Vitamin B12-Independent Synechococcus sp. PCC 11901 Strain
3.6. The CodA and SacB Negative Selectable Markers Were Not Successfully Used in Synechococcus sp. PCC 11901 for Generating Unmarked Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lea-Smith, D.J.; Howe, C.J. The Use of Cyanobacteria for Biofuel Production. In Biofuels and Bioenergy; Love, J., Bryant, J.A., Eds.; Wiley: New York, NY, USA, 2017. [Google Scholar]
- Ducat, D.C.; Way, J.C.; Silver, P.A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 2011, 29, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.J.; Liberton, M.; Cliften, P.F.; Head, R.D.; Jacobs, J.M.; Smith, R.D.; Koppenaal, D.W.; Brand, J.J.; Pakrasi, H.B. Synechococcus elongatus UTEX 2973, a fast growing cyanobacterial chassis for biosynthesis using light and CO2. Sci. Rep. 2015, 5, 8132. [Google Scholar] [CrossRef] [PubMed]
- Sezonov, G.; Joseleau-Petit, D.; D’Ari, R. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 2007, 189, 8746–8749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoep, J.L.; Mrwebi, M.; Schuurmans, J.M.; Rohwer, J.M.; Teixeira de Mattos, M.J. Control of specific growth rate in Saccharomyces cerevisiae. Microbiology 2009, 155, 1699–1707. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, D.; Sengupta, A.; Sohoni, S.; Sengupta, S.; Phadnavis, A.G.; Pakrasi, H.B.; Wangikar, P.P. Genome Features and Biochemical Characteristics of a Robust, Fast Growing and Naturally Transformable Cyanobacterium Synechococcus elongatus PCC 11801 Isolated from India. Sci. Rep. 2018, 8, 16632. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Selao, T.T.; Norling, B.; Nixon, P.J. Newly discovered Synechococcus sp. PCC 11901 is a robust cyanobacterial strain for high biomass production. Commun. Biol. 2020, 3, 215. [Google Scholar] [CrossRef]
- Vasudevan, R.; Gale, G.A.R.; Schiavon, A.A.; Puzorjov, A.; Malin, J.; Gillespie, M.D.; Vavitsas, K.; Zulkower, V.; Wang, B.J.; Howe, C.J.; et al. CyanoGate: A Modular Cloning Suite for Engineering Cyanobacteria Based on the Plant MoClo Syntax. Plant Physiol. 2019, 180, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Perez, A.A.; Liu, Z.F.; Rodionov, D.A.; Li, Z.K.; Bryant, D.A. Complementation of Cobalamin Auxotrophy in Synechococcus sp. Strain PCC 7002 and Validation of a Putative Cobalamin Riboswitch In Vivo. J. Bacteriol. 2016, 198, 2743–2752. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.S.; Covert, M.; Palsson, B. Metabolic modelling of microbes: The flux-balance approach. Environ. Microbiol. 2002, 4, 133–140. [Google Scholar] [CrossRef]
- Steuer, R. Fast-growing phototrophic microorganisms and the productivity of phototrophic cultures. Biotechnol. Bioeng. 2022. [Google Scholar] [CrossRef]
- Young, R.E.B.; Purton, S. Cytosine deaminase as a negative selectable marker for the microalgal chloroplast: A strategy for the isolation of nuclear mutations that affect chloroplast gene expression. Plant J. 2014, 80, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lea-Smith, D.J.; Vasudevan, R.; Howe, C.J. Generation of marked and markerless mutants in model cyanobacterial species. J. Vis. Exp. 2016, 111, e54001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.G.K. Construction of specific mutations in photosystem-II photosynthetic reaction center by genetic-engineering methods in Synechocystis-6803. Methods Enzymol. 1988, 167, 766–778. [Google Scholar]
- Ungerer, J.; Wendt, K.E.; Hendry, J.I.; Maranas, C.D.; Pakrasi, H.B. Comparative genomics reveals the molecular determinants of rapid growth of the cyanobacterium Synechococcus elongatus UTEX 2973. Proc. Natl. Acad. Sci. USA 2018, 115, E11761–E11770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Baers, L.L.; Breckels, L.M.; Mills, L.A.; Gatto, L.; Deery, M.; Stevens, T.J.; Howe, C.J.; Lilley, K.S.; Lea-Smith, D.J. Proteome mapping of a cyanobacterium reveals distinct compartment organisation and cell-dispersed metabolism. Plant Physiol. 2019, 181, 1721–1738. [Google Scholar] [CrossRef] [Green Version]
- Mills, L.A.; McCormick, A.J.; Lea-Smith, D.J. Current knowledge and recent advances in understanding metabolism of the model cyanobacterium Synechocystis sp. PCC 6803. Biosci. Rep. 2020, 40, BSR20193325. [Google Scholar] [CrossRef] [Green Version]
- Lea-Smith, D.J.; Ross, N.; Zori, M.; Bendall, D.S.; Dennis, J.S.; Scott, S.a.; Smith, A.G.; Howe, C.J. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 2013, 162, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Porra, R.J.; Thompson, W.A.; Kriedemann, P.E. Determination of Accurate Extinction Coefficients and Simultaneous-Equations for Assaying Chlorophyll-a and Chlorophyll-B Extracted with 4 Different Solvents—Verification of the Concentration of Chlorophyll Standards by Atomic-Absorption Spectroscopy. Biochim. Biophys. Acta 1989, 975, 384–394. [Google Scholar] [CrossRef]
- Kostylev, M.; Otwell, A.E.; Richardson, R.E.; Suzuki, Y. Cloning Should Be Simple: Escherichia coli DH5 alpha-Mediated Assembly of Multiple DNA Fragments with Short End Homologies. PLoS ONE 2015, 10, e0137466. [Google Scholar] [CrossRef] [Green Version]
- Gale, G.A.R.; Osorio, A.A.S.; Puzorjov, A.; Wang, B.J.; McCormick, A.J. Genetic Modification of Cyanobacteria by Conjugation Using the CyanoGate Modular Cloning Toolkit. JoVE-J. Vis. Exp. 2019, 152, e60451. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Lucius, S.; Kern, R.; Bauwe, H.; Kaplan, A.; Kopka, J.; Hagemann, M. The Synechocystis sp. PCC 6803 Genome Encodes Up to Four 2-Phosphoglycolate Phosphatases. Front. Plant Sci. 2018, 9, 1718. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Perez, D.; Begemann, M.B.; Pfleger, B.F. Modular synthase-encoding gene involved in α-olefin biosynthesis in Synechococcus sp. strain PCC 7002. Appl. Environ. Microbiol. 2011, 77, 4264–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checchetto, V.; Segalla, A.; Allorent, G.; La Rocca, N.; Leanza, L.; Giacometti, G.M.; Uozumi, N.; Finazzi, G.; Bergantino, E.; Szabo, I. Thylakoid potassium channel is required for efficient photosynthesis in cyanobacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 11043–11048. [Google Scholar] [CrossRef] [Green Version]
- Lea-Smith, D.J.; Hanke, G.T. Electron transport in Cyanobacteria and its Potential in Bioproduction. In Cyanobacteria Biotechnology; Hudson, P., Ed.; Wiley-VCH: New York, NY, USA, 2021; Volume 12, pp. 33–63. [Google Scholar]
- Nomura, C.; Bryant, D. Cytochrome c6 from Synechococcus sp. PCC 7002. In The Phototrophic Prokaryotes; Peschek, G., Loffelhardt, W., Schmetterer, G., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1997; pp. 269–274. [Google Scholar]
- Solymosi, D.; Nikkanen, L.; Muth-Pawlak, D.; Fitzpatrick, D.; Vasudevan, R.; Howe, C.J.; Lea-Smith, D.J.; Allahverdiyeva, Y. Cytochrome c M Decreases Photosynthesis under Photomixotrophy in Synechocystis sp. PCC 6803. Plant Physiol. 2020, 183, 700–716. [Google Scholar] [CrossRef] [Green Version]
- Dann, M.; Leister, D. Evidence that cyanobacterial Sll1217 functions analogously to PGRL1 in enhancing PGR5-dependent cyclic electron flow. Nat. Commun. 2019, 10, 5299. [Google Scholar] [CrossRef] [Green Version]
- Schorsch, M.; Kramer, M.; Goss, T.; Eisenhut, M.; Robinson, N.; Osman, D.; Wilde, A.; Sadaf, S.; Bruckler, H.; Walder, L.; et al. A unique ferredoxin acts as a player in the low-iron response of photosynthetic organisms. Proc. Natl. Acad. Sci. USA 2018, 115, E12111–E12120. [Google Scholar] [CrossRef] [Green Version]
- Battchikova, N.; Eisenhut, M.; Aro, E.-M.M. Cyanobacterial NDH-1 complexes: Novel insights and remaining puzzles. Biochim. Biophys. Acta-Bioenerg. 2011, 1807, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Xiong, F.; LoBrutto, R.; Vermaas, W. The Synechocystis sp. PCC 6803 open reading frame slr0201 that is homologous to sdhC from Archaea codes for a [2Fe-2S] protein. bioRXiv 2021. [Google Scholar] [CrossRef]
- Nomura, C.T.; Persson, S.; Shen, G.; Inoue-Sakamoto, K.; Bryant, D.A. Characterization of two cytochrome oxidase operons in the marine cyanobacterium Synechococcus sp. PCC 7002: Inactivation of ctaDI affects the PS I:PS II ratio. Photosynth. Res. 2006, 87, 215–228. [Google Scholar] [CrossRef]
- Lea-Smith, D.J.; Bombelli, P.; Dennis, J.S.; Scott, S.A.; Smith, A.G.; Howe, C.J. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 2014, 165, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lea-Smith, D.J.; Ortiz-Suarez, M.L.; Lenn, T.; Nurnberg, D.J.; Baers, L.L.; Davey, M.P.; Parolini, L.; Huber, R.G.; Cotton, C.A.R.; Mastroianni, G.; et al. Hydrocarbons are essential for optimal cell size, division and growth of cyanobacteria. Plant Physiol. 2016, 172, 1928–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begemann, M.B.; Zess, E.K.; Walters, E.M.; Schmitt, E.F.; Markley, A.L.; Pfleger, B.F. An Organic Acid Based Counter Selection System for Cyanobacteria. PLoS ONE 2013, 8, e76594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubeau, M.P.; Ghinet, M.G.; Jacques, P.E.; Clermont, N.; Beaulieu, C.; Brzezinski, R. Cytosine Deaminase as a Negative Selection Marker for Gene Disruption and Replacement in the Genus Streptomyces and Other Actinobacteria. Appl. Environ. Microbiol. 2009, 75, 1211–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, A.I.M.; Lale, R.; Hohmann-Marriott, M.F. Streamlining recombination-mediated genetic engineering by validating three neutral integration sites in Synechococcus sp. PCC 7002. J. Biol. Eng. 2017, 11, 19. [Google Scholar] [CrossRef]
- Klahn, S.; Hagemann, M. Compatible solute biosynthesis in cyanobacteria. Environ. Microbiol. 2011, 13, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Gale, G.A.R.; Schiavon Osorio, A.A.; Mills, L.A.; Wang, B.; Lea-Smith, D.J.; McCormick, A.J. Emerging Species and Genome Editing Tools: Future Prospects in Cyanobacterial Synthetic Biology. Microorganisms 2019, 7, 409. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.M.; Parrish, S.; Nielsen, D.R. Exploiting Polyploidy for Markerless and Plasmid-Free Genome Engineering in Cyanobacteria. ACS Synth. Biol. 2021, 10, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, J.; Pakrasi, H.B. Cpf1 Is A Versatile Tool for CRISPR Genome Editing Across Diverse Species of Cyanobacteria. Sci. Rep. 2016, 6, 39681. [Google Scholar] [CrossRef]
- Zerulla, K.; Ludt, K.; Soppa, J. The ploidy level of Synechocystis sp. PCC 6803 is highly variable and is influenced by growth phase and by chemical and physical external parameters. Microbiol.-Sgm 2016, 162, 730–739. [Google Scholar] [CrossRef]
- Pope, M.A.; Hodge, J.A.; Nixon, P.J. An Improved Natural Transformation Protocol for the Cyanobacterium Synechocystis sp. PCC 6803. Front. Plant Sci. 2020, 11, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, J.C.; Banerjee, R.V.; Huang, S.; Sumner, J.S.; Matthews, R.G. Comparison of Cobalamin-Independent and Cobalamin-Dependent Methionine Synthases from Escherichia-Coli—2 Solutions to the Same Chemical Problem. Biochemistry 1992, 31, 6045–6056. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Goodger, C.; Merino, E. RibEx: A web server for locating riboswitches and other conserved bacterial regulatory elements. Nucleic Acids Res. 2005, 33, W690–W692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.N.; Crawford, T.S.; Jeffs, A.; Stockwell, P.A.; Eaton-Rye, J.J.; Summerfield, T.C. Whole genome re-sequencing of two ‘wild-type’ strains of the model cyanobacterium Synechocystis sp. PCC 6803. N. Z. J. Bot. 2014, 52, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Kirst, H.; Formighieri, C.; Melis, A. Maximizing photosynthetic efficiency and culture productivity in cyanobacteria upon minimizing the phycobilisome light-harvesting antenna size. Biochim. Biophys. Acta-Bioenergy 2014, 1837, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Page, L.E.; Liberton, M.; Pakrasi, H.B. Reduction of photoautotrophic productivity in the cyanobacterium Synechocystis sp. strain PCC 6803 by phycobilisome antenna truncation. Appl. Environ. Microbiol. 2012, 78, 6349–6351. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.H.; Bernat, G.; Wagner, H.; Rogner, M.; Rexroth, S. Reduced light-harvesting antenna: Consequences on cyanobacterial metabolism and photosynthetic productivity. Algal Res.-Biomass Biofuels Bioprod. 2013, 2, 188–195. [Google Scholar] [CrossRef]
- Collins, A.M.; Liberton, M.; Jones, H.D.; Garcia, O.F.; Pakrasi, H.B.; Timlin, J.A. Photosynthetic pigment localization and thylakoid membrane morphology are altered in Synechocystis 6803 phycobilisome mutants. Plant Physiol. 2012, 158, 1600–1609. [Google Scholar] [CrossRef] [Green Version]
- Liberton, M.; Collins, A.M.; Page, L.E.; O’Dell, W.B.; O’Neill, H.; Urban, V.S.; Timlin, J.A.; Pakrasi, H.B. Probing the consequences of antenna modification in cyanobacteria. Photosynth. Res. 2013, 118, 17–24. [Google Scholar] [CrossRef]
- Liberton, M.; Chrisler, W.B.; Nicora, C.D.; Moore, R.J.; Smith, R.D.; Koppenaal, D.W.; Pakrasi, H.B.; Jacobs, J.M. Phycobilisome truncation causes widespread proteome changes in Synechocystis sp PCC 6803. PLoS ONE 2017, 12, e0173251. [Google Scholar] [CrossRef] [Green Version]
- Savir, Y.; Noor, E.; Milo, R.; Tlusty, T. Cross-species analysis traces adaptation of Rubisco toward optimality in a low-dimensional landscape. Proc. Natl. Acad. Sci. USA 2010, 107, 3475–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, C.; Ogata, K.; Shikata, M.; Jikuya, H.; Takano, J.; Furumichi, M.; Kanehisa, M.; Omata, T.; Sugiura, M.; Sugita, M. Complete nucleotide sequence of the freshwater unicellular cyanobacterium Synechococcus elongatus PCC 6301 chromosome: Gene content and organization. Photosynth. Res. 2007, 93, 55–67. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mills, L.A.; Moreno-Cabezuelo, J.Á.; Włodarczyk, A.; Victoria, A.J.; Mejías, R.; Nenninger, A.; Moxon, S.; Bombelli, P.; Selão, T.T.; McCormick, A.J.; et al. Development of a Biotechnology Platform for the Fast-Growing Cyanobacterium Synechococcus sp. PCC 11901. Biomolecules 2022, 12, 872. https://doi.org/10.3390/biom12070872
Mills LA, Moreno-Cabezuelo JÁ, Włodarczyk A, Victoria AJ, Mejías R, Nenninger A, Moxon S, Bombelli P, Selão TT, McCormick AJ, et al. Development of a Biotechnology Platform for the Fast-Growing Cyanobacterium Synechococcus sp. PCC 11901. Biomolecules. 2022; 12(7):872. https://doi.org/10.3390/biom12070872
Chicago/Turabian StyleMills, Lauren A., José Ángel Moreno-Cabezuelo, Artur Włodarczyk, Angelo J. Victoria, Rebeca Mejías, Anja Nenninger, Simon Moxon, Paolo Bombelli, Tiago T. Selão, Alistair J. McCormick, and et al. 2022. "Development of a Biotechnology Platform for the Fast-Growing Cyanobacterium Synechococcus sp. PCC 11901" Biomolecules 12, no. 7: 872. https://doi.org/10.3390/biom12070872
APA StyleMills, L. A., Moreno-Cabezuelo, J. Á., Włodarczyk, A., Victoria, A. J., Mejías, R., Nenninger, A., Moxon, S., Bombelli, P., Selão, T. T., McCormick, A. J., & Lea-Smith, D. J. (2022). Development of a Biotechnology Platform for the Fast-Growing Cyanobacterium Synechococcus sp. PCC 11901. Biomolecules, 12(7), 872. https://doi.org/10.3390/biom12070872