
Tertiary and Quaternary Structure Organization in GMP Synthetases: Implications for Catalysis

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of PfGMPS and Mutagenesis
2.2. Protein Purification
2.3. Crystallization, Data Collection and Structure Determination
2.4. Small Angle X-ray Scattering
2.5. Characterization of GATase Domain Rotation
2.6. Phylogenetic Data Analysis
2.7. Bioinformatic Analysis
2.8. Figure Rendering
3. Results
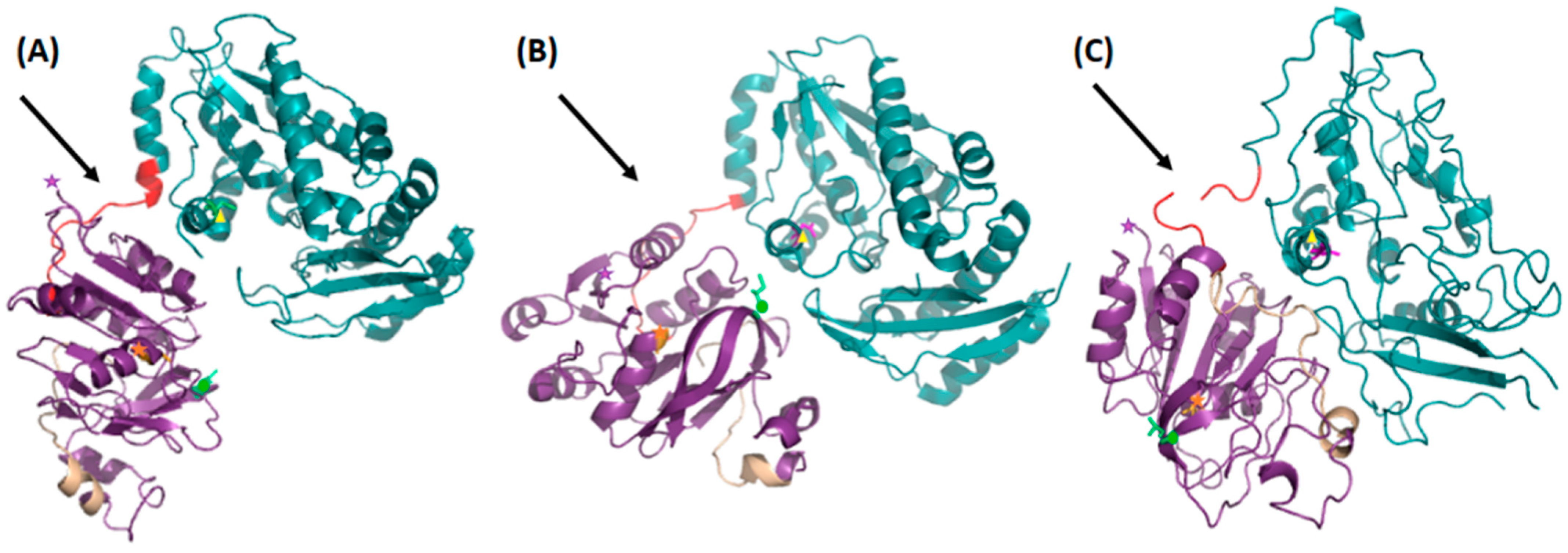
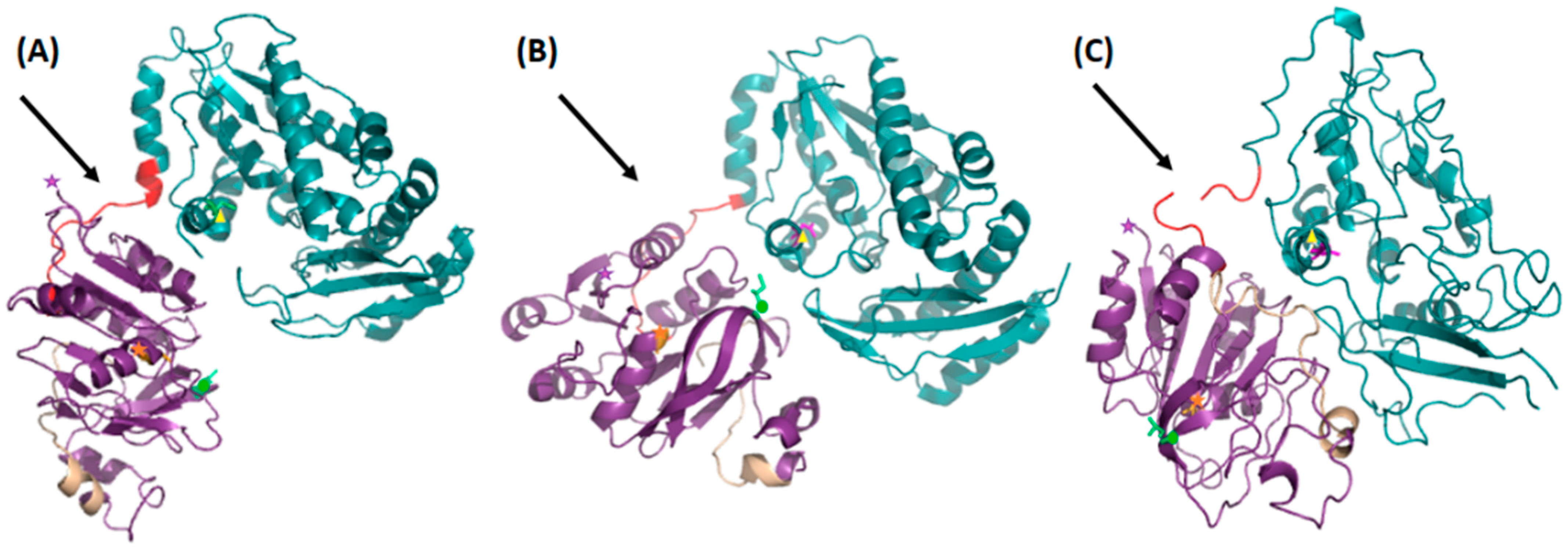
3.1. Crystal- and Solution Structures

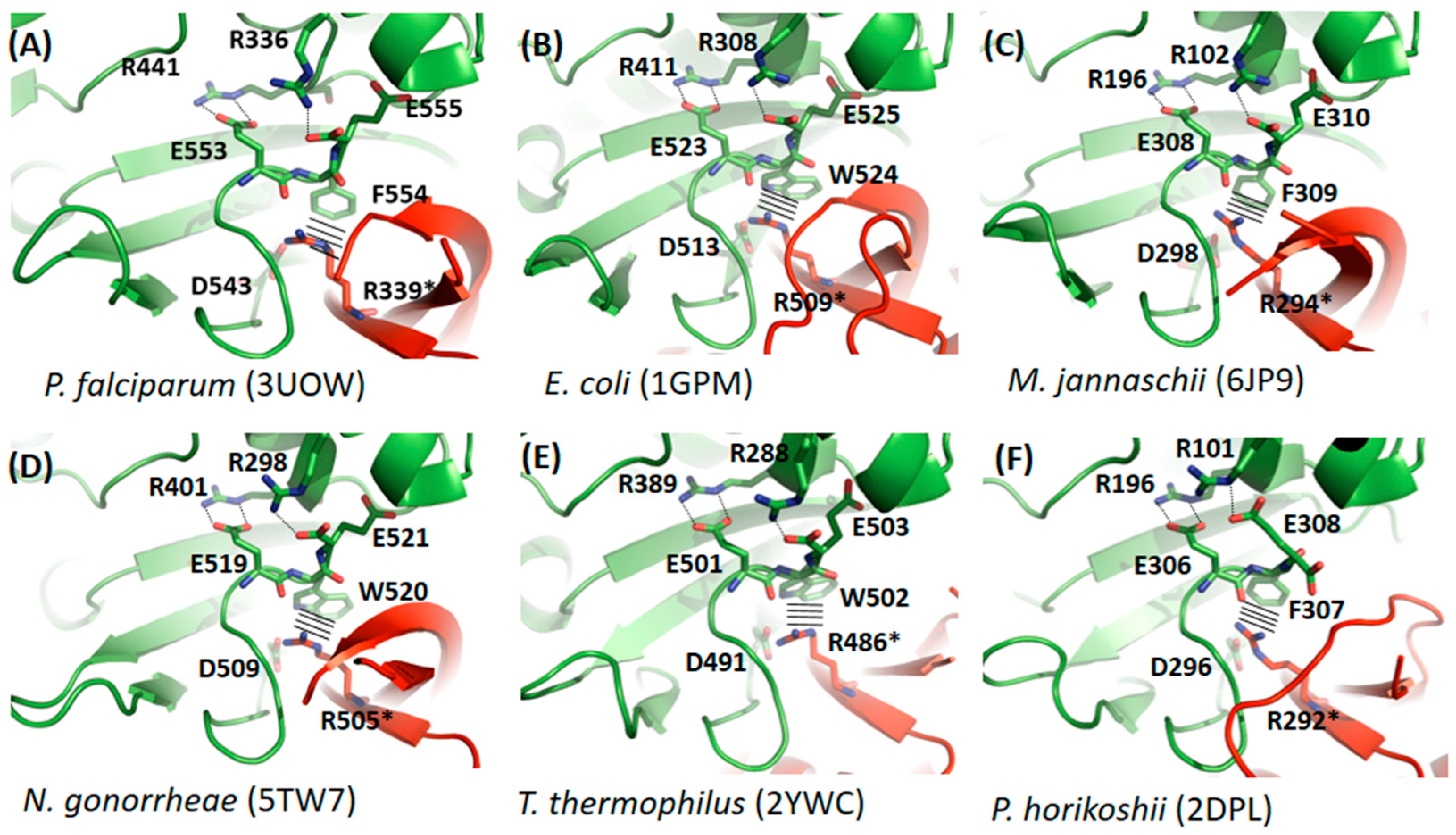
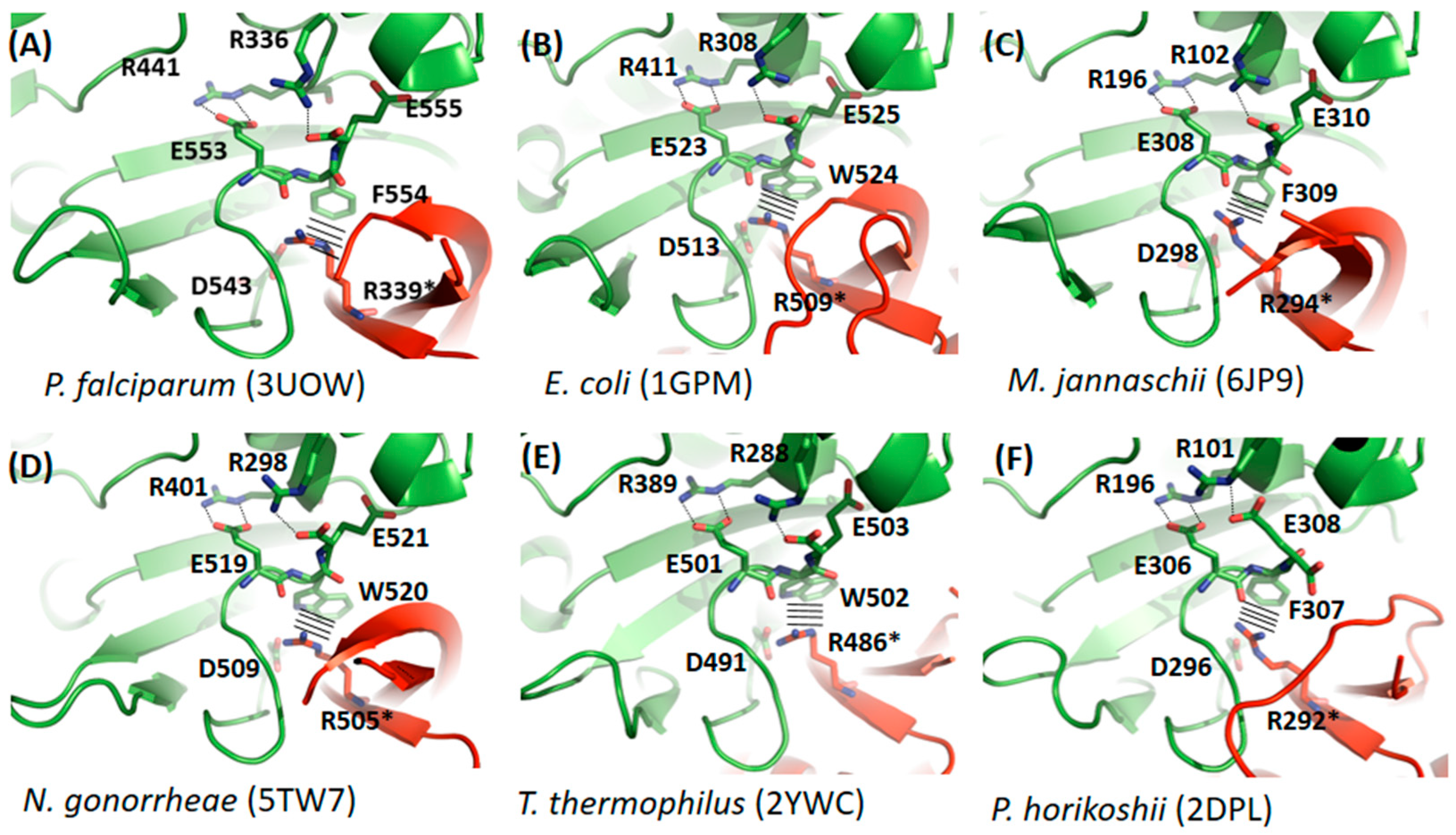
3.2. Comparative Studies of Dimerization Interfaces
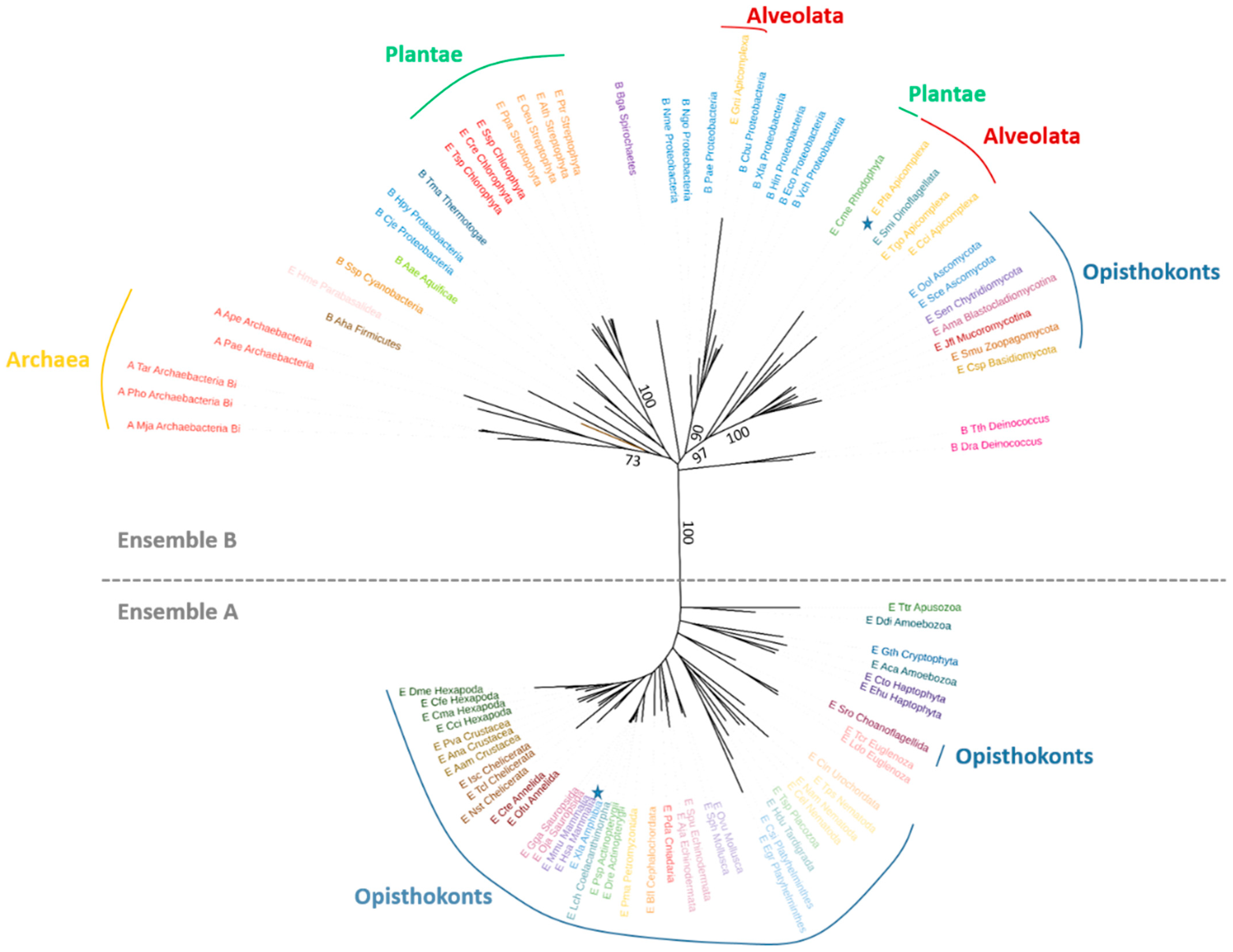
3.3. Phylogenetic Analyses
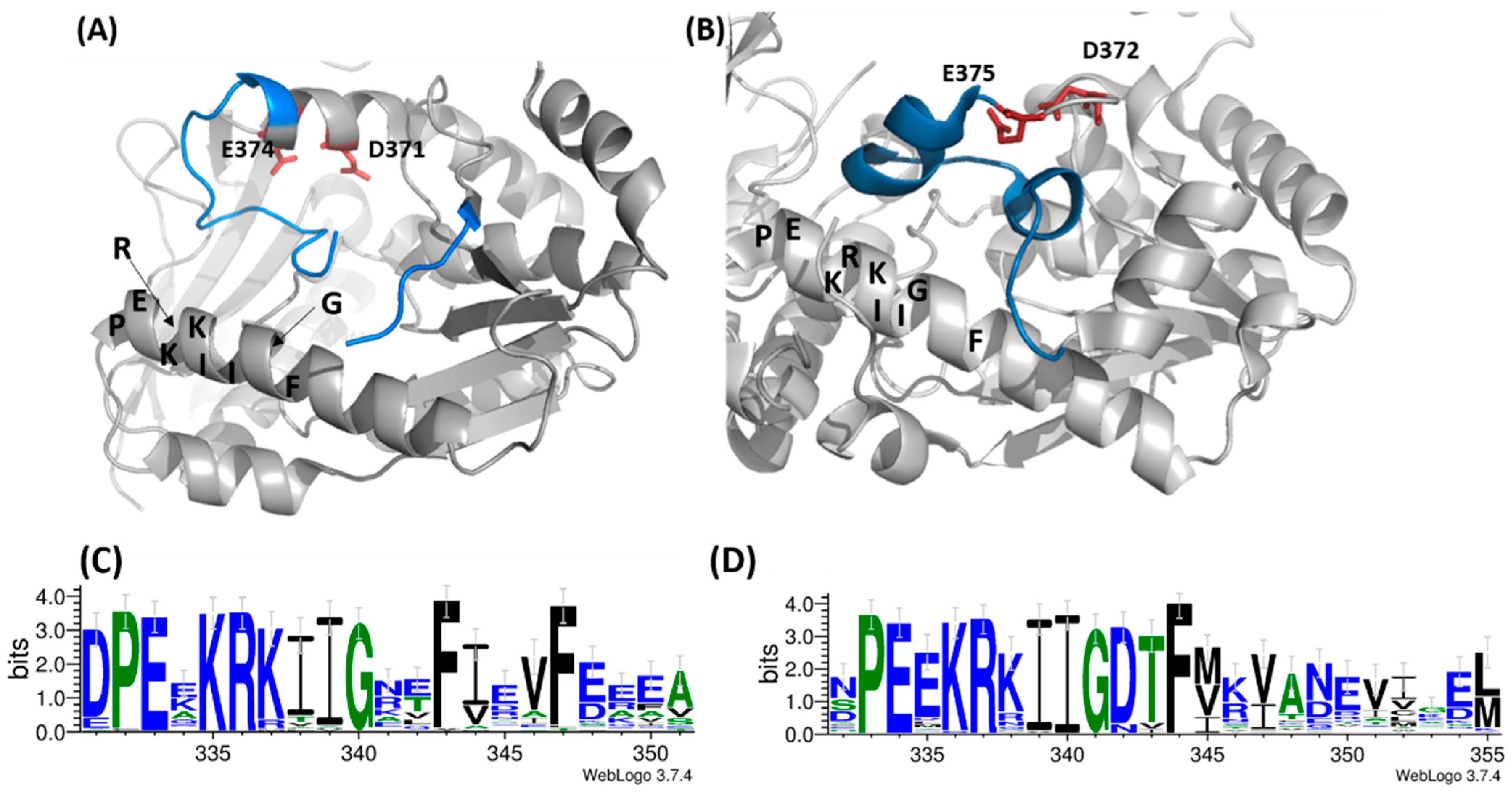
3.4. Comparative Studies of the Catalytic Loop Region in the ATPPase Domain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi-Smiraglia, A.; Wawrzyniak, J.A.; Bagati, A.; Marvin, E.K.; Ackroyd, J.; Moparthy, S.; Bshara, W.; Fink, E.E.; Foley, C.E.; Morozevich, G.E.; et al. Pharmacological Targeting of Guanosine Monophosphate Synthase Suppresses Melanoma Cell Invasion and Tumorigenicity. Cell Death Differ. 2015, 22, 1858–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wu, Y.; Li, Y.; Wang, Y.; Shen, F.; Zhou, J.; Chen, Y. Guanosine Monophosphate Synthase Upregulation Mediates Cervical Cancer Progression by Inhibiting the Apoptosis of Cervical Cancer Cells via the Stat3/P53 Pathway. Int. J. Oncol. 2021, 58, 3. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, J.A.; Kozhevnikova, E.; Langenberg, K.; Moshkin, Y.M.; Verrijzer, C.P. Biosynthetic Enzyme GMP Synthetase Cooperates with Ubiquitin-Specific Protease 7 in Transcriptional Regulation of Ecdysteroid Target Genes. Mol. Cell. Biol. 2010, 30, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Leija, C.; Rijo-Ferreira, F.; Chen, J.; Cestari, I.; Stuart, K.; Tu, B.P.; Phillips, M.A. GMP Synthase Is Essential for Viability and Infectivity of Trypanosoma Brucei despite a Redundant Purine Salvage Pathway. Mol. Microbiol. 2015, 97, 1006–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerchl, J.; Erhardt, T.; Sonnewald, U.; Boldt, R. GMP Synthetase Derived from Plants 2001. Google Patents AU7658700A, 1 October 2010. [Google Scholar]
- Chitty, J.L.; Tatzenko, T.L.; Williams, S.J.; Koh, Y.Q.A.E.; Corfield, E.C.; Butler, M.S.; Robertson, A.A.B.; Cooper, M.A.; Kappler, U.; Kobe, B.; et al. GMP Synthase Is Required for Virulence Factor Production and Infection by Cryptococcus neoformans. J. Biol. Chem. 2017, 292, 3049–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Peter, E.; Séguin, D.L.; St-Pierre, É.; Sekulovic, O.; Jeanneau, S.; Tremblay-Tétreault, C.; Lamontagne, A.-M.; Jacques, P.-É.; Lafontaine, D.A.; Fortier, L.-C. Inactivation of the Riboswitch-Controlled GMP Synthase GuaA in Clostridioides difficile Is Associated with Severe Growth Defects and Poor Infectivity in a Mouse Model of Infection. RNA Biol. 2021, 18, 699–710. [Google Scholar] [CrossRef]
- Lou, L.; Nakamura, J.; Tsing, S.; Nguyen, B.; Chow, J.; Straub, K.; Chan, H.; Barnett, J. High-Level Production from a Baculovirus Expression System and Biochemical Characterization of Human GMP Synthetase. Protein Expr. Purif. 1995, 6, 487–495. [Google Scholar] [CrossRef]
- Ballut, L.; Violot, S.; Shivakumaraswamy, S.; Thota, L.P.; Sathya, M.; Kunala, J.; Dijkstra, B.W.; Terreux, R.; Haser, R.; Balaram, H.; et al. Active Site Coupling in Plasmodium falciparum GMP Synthetase Is Triggered by Domain Rotation. Nat. Commun. 2015, 6, 8930. [Google Scholar] [CrossRef] [Green Version]
- Shivakumaraswamy, S.; Pandey, N.; Ballut, L.; Violot, S.; Aghajari, N.; Balaram, H. Helices on Interdomain Interface Couple Catalysis in the ATPPase Domain with Allostery in Plasmodium falciparum GMP Synthetase. Chembiochem 2020, 21, 2805–2817. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Klem, T.J.; Deras, M.L.; Davisson, V.J.; Smith, J.L. The Crystal Structure of GMP Synthetase Reveals a Novel Catalytic Triad and Is a Structural Paradigm for Two Enzyme Families. Nat. Struct. Biol. 1996, 3, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.C.; Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.; Gary, E.; Hillerich, B.; Yao, Z.-K.; Carlier, P.R.; Totrov, M.; et al. Structural Genomics for Drug Design against the Pathogen Coxiella burnetii. Proteins 2015, 83, 2124–2136. [Google Scholar] [CrossRef] [PubMed]
- Bhat, J.Y.; Venkatachala, R.; Singh, K.; Gupta, K.; Sarma, S.P.; Balaram, H. Ammonia Channeling in Plasmodium falciparum GMP Synthetase: Investigation by NMR Spectroscopy and Biochemical Assays. Biochemistry 2011, 50, 3346–3356. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative Model Building, Structure Refinement and Density Modification with the PHENIX AutoBuild Wizard. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 Dictionary: Organization of Prior Chemical Knowledge and Guidelines for Its Use. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2184–2195. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded Functionality and New Tools for Small-Angle Scattering Data Analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Qi, G.; Lee, R.; Hayward, S. A Comprehensive and Non-Redundant Database of Protein Domain Movements. Bioinformatics 2005, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Yanai, I.; Wolf, Y.I.; Koonin, E.V. Evolution of Gene Fusions: Horizontal Transfer versus Independent Events. Genome Biol. 2002, 3, 1–13. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A New Software for Selection of Phylogenetic Informative Regions from Multiple Sequence Alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Henikoff, J.G. Amino Acid Substitution Matrices from Protein Blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. PyMOL 0.99.Rc6; DeLano Scientific LLC.: Palo Alto, CA, USA, 2002. [Google Scholar]
- Bhat, J.Y.; Shastri, B.G.; Balaram, H. Kinetic and Biochemical Characterization of Plasmodium falciparum GMP Synthetase. Biochem. J. 2008, 409, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Svergun, D.I. DAMMIF, a Program for Rapid ab-initio Shape Determination in Small-Angle Scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Welin, M.; Lehtiö, L.; Johansson, A.; Flodin, S.; Nyman, T.; Trésaugues, L.; Hammarström, M.; Gräslund, S.; Nordlund, P. Substrate Specificity and Oligomerization of Human GMP Synthetase. J. Mol. Biol. 2013, 425, 4323–4333. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.D.; Stephens, R.M. Sequence Logos: A New Way to Display Consensus Sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Nguyen, S.; Jovcevski, B.; Pukala, T.L.; Bruning, J.B. Structural Insights into the Antifungal Drug Target Guanosine Monophosphate Synthase from Aspergillus fumigatus. Acta Crystallogr. D Struct. Biol. 2022, 78, 248–259. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PfGMPS_C89A_C113A | |
|---|---|
| Data collection | |
| Beamline | ID30-A ESRF |
| Wavelength (Å) | 0.87290 |
| Resolution range (Å) | 30.0–2.80 |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 111.65, 64.54, 56.33 |
| α, β, γ (°) | 90.00, 90.03, 90.00 |
| number of reflections | 24,244 |
| number of unique reflections | 9228 |
| Rmeas (%) | 11.8 (46.5) |
| CC ½ | 0.99 (0.74) |
| I/σ(I) | 9.3 (3.6) |
| Multiplicity | 2.6 (2.7) |
| Completeness (%) | 92.3 (93.5) |
| number of molecules/asymm. unit | 1 |
| Refinement | |
| R/Rfree 1 (%) | 27.4/31.4 |
| number of atoms | |
| protein | 4070 |
| water | 10 |
| Average B-factor (Å2) | 48.7 |
| protein | 48.8 |
| water | 21.5 |
| RMSD | |
| Bond lengths (Å) | 0.005 |
| Angles (°) | 1.005 |
| Ramachandran | |
| Favored (%) | 76.3 |
| Allowed (%) | 18.5 |
| Outliers (%) | 5.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballut, L.; Violot, S.; Galisson, F.; Gonçalves, I.R.; Martin, J.; Shivakumaraswamy, S.; Carrique, L.; Balaram, H.; Aghajari, N. Tertiary and Quaternary Structure Organization in GMP Synthetases: Implications for Catalysis. Biomolecules 2022, 12, 871. https://doi.org/10.3390/biom12070871
Ballut L, Violot S, Galisson F, Gonçalves IR, Martin J, Shivakumaraswamy S, Carrique L, Balaram H, Aghajari N. Tertiary and Quaternary Structure Organization in GMP Synthetases: Implications for Catalysis. Biomolecules. 2022; 12(7):871. https://doi.org/10.3390/biom12070871
Chicago/Turabian StyleBallut, Lionel, Sébastien Violot, Frédéric Galisson, Isabelle R. Gonçalves, Juliette Martin, Santosh Shivakumaraswamy, Loïc Carrique, Hemalatha Balaram, and Nushin Aghajari. 2022. "Tertiary and Quaternary Structure Organization in GMP Synthetases: Implications for Catalysis" Biomolecules 12, no. 7: 871. https://doi.org/10.3390/biom12070871
APA StyleBallut, L., Violot, S., Galisson, F., Gonçalves, I. R., Martin, J., Shivakumaraswamy, S., Carrique, L., Balaram, H., & Aghajari, N. (2022). Tertiary and Quaternary Structure Organization in GMP Synthetases: Implications for Catalysis. Biomolecules, 12(7), 871. https://doi.org/10.3390/biom12070871