
Immunohistochemical Demonstration of the pGlu79 α-Synuclein Fragment in Alzheimer’s Disease and Its Tg2576 Mouse Model


 , ,
, , 
Abstract
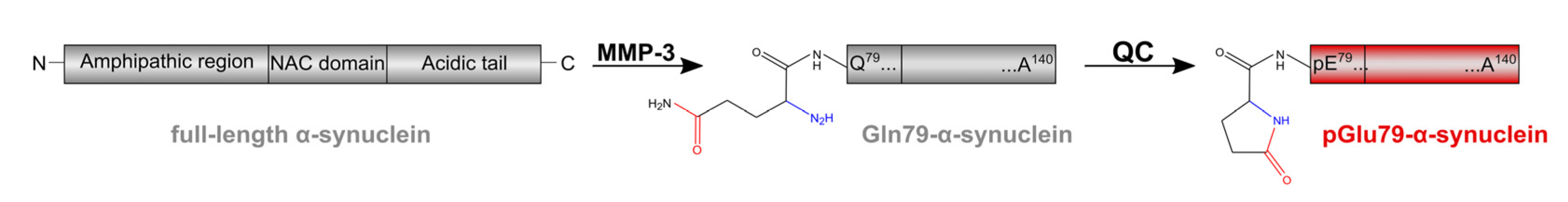
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Mouse Brain Study
2.2.1. Experimental Animals
2.2.2. Tissue Preparation
2.2.3. Immunohistochemistry
Single Labeling Immunohistochemistry
Triple Immunofluorescent Labelings
2.3. Human Brain Study
2.3.1. Human Brain Tissue
2.3.2. Human Brain Tissue Preparation
2.3.3. Immunohistochemistry
Single Labeling Immunohistochemistry
Double Labeling Procedures
2.4. Microscopy
2.4.1. Light Microscopy
2.4.2. Confocal Laser Scanning Microscopy
3. Results
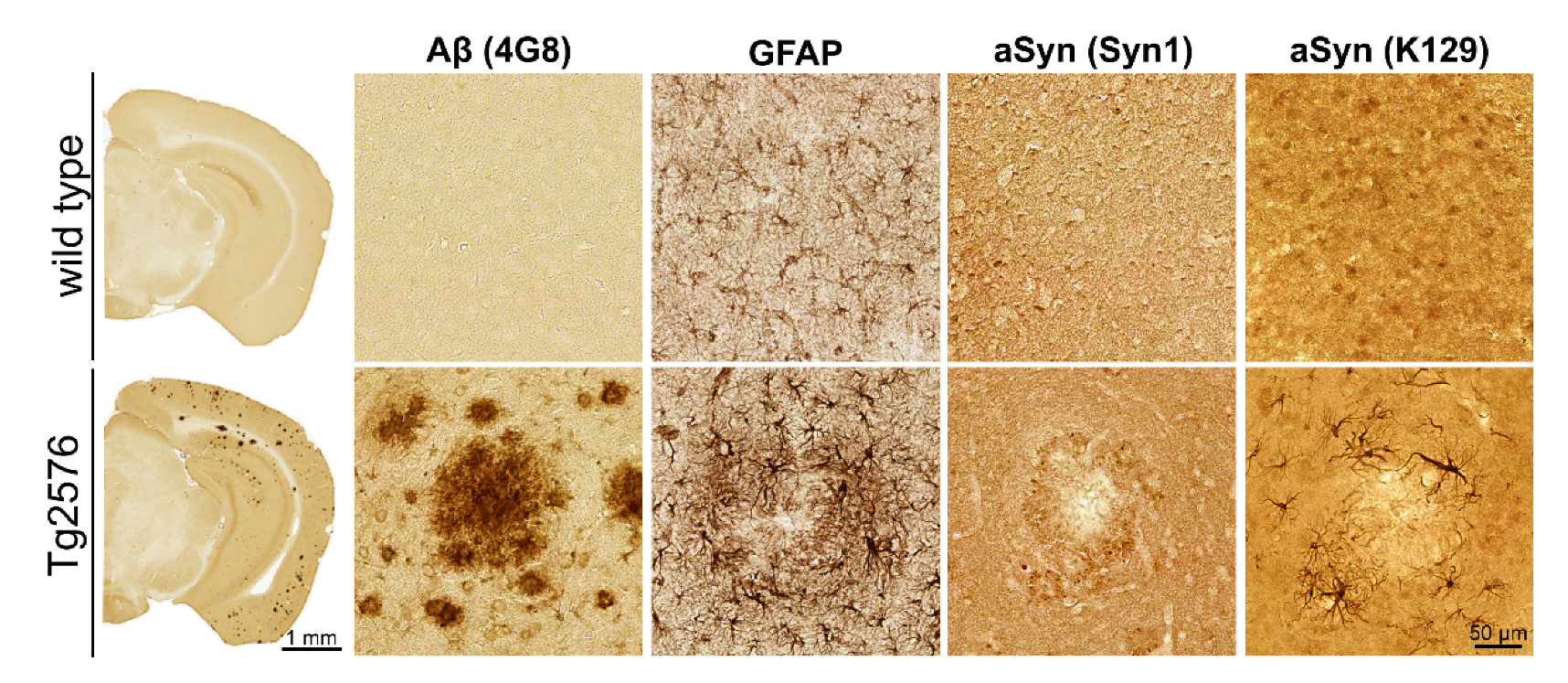
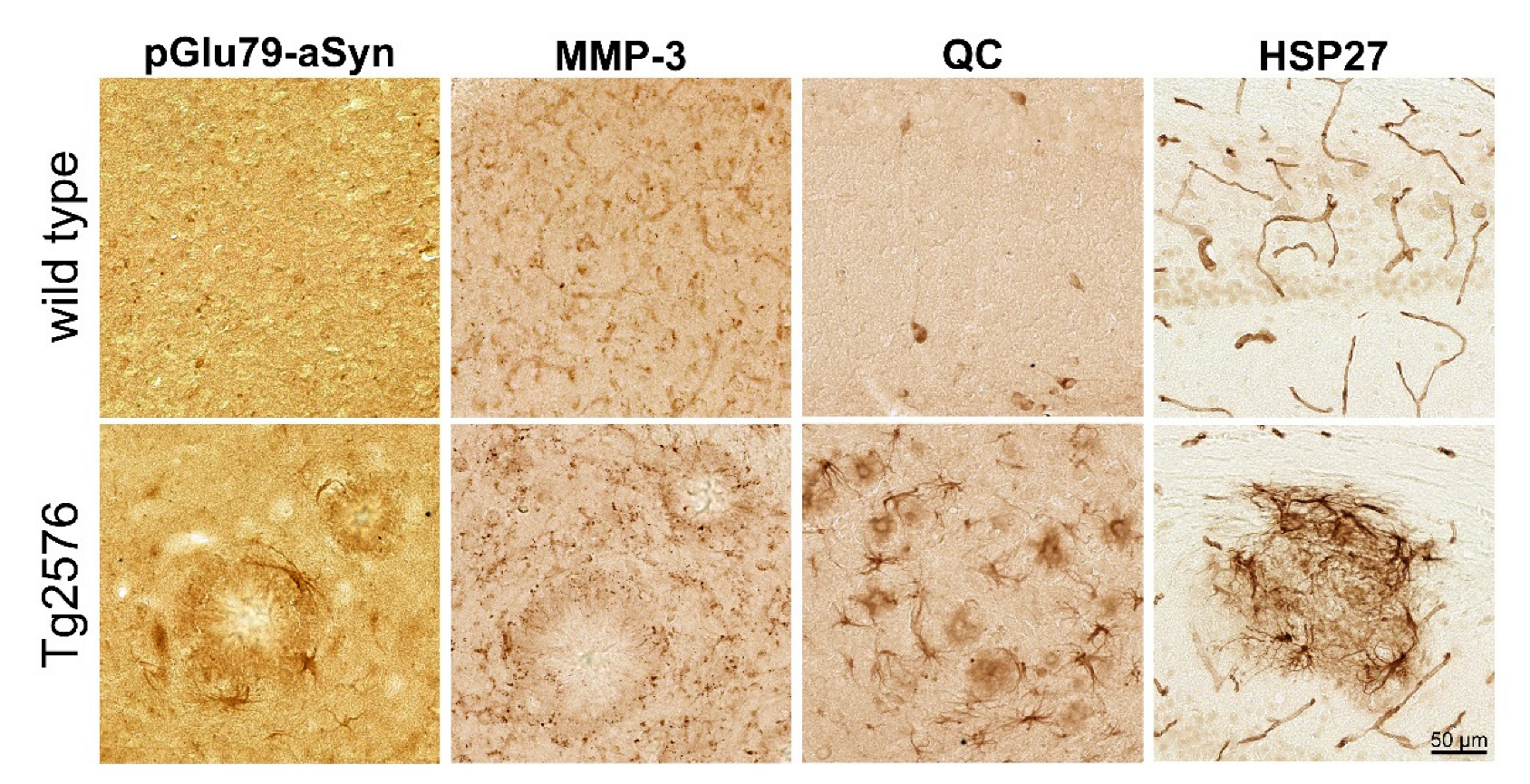
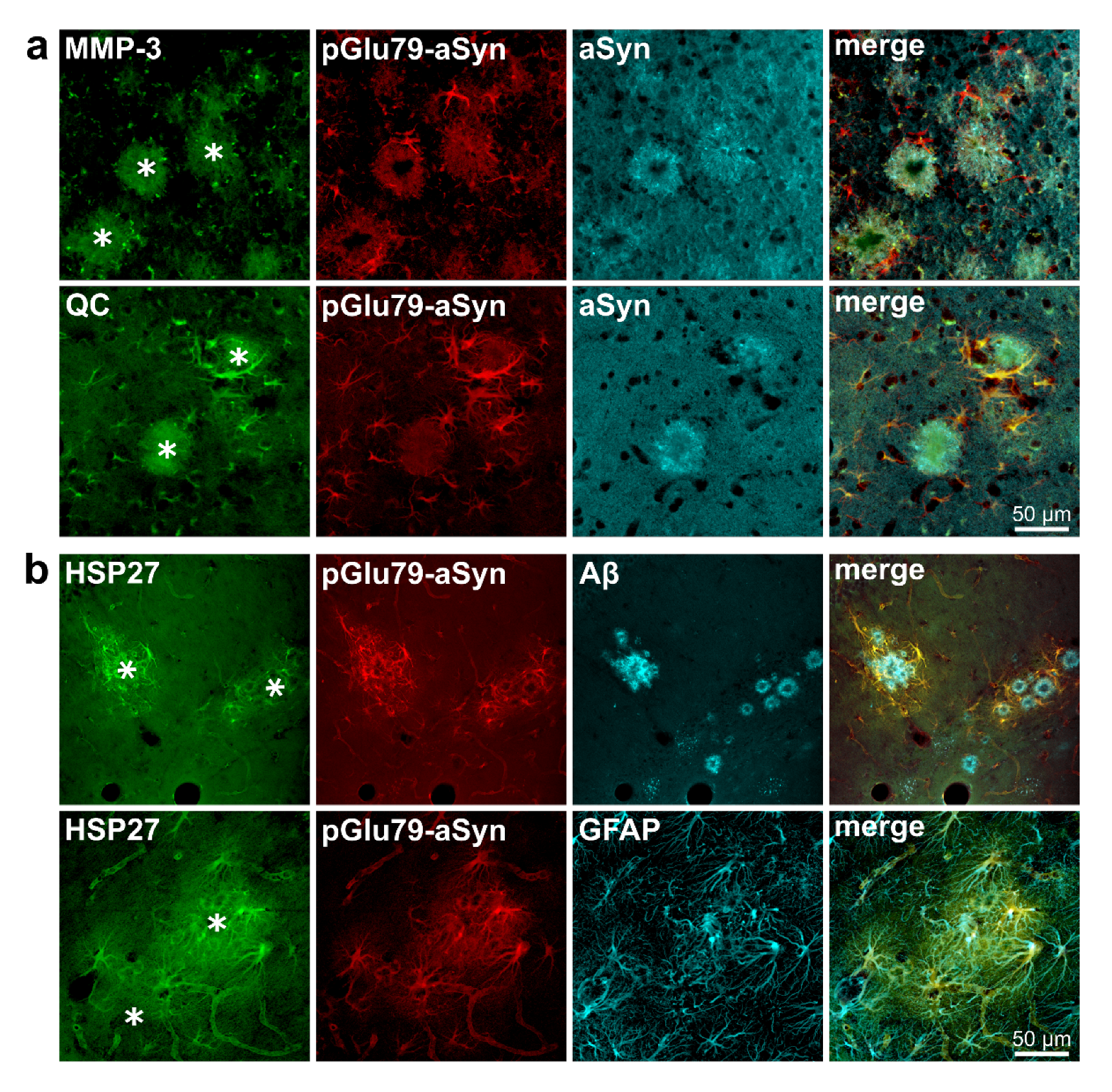
3.1. Association of pGlu79-aSyn with Aβ Plaques in Tg2576 Mouse Brain
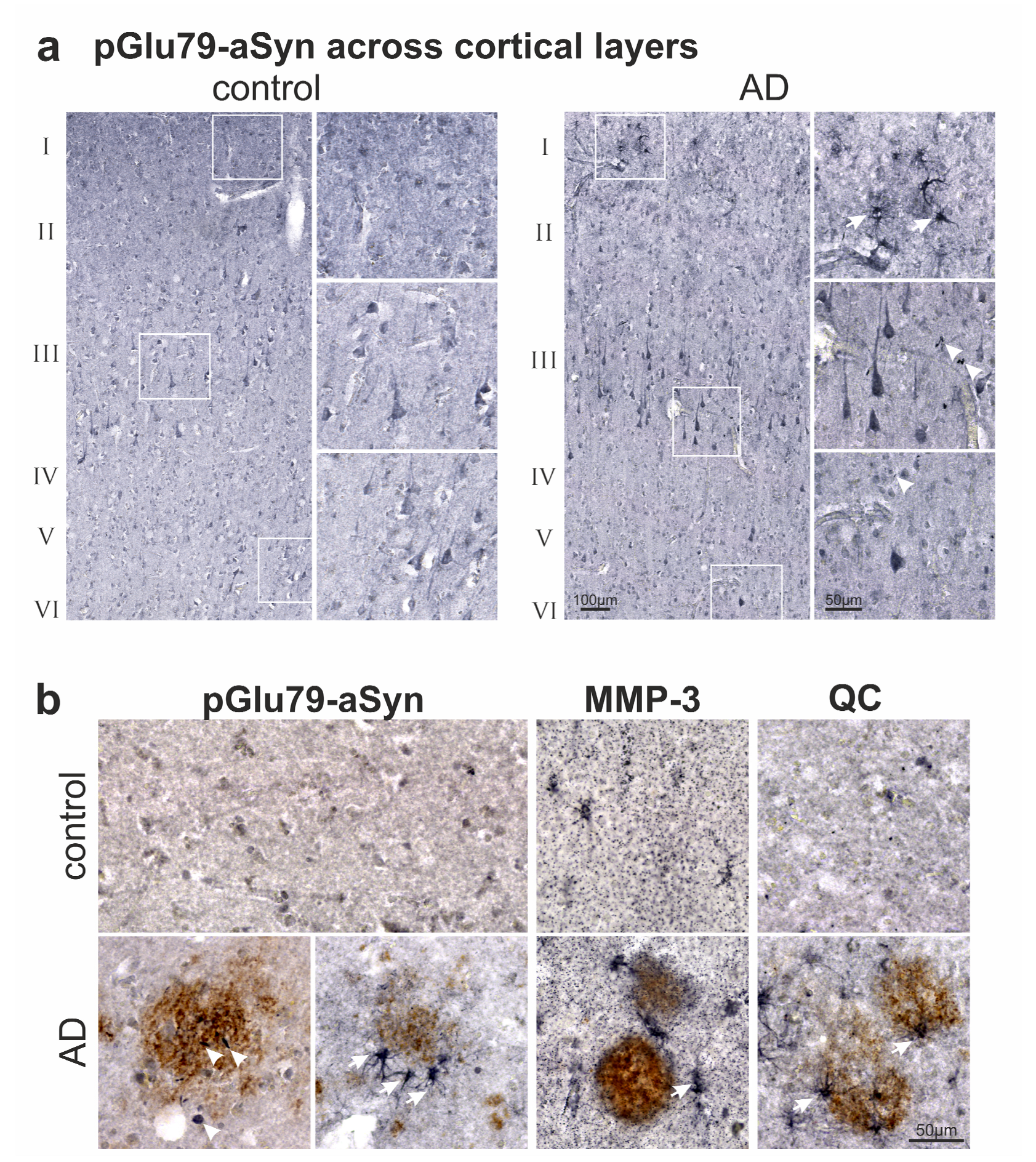
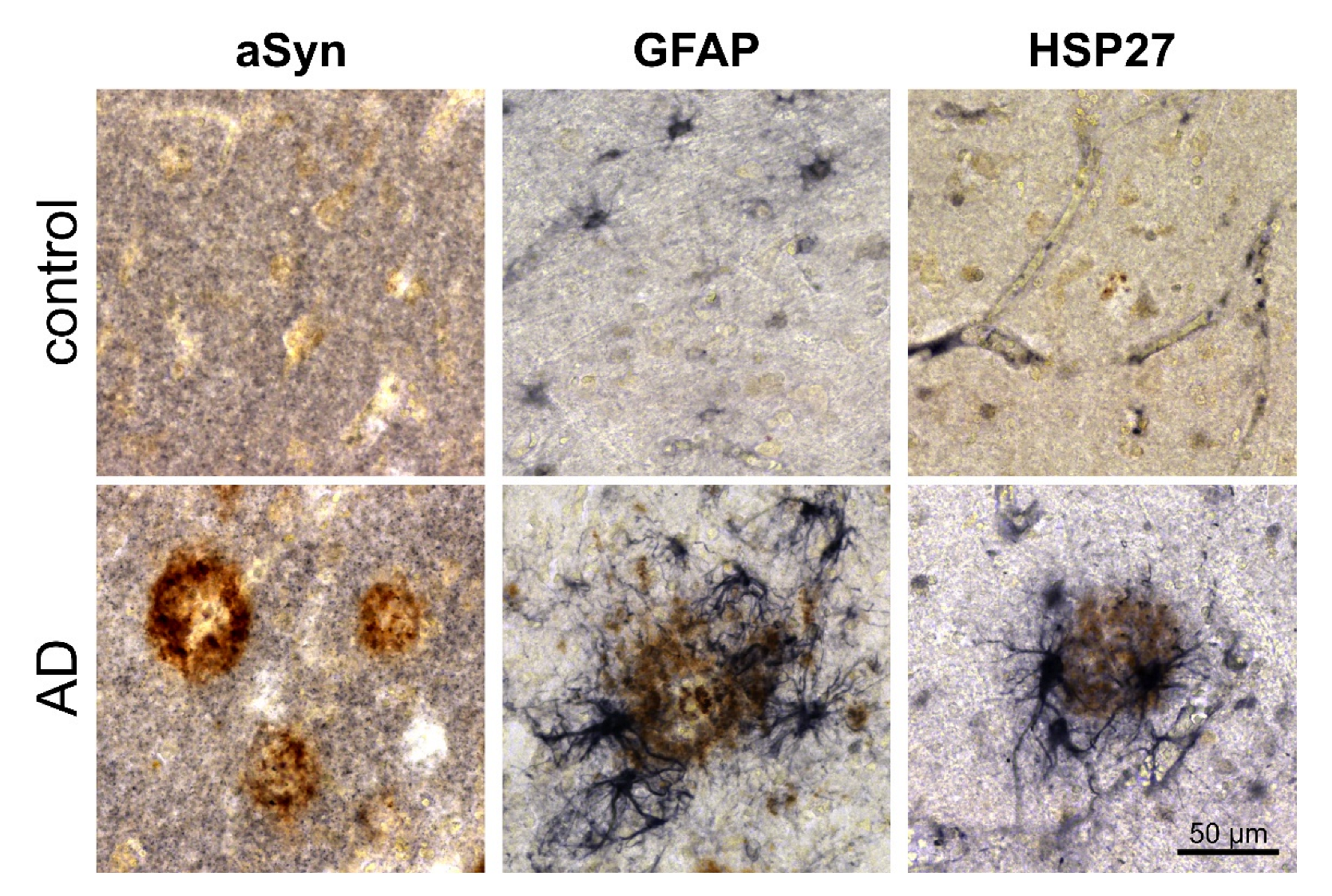
3.2. Association of pGlu79-aSyn with Aβ Plaques in AD Brain
4. Discussion
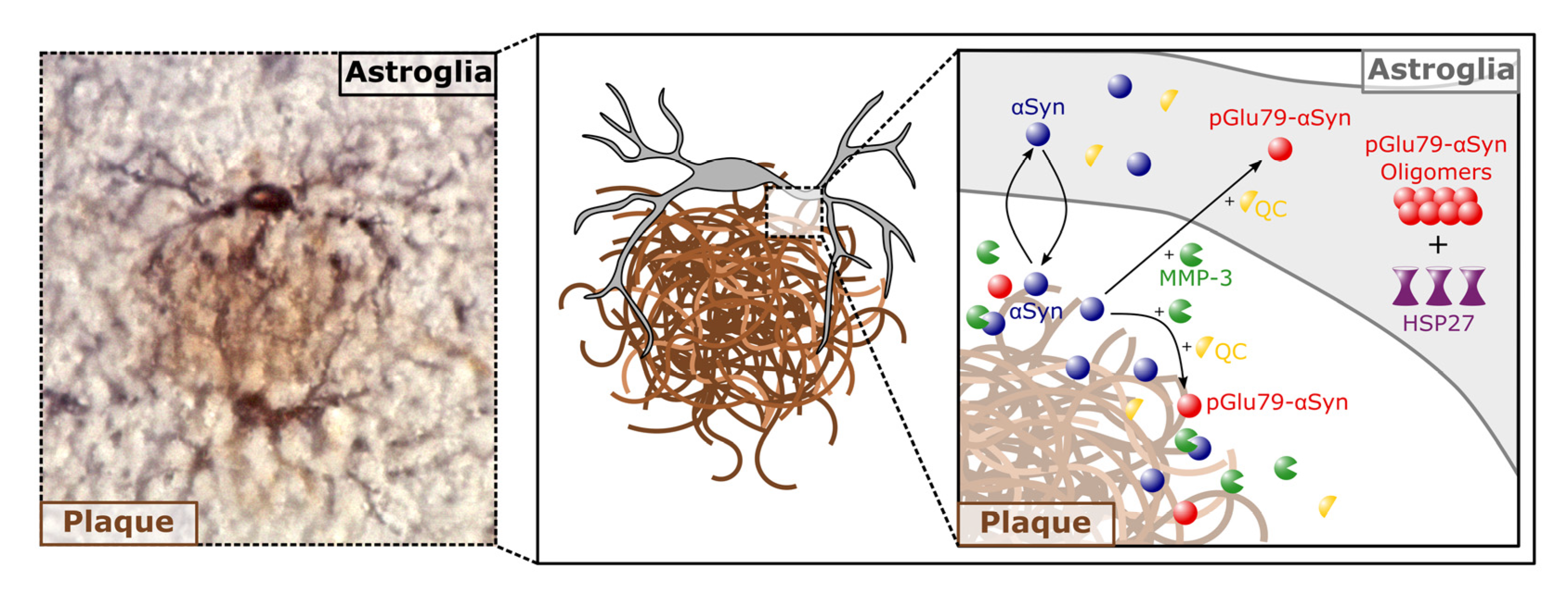
4.1. aSyn and pGlu79-aSyn Aggregates in Proximity to Aβ Plaques
4.2. pGlu79-aSyn, QC and HSP27 in Reactive Astrocytes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barthelemy, N.R.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; Morris, J.C.; et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid ß deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer’s disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef]
- Coyle, J.T.; Price, D.L.; DeLong, M.R. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science 1983, 219, 1184–1190. [Google Scholar] [CrossRef]
- Bondareff, W.; Mountjoy, C.Q.; Rossor, R.M.; Iversen, L.L.; Reynolds, G.P.; Hauser, D.L. Neuronal degeneration in the locus coeruleus and cortical correlates of Alzheimer’s disease. Alzheimer Dis. Assoc. Discord. 1987, 1, 256–262. [Google Scholar] [CrossRef]
- Busch, C.; Bohl, J.; Ohm, T.G. Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol. Aging 1997, 18, 401–406. [Google Scholar] [CrossRef]
- Weinshenker, D. Functional consequences of locus coeruleus degeneration in Alzheimer’s disease. Curr. Alzheimer. Res. 2008, 5, 342–345. [Google Scholar] [CrossRef]
- Rüb, U.; Del Tredici, K.; Schultz, C.; Büttner-Ennever, J.A.; Braak, H. The premotor region essential for rapid vertical eye movements shows early involvement in Alzheimer’s disease-related cytoskeletal pathology. Vis. Res 2001, 41, 2149–2156. [Google Scholar] [CrossRef]
- Scinto, L.F.; Wu, C.K.; Firla, K.M.; Daffner, K.R.; Saroff, D.; Geula, C. Focal pathology in Edinger-Westphal nucleus explains pupillary hypersensitivity in Alzheimer’s disease. Acta Neuropathol. 1999, 97, 557–564. [Google Scholar] [CrossRef]
- Scinto, L.F.; Frosch, M.; Wu, C.K.; Daffner, K.R.; Gedi, N.; Geula, C. Selective cell loss in Edinger-Westphal in asymptomatic elders and Alzheimer’s patients. Neurobiol. Aging 2001, 22, 729–736. [Google Scholar] [CrossRef]
- Hirsch, E.C. Iron transport in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S209–S211. [Google Scholar] [CrossRef]
- Kostka, M.; Högen, T.; Danzer, K.M.; Levin, J.; Habeck, M.; Wirth, A.; Wagner, R.; Glabe, C.G.; Finger, S.; Heinzelmann, U.; et al. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J. Biol. Chem. 2008, 283, 10992–11003. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Schlachetzki, J.C.M.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Vicente Miranda, H.; Szego, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- González, N.; Arcos-López, T.; König, A.; Quintanar, L.; Menacho Márquez, M.; Outeiro, T.F.; Fernández, C.O. Effects of alpha-synuclein post-translational modifications on metal binding. J. Neurochem. 2019, 150, 507–521. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Cortico-basal ganglia-cortical circuitry in Parkinson’s disease reconsidered. Exp. Neurol. 2008, 212, 226–229. [Google Scholar] [CrossRef]
- Galvan, A.; Wichmann, T. Pathophysiology of Parkinsonism. Clin. Neurophysiol. 2008, 119, 1459–1474. [Google Scholar] [CrossRef]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nature Med. 2008, 14, 1106–1111. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Bluhm, A.; Moceri, S.; Machner, L.; Köppen, J.; Schenk, M.; Hilbrich, I.; Holzer, M.; Weidenfeller, M.; Richter, F.; et al. A glutaminyl cyclase-catalyzed alpha-synuclein modification identified in human synucleinopathies. Acta Neuropathol. 2021, 142, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Birkedal-Hansen, H.; Moore, W.G.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix metalloproteinases: A review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar] [CrossRef] [PubMed]
- Madzharova, E.; Kastl, P.; Sabino, F.; auf dem Keller, U. Post-translational modification-dependent activity of matrix metalloproteinases. Int. J. Molec. Sci. 2019, 20, 3077. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Hwang, O. Role of matrix metalloproteinase-3 in neurodegeneration. J. Neurochem. 2011, 116, 22–32. [Google Scholar] [CrossRef]
- Sung, J.Y.; Park, S.M.; Lee, C.H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.T.; Paik, S.R.; Chung, K.C. Proteolytic cleavage of extracellular secreted alpha-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef]
- Levin, J.; Giese, A.; Boetzel, K.; Israel, L.; Högen, T.; Nübling, G.; Kretzschmar, H.; Lorenzl, S. Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp. Neurol. 2009, 215, 201–208. [Google Scholar] [CrossRef]
- Bluhm, A.; Schrempel, S.; von Hörsten, S.; Schulze, A.; Roßner, S. Proteolytic α-synuclein cleavage in health and disease. Int. J. Mol. Sci. 2021, 22, 5450. [Google Scholar] [CrossRef]
- Böckers, T.M.; Kreutz, M.R.; Pohl, T. Glutaminyl-cyclase expression in the bovine/porcine hypothalamus and pituitary. J. Neuroendocrinol. 1995, 7, 445–453. [Google Scholar] [CrossRef]
- Busby, W.H.; Quackenbush, G.E.; Humm, J.; Youngblood, W.W.; Kizer, J.S. An enzyme(s) that converts glutaminyl-peptides into pyroglutamyl-peptides. J. Biol. Chem. 1987, 262, 8532–8536. [Google Scholar] [CrossRef]
- Fischer, W.H.; Spiess, J. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3628–3632. [Google Scholar] [CrossRef]
- Pohl, T.; Zimmer, M.; Mugele, K.; Spiess, J. Primary structure and functional expression of a glutaminyl cyclase. Proc. Natl. Acad. Sci. USA 1991, 88, 10059–10063. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Staffa, K.; Waniek, A.; Wermann, M.; Hoffmann, T.; Cynis, H.; Schilling, S.; Demuth, H.U.; Roßner, S. Developmental expression and subcellular localization of glutaminyl cyclase in mouse brain. Int. J. Devl. Neurosci. 2009, 27, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Hartlage-Rübsamen, M.; Morawski, M.; Waniek, A.; Jäger, C.; Zeitschel, U.; Koch, B.; Cynis, H.; Schilling, S.; Schliebs, R.; Demuth, H.U.; et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011, 121, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Morawski, M.; Schilling, S.; Kreuzberger, M.; Waniek, A.; Jäger, C.; Koch, B.; Cynis, H.; Kehlen, A.; Arendt, T.; Hartlage-Rübsamen, M.; et al. Glutaminyl cyclase in human cortex: Correlation with (pGlu)-amyloid-β load and cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Morawski, M.; Hartlage-Rübsamen, M.; Jäger, C.; Waniek, A.; Schilling, S.; Schwab, C.; McGeer, P.; Arendt, T.; Demuth, H.U.; Roßner, S. Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Aβ pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 120, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef]
- Debatin, L.; Streffer, J.; Geissen, M.; Matschke, J.; Aguzzi, A.; Glatzel, M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener Dis. 2008, 5, 347–354. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B.; Ribera, R.; Rey, M.J.; Ribalta, T. Prion protein expression in senile plaques in Alzheimer’s disease. Acta Neuropathol. 2001, 101, 49–56. [Google Scholar] [CrossRef]
- Hainfeller, J.A.; Wanschitz, J.; Jellinger, K.; Liberski, P.P.; Gullotta, F.; Budka, H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998, 96, 116–122. [Google Scholar] [CrossRef]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of beta-amyloid by intracerebral infusion of Alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Schlüter, O.M.; Südhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to APOE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [CrossRef]
- Lashley, T.; Holton, J.L.; Gray, E.; Kirkham, K.; O’Sullivan, S.S.; Hilbig, A.; Wood, N.W.; Lees, A.J.; Revesz, T. Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol. 2008, 115, 417–425. [Google Scholar] [CrossRef]
- Köppen, J.; Schulze, A.; Machner, L.; Wermann, M.; Eichentopf, R.; Guthardt, M.; Hähnel, A.; Klehm, J.; Kriegeskorte, M.; Hartlage-Rübsamen, M.; et al. Amyloid-beta peptides trigger aggregation of alpha-synuclein by in vitro. Molecules 2020, 25, 580. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic interactions between Aß, Tau, and α-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef]
- Masliah, E.; Iwai, A.; Mallory, M.; Uéda, K.; Saitoh, T. Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer’s disease. Am. J. Pathol. 1996, 148, 201–210. [Google Scholar]
- Wirths, O.; Weickert, S.; Majtenyi, K.; Havas, L.; Kahle, P.J.; Okochi, M.; Haass, C.; Multhaup, G.; Beyreuther, K.; Bayer, T.A. Lewy body variant of Alzheimer’s disease: Alpha-synuclein in dystrophic neurites of A beta plaques. Neuroreport 2000, 11, 3737–3741. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβ. pathology. Neuron 2020, 105, 260–275.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Uéda, K.; Chen, P.; Ashe, K.H.; Cole, G.M. Plaque-associated alpha-synuclein (NACP) pathology in aged transgenic mice expressing amyloid precursor protein. Brain Res. 2000, 853, 381–383. [Google Scholar] [CrossRef]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune signaling in neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk-Wegrzynowicz, M.; Gerber, Y.N.; Ries, M.; Sastre, M.; Tolkovsky, A.M.; Spillantini, M.G. Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol. Commun. 2017, 5, 89. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Fellner, L.; Jellinger, K.A.; Wenning, G.K.; Stefanova, N. Glial dysfunction in the pathogenesis of α-synucleinopathies: Emerging concepts. Acta Neuropathol. 2011, 121, 675–693. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Zeitschel, U.; Apelt, J.; Gärtner, U.; Franke, H.; Stahl, T.; Günther, A.; Schliebs, R.; Penkowa, M.; Bigl, V.; et al. Astrocytic expression of the Alzheimer’s disease beta-secretase (BACE1) is stimulus-dependent. Glia 2003, 41, 169–179. [Google Scholar] [CrossRef]
- Kim, H.J.; Hwang, N.R.; Lee, K.J. Heat shock responses for understanding diseases of protein denaturation. Mol. Cells 2007, 23, 123–131. [Google Scholar]
- Liberek, K.; Lewandowska, A.; Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 2008, 27, 328–335. [Google Scholar] [CrossRef]
- Navarro-Zaragoza, J.; Cuenca-Bermejo, L.; Almela, P.; Laorden, M.L.; Herrero, M.T. Could small heat shock protein HSP27 be a first-line target for preventing protein aggregation in Parkinson’s disease? Int. J. Mol. Sci. 2021, 22, 3038. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.E.; Szegedi, V.; Varga, E.; Juhász, G.; Horváth, J.; Borbély, E.; Csibrány, B.; Alföldi, R.; Lénárt, N.; Penke, B.; et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones 2013, 18, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Carson, K.; Rice-Ficht, A.; Good, T. Small heat shock proteins differentially affect Abeta aggregation and toxicity. Biochem. Biophys. Res. Commun. 2006, 347, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Carver, J.A.; Ecroyd, H. Preventing α-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta 2014, 1842, 1830–1843. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Bluhm, A.; Piechotta, A.; Linnert, M.; Rahfeld, J.U.; Demuth, H.U.; Lues, I.; Kuhn, P.H.; Lichtenthaler, S.F.; Roßner, S.; et al. Immunohistochemical evidence from APP-transgenic mice for glutaminyl cyclase as drug target to diminish pE-Abeta formation. Molecules 2018, 23, 924. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- The National Institute on Aging; Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol. Aging 1997, 18, S1–S2. [Google Scholar] [CrossRef]
- Walker, L.; McAleese, K.E.; Thomas, A.J.; Johnson, M.; Martin-Ruiz, C.; Parker, C.; Colloby, S.J.; Jellinger, K.; Attems, J. Neuropathologically mixed Alzheimer’s and Lewy body disease: Burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015, 129, 729–748. [Google Scholar] [CrossRef]
- Candreva, J.; Chau, E.; Rice, M.E.; Kim, J.R. Interactions between soluble species of beta-amyloid and alpha-synuclein promote oligomerization while inhibiting fibrillization. Biochemistry 2020, 59, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, N.; Hanakawa, T. It is time to study overlapping molecular and circuit pathophysiologies in Alzheimer’s and Lewy body disease spectra. Front. Syst. Neurosci. 2021, 15, 777706. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Pettegrew, J.W.; Masliah, E.; Hamilton, R.L.; Mandal, R. Interaction between Abeta peptide and α-synuclein: Molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy bodies. Neurochem. Res. 2006, 31, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Bachhuber, T.; Katzmarski, N.; McCarter, J.F.; Loreth, D.; Tahirovic, S.; Kamp, F.; Abou-Ajram, C.; Nuscher, B.; Serrano-Pozo, A.; Müller, A.; et al. Inhibition of amyloid-β plaque formation by α-synuclein. Nat. Med. 2015, 21, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef]
- Bayer, T.A.; Jäkälä, P.; Hartmann, T.; Havas, L.; McLean, C.; Culvenor, J.G.; Li, Q.X.; Masters, C.L.; Falkai, P.; Beyreuther, K. Alpha-synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies but not in Alzheimer’s disease beta-amyloid plaque cores. Neurosci. Lett. 1999, 266, 213–216. [Google Scholar] [CrossRef]
- Culvenor, J.G.; McLean, C.A.; Cutt, S.; Campbell, B.C.; Maher, F.; Jäkälä, P.; Hartmann, T.; Beyreuther, K.; Masters, C.L.; Li, Q.X. Non-Abeta component of Alzheimer’s disease amyloid (NAC) revisited. NAC and alpha-synuclein are not associated with Abeta amyloid. Am. J. Pathol. 1999, 155, 1173–1181. [Google Scholar] [CrossRef]
- Wirths, O.; Bayer, T.A. α-synuclein, Aβ and Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 103–108. [Google Scholar] [CrossRef]
- Smit, T.; Deshayes, N.A.C.; Borchelt, D.R.; Kamphuis, W.; Middeldorp, J.; Hol, E.M. Reactive astrocytes as treatment targets in Alzheimer’s disease-Systematic review of studies using the APPswePS1dE9 mouse model. Glia 2021, 69, 1852–1881. [Google Scholar] [CrossRef]
- Wang, P.; Ye, Y. Astrocytes in neurodegenerative diseases: A perspective from tauopathy and a-synucleinopathy. Life 2021, 11, 938. [Google Scholar] [CrossRef]
- Lan, G.; Wang, P.; Chan, R.B.; Liu, Z.; Yu, Z.; Liu, X.; Yang, Y.; Zhang, J. Astrocytic VEGFA: An essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia 2022, 70, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef]
- Chavarría, C.; Rodríguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Molec. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef]
- Sheng, L.; Stewart, T.; Yang, D.; Thorland, E.; Soltys, D.; Aro, P.; Khrisat, T.; Xie, Z.; Li, N.; Liu, Z.; et al. Erythrocytic α-synuclein contained in microvesicles regulates astrocytic glutamate homeostasis: A new perspective on Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 102. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, Y.; Lee, J.; Jarnik, M.; Wu, X.; Bonifacino, J.S.; Shen, J.; Ye, Y. A myosin-7B-dependent endocytosis pathway mediates cellular entry of alpha-synuclein fibrils and polycation-bearing cargos. Proc. Natl. Acad. Sci. USA 2020, 117, 10865–10875. [Google Scholar] [CrossRef]
- Rostami, J.; Holmqvist, S.; Lindstrom, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergstrom, J.; Roybon, L.; Erlandsson, A. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. Alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Waniek, A.; Hartlage-Rübsamen, M.; Höfling, C.; Kehlen, A.; Schilling, S.; Demuth, H.U.; Roßner, S. Identification of thyrotropin-releasing hormone as hippocampal glutaminyl cyclase substrate in neurons and reactive astrocytes. Biochim. Biophys. Acta 2015, 1852, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Boros, S.; Kamps, B.; Wunderink, L.; de Bruijn, W.; de Jong, W.W.; Boelens, W.C. Transglutaminase catalyzes differential crosslinking of small heat shock proteins and amyloid-beta. FEBS Lett. 2004, 576, 57–62. [Google Scholar] [CrossRef][Green Version]
- Chiu, Y.J.; Hsieh, Y.H.; Lin, T.H.; Lee, G.C.; Hsieh-Li, H.M.; Sun, Y.C.; Chen, C.M.; Chang, K.H.; Lee-Chen, G.J. Novel compound VB-037 inhibits Abeta aggregation and promotes neurite outgrowth through enhancement of HSP27 and reduction of P38 and JNK-mediated inflammation in cell models for Alzheimer’s disease. Neurochem. Int. 2019, 125, 175–186. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Otte-Höller, I.; Wesseling, P.; de Waal, R.M.; Boelens, W.C.; Verbeek, M.M. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2006, 32, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Nafar, F.; Williams, J.B.; Mearow, K.M. Astrocytes release HspB1 in response to amyloid-β exposure in vitro. J. Alzheimers Dis. 2016, 49, 251–263. [Google Scholar] [CrossRef]
- Ojha, J.; Masilamoni, G.; Dunlap, D.; Udoff, R.A.; Cashikar, A.G. Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol. Cell. Biol. 2011, 31, 3146–3157. [Google Scholar] [CrossRef]
- Sharma, S.K.; Priya, S. Expanding role of molecular chaperones in regulating α-synuclein misfolding; implications in Parkinson’s disease. Cell. Mol. Life Sci. 2017, 74, 617–629. [Google Scholar] [CrossRef]
- Bruinsma, I.B.; Bruggink, K.A.; Kinast, K.; Versleijen, A.A.; Segers-Nolten, I.M.; Subramaniam, V.; Kuiperij, H.B.; Boelens, W.; de Waal, R.M.; Verbeek, M.M. Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 2011, 79, 2956–2967. [Google Scholar] [CrossRef]
- Zourlidou, A.; Payne Smith, M.D.; Latchman, D.S. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J. Neurochem. 2004, 88, 1439–1448. [Google Scholar] [CrossRef]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Klucken, J.; Strathearn, K.E.; Liu, F.; Nguyen, P.; Rochet, J.C.; Hyman, B.T.; McLean, P.J. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem. Biophys. Res. Commun. 2006, 351, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Vicente Miranda, H.; Chegão, A.; Oliveira, M.S.; Fernandes Gomes, B.; Enguita, F.J.; Outeiro, T.F. Hsp27 reduces glycation-induced toxicity and aggregation of alpha-synuclein. FASEB J. 2020, 34, 6718–6728. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody (Clone/cat. #) | Company | Host | Species Reactivity | Dilution | |
|---|---|---|---|---|---|
| Mouse | Human | ||||
| Aβ (4G8/800710) | Biolegend | Mouse | Mouse, human | 1:8000 | 1:16,000 |
| GFAP (-/173004) | Synaptic Systems | Guinea pig | Mouse, rat, human | 1:200 | 1:6000 |
| aSyn; Syn1 (42/610787) | BD Transd. Labs | Mouse | Mouse, rat, human | 1:8000 | - |
| aSyn, K129 (-/0103001901) | Roboscreen | Rabbit | Mouse, human | 1:200 | - |
| aSyn (15G7/ ALX-804-258) | Enzo Life Sciences | Rat | Human | - | 1:1000 |
| MMP-3 (-/sc6839) | Santa Cruz | Goat | Mouse, rat, human | 1:50 | - |
| MMP-3 (EP1186Y/ab52915) | Abcam | Rabbit | Mouse, rat, human | - | 1:3,000 |
| QC (-/77101) | Fraunhofer IZI (Halle, Germany) | Goat | Mouse, human | 1:200 | 1:500 |
| pGlu79-aSyn | Fraunhofer IZI | Rabbit | Mouse, human | 1:100 | 1:500 |
| HSP27 (-/sc1049) | Santa Cruz | Goat | Mouse, human | 1:200 | 1:2000 |
| Primary Antibody | Host | Dilution | Secondary Antibody |
|---|---|---|---|
| MMP-3 | Goat | 1:100 | Donkey anti-goat Cy2 |
| pGlu79-aSyn | Rabbit | 1:100 | Donkey anti-rabbit Cy3 |
| aSyn (Syn1) | Mouse | 1:4000 | Donkey anti-mouse Cy5 |
| QC | Goat | 1:100 | Donkey anti-goat Cy2 |
| pGlu79-aSyn | Rabbit | 1:100 | Donkey anti-rabbit Cy3 |
| aSyn (Syn1) | Mouse | 1:4000 | Donkey anti-mouse Cy5 |
| HSP27 | Goat | 1:100 | Donkey anti-goat Cy2 |
| pGlu79-aSyn | Rabbit | 1:100 | Donkey anti-rabbit Cy3 |
| GFAP | Guinea pig | 1:200 | Donkey anti-guinea pig Cy5 |
| HSP27 | Goat | 1:100 | Donkey anti-goat Cy2 |
| pGlu79-aSyn | Rabbit | 1:100 | Donkey anti-rabbit Cy3 |
| Aβ (4G8) | Mouse | 1:4000 | Donkey anti-mouse Cy5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bluhm, A.; Schrempel, S.; Schilling, S.; von Hörsten, S.; Schulze, A.; Roßner, S.; Hartlage-Rübsamen, M. Immunohistochemical Demonstration of the pGlu79 α-Synuclein Fragment in Alzheimer’s Disease and Its Tg2576 Mouse Model. Biomolecules 2022, 12, 1006. https://doi.org/10.3390/biom12071006
Bluhm A, Schrempel S, Schilling S, von Hörsten S, Schulze A, Roßner S, Hartlage-Rübsamen M. Immunohistochemical Demonstration of the pGlu79 α-Synuclein Fragment in Alzheimer’s Disease and Its Tg2576 Mouse Model. Biomolecules. 2022; 12(7):1006. https://doi.org/10.3390/biom12071006
Chicago/Turabian StyleBluhm, Alexandra, Sarah Schrempel, Stephan Schilling, Stephan von Hörsten, Anja Schulze, Steffen Roßner, and Maike Hartlage-Rübsamen. 2022. "Immunohistochemical Demonstration of the pGlu79 α-Synuclein Fragment in Alzheimer’s Disease and Its Tg2576 Mouse Model" Biomolecules 12, no. 7: 1006. https://doi.org/10.3390/biom12071006
APA StyleBluhm, A., Schrempel, S., Schilling, S., von Hörsten, S., Schulze, A., Roßner, S., & Hartlage-Rübsamen, M. (2022). Immunohistochemical Demonstration of the pGlu79 α-Synuclein Fragment in Alzheimer’s Disease and Its Tg2576 Mouse Model. Biomolecules, 12(7), 1006. https://doi.org/10.3390/biom12071006