
Calpain-Independent Intracellular Protease Activity Is Elevated in Excitotoxic Cortical Neurons Prior to Delayed Calcium Deregulation and Mitochondrial Dysfunction



Abstract
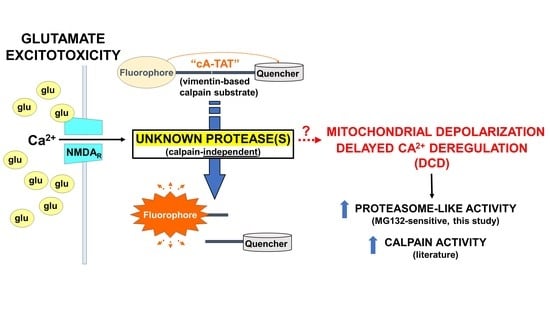
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation and Culture of Primary Neurons
2.3. Culture of Mouse Embryonic Fibroblasts (MEFs)
2.4. Live-Cell Imaging
2.5. Gel Electrophoresis and Endogenous Protein or Biotin Detection
2.6. Statistical Analysis
3. Results and Discussion
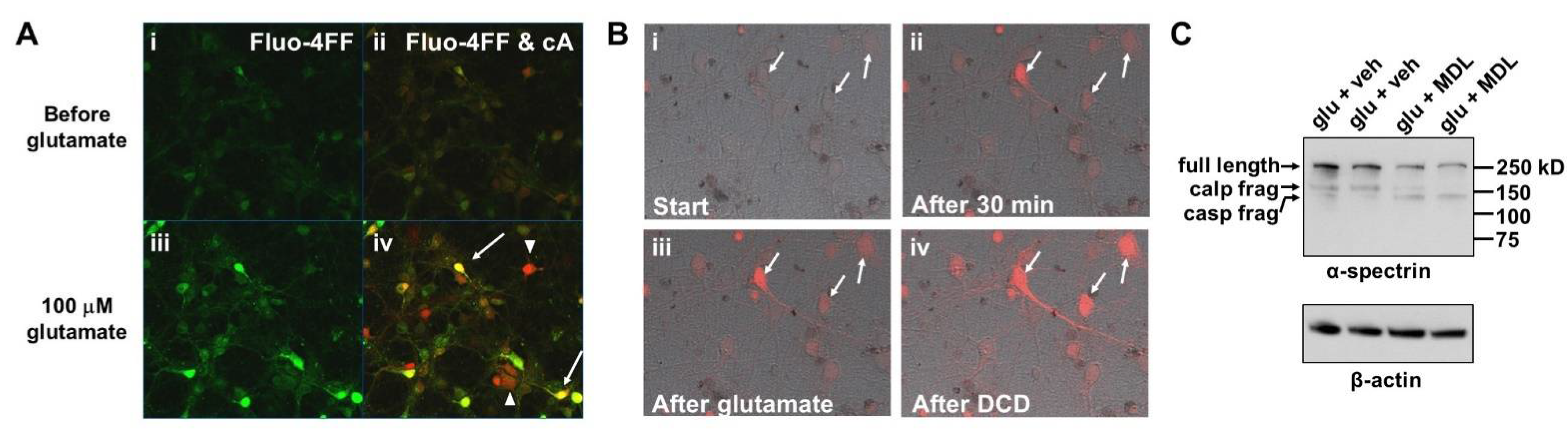
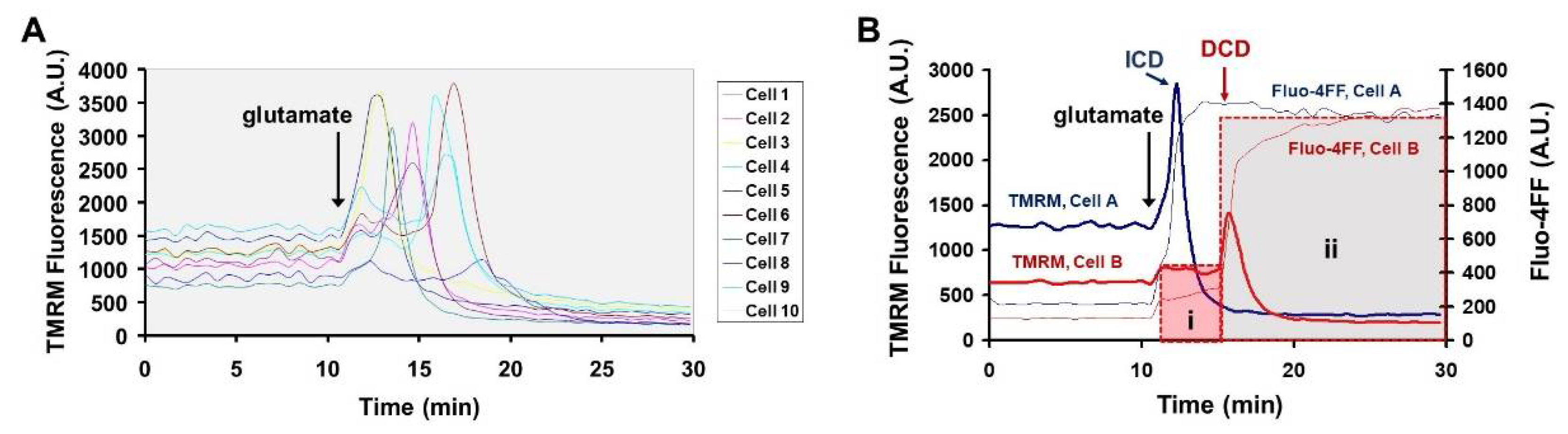
3.1. Glutamate Stimulates the Rate of cA Fluoresescent Product Accumulation Prior to DCD in Calpain Inhibitor-Insensitive Fashion
3.2. Sustained Mitochondrial Depolarization Occurs Simultaneously with Ca2+ Deregulation
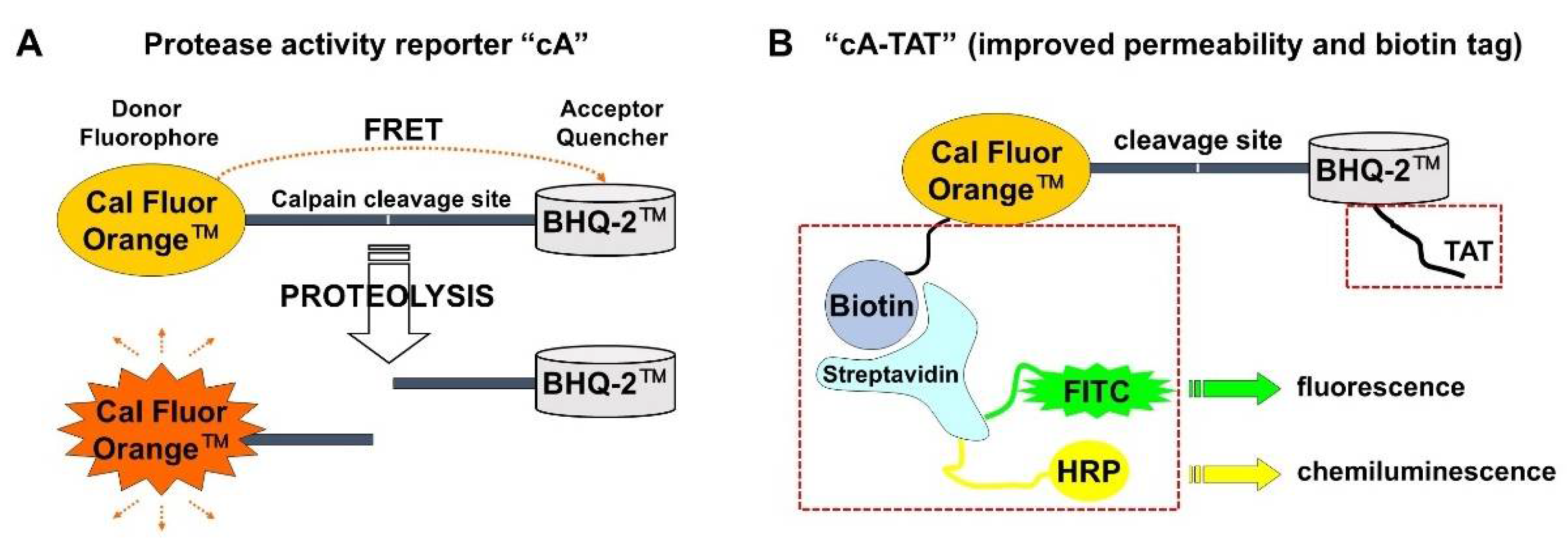
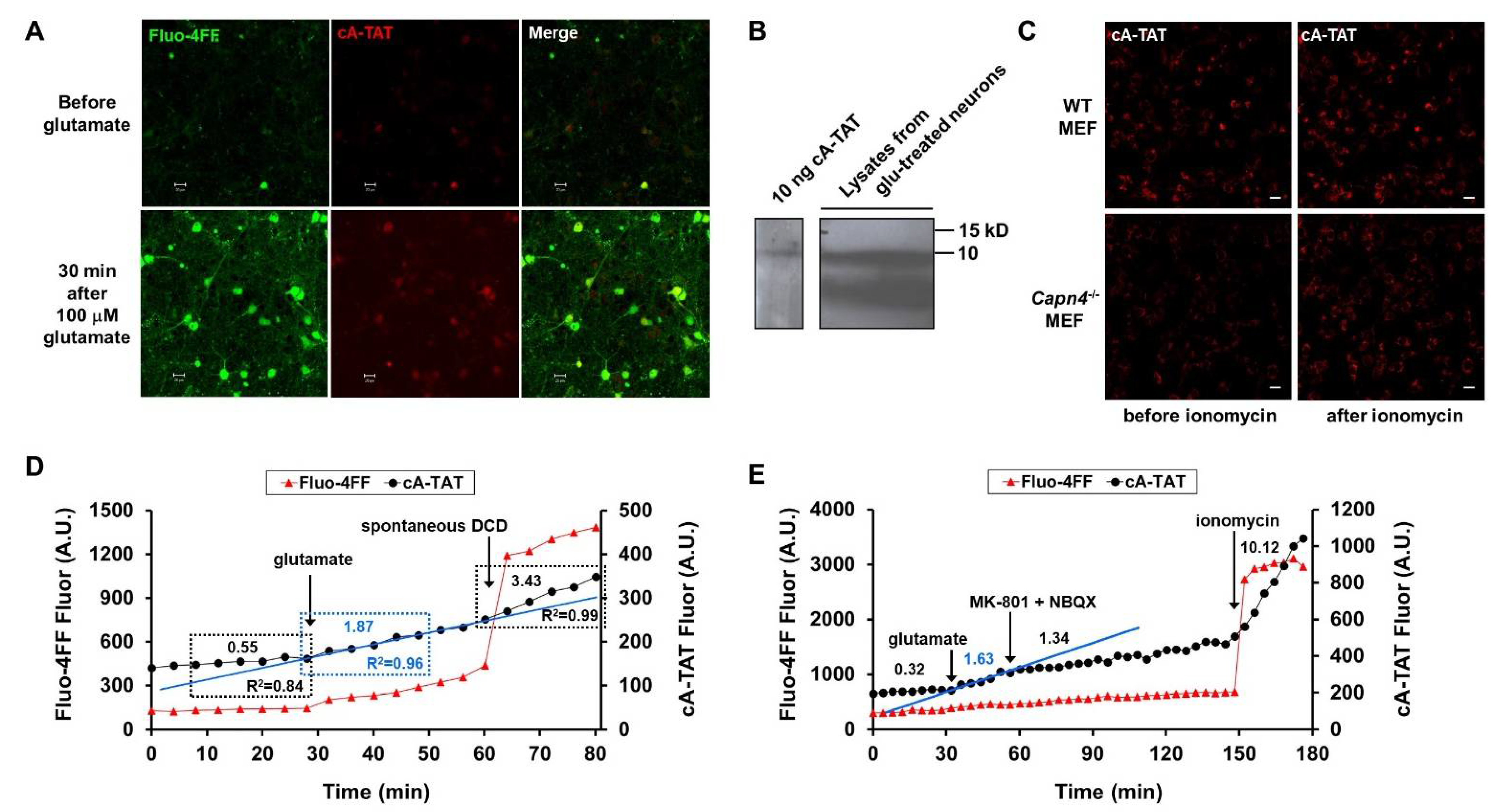
3.3. Description of the Second-Generation Fluorogenic Protease Reporter cA-TAT
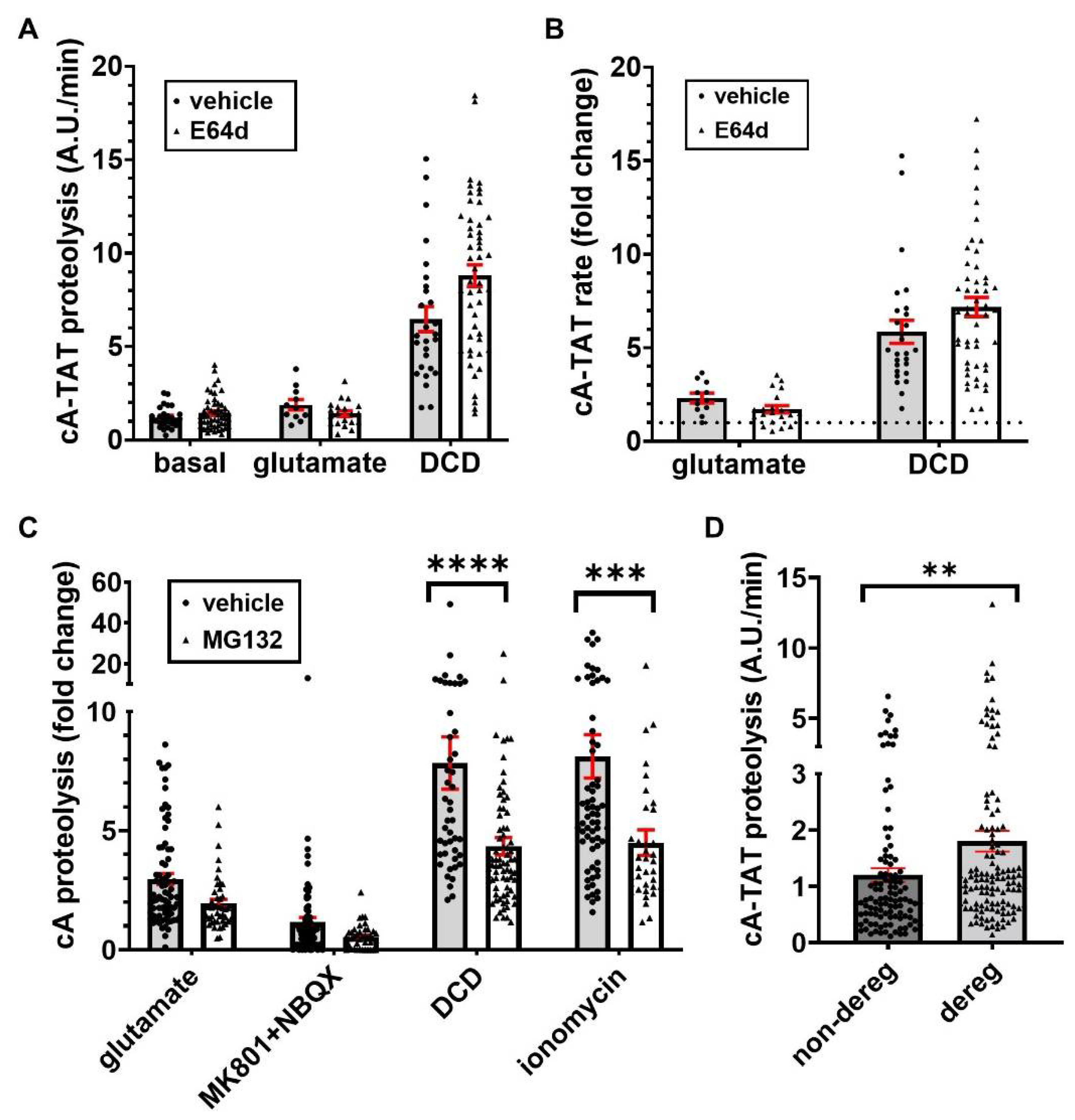
3.4. Evidence That Glutamate-Induced cA-TAT Fluorescence Accumulation Is Due to Calpain-Independent Cleavage
3.5. Glutamate-Stimulated cA-TAT Proteolysis Is Acutely Inhibited by Glutamate Receptor Antagonists
3.6. DCD- and Ionomycin-Stimulated cA-TAT Proteolysis, but Not Glutamate-Stimulated Proteolysis, Are Partly Attenuated by Proteasome Inhibition
3.7. The Neuronal Subpopulation That Undergoes DCD Exhibits a Significantly Higher Baseline cA-TAT Proteolysis Rate
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPA | A-amino-3-hydroxy-5-methylisoxazole-4-propionate |
| ANOVA | Analysis of variance |
| A.U. | Arbitrary units |
| Ca2+ | Calcium |
| DCD | Delayed calcium deregulation |
| DMSO | Dimethyl sulfoxide |
| FRET | Fluorescence resonance energy transfer |
| ICD | Immediate calcium deregulation |
| MEF | Mouse Embryonic Fibroblast |
| NBQX | 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione |
| NMDA | N-methyl-D-aspartate |
| PVDF | Polyvinylidene difluoride |
| REML | Restricted Maximum Likelihood |
| SDS–PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEM | Standard error of the mean |
| TBS–T | Tris-buffered saline containing 0.05% Tween-20 |
| TMRM+ | Tetramethylrhodamine methyl ester |
Appendix A
- The noise-to-signal ratio precluded either a linear fit of the baseline rate used for normalization or an ionomycin response. Most excluded cells fell into this category. Across all treatment conditions, 118 of 227 cells failing to show Ca2+ deregulation and 42 of 244 DCD cells were Criterion 1 exclusions. Possible explanations for high noise-to-signal ratio include poor cA-TAT loading, consistent with low ionomycin response (Figure A1A,B), and subtle shifts of the cells out of the focal plane over the time course of the experiment.
- The cell underwent lysis and lost the fluorescent indicator before any post-baseline rate could be calculated. Only one cell fell into this category. A few additional cells underwent lysis after at least a glutamate rate could be obtained. These cells were included in the analysis.
- The cell spontaneously underwent Ca2+ deregulation prior to glutamate addition. This was a rare event that was seen only in the presence of MG132. Four cells in total fell into this category.
- The cell recovered calcium homeostasis following DCD. This was a rare event that was seen only in the presence of MG132. Three cells in total fell into this category.

References
- Riedel, G.; Platt, B.; Micheau, J. Glutamate Receptor Function in Learning and Memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Tymianski, M.; Charlton, M.P.; Carlen, P.L.; Tator, C.H. Source Specificity of Early Calcium Neurotoxicity in Cultured Embryonic Spinal Neurons. J. Neurosci. 1993, 13, 2085–2104. [Google Scholar] [CrossRef]
- Choi, D.W. Ionic Dependence of Glutamate Neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G. Mitochondrial Dysfunction and Glutamate Excitotoxicity Studied in Primary Neuronal Cultures. Curr. Mol. Med. 2004, 4, 149–177. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics 4, 4th ed.; Academic Press: London, UK, 2013. [Google Scholar]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A Forty-Kilodalton Protein of the Inner Membrane is the Mitochondrial Calcium Uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Gerencser, A.A.; Mark, K.A.; Hubbard, A.E.; Divakaruni, A.S.; Mehrabian, Z.; Nicholls, D.G.; Polster, B.M. Real-Time Visualization of Cytoplasmic Calpain Activation and Calcium Deregulation in Acute Glutamate Excitotoxicity. J. Neurochem. 2009, 110, 990–1004. [Google Scholar] [CrossRef] [Green Version]
- D’Orsi, B.; Bonner, H.; Tuffy, L.P.; Dussmann, H.; Woods, I.; Courtney, M.J.; Ward, M.W.; Prehn, J.H. Calpains are Downstream Effectors of Bax-Dependent Excitotoxic Apoptosis. J. Neurosci. 2012, 32, 1847–1858. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Cheung, E.C.; Melanson-Drapeau, L.; Cregan, S.P.; Vanderluit, J.L.; Ferguson, K.L.; McIntosh, W.C.; Park, D.S.; Bennett, S.A.; Slack, R.S. Apoptosis-Inducing Factor is a Key Factor in Neuronal Cell Death Propagated by BAX-Dependent and BAX-Independent Mechanisms. J. Neurosci. 2005, 25, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polster, B.M.; Basanez, G.; Etxebarria, A.; Hardwick, J.M.; Nicholls, D.G. Calpain I Induces Cleavage and Release of Apoptosis-Inducing Factor from Isolated Mitochondria. J. Biol. Chem. 2005, 280, 6447–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.; Xing, J.; Xiao, X.; Liou, A.K.; Gao, Y.; Yin, X.M.; Clark, R.S.; Graham, S.H.; Chen, J. Critical Role of Calpain I in Mitochondrial Release of Apoptosis-Inducing Factor in Ischemic Neuronal Injury. J. Neurosci. 2007, 27, 9278–9293. [Google Scholar] [CrossRef]
- Yuste, V.J.; Moubarak, R.S.; Delettre, C.; Bras, M.; Sancho, P.; Robert, N.; d’Alayer, J.; Susin, S.A. Cysteine Protease Inhibition Prevents Mitochondrial Apoptosis-Inducing Factor (AIF) Release. Cell Death Differ. 2005, 12, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M.; Arze, R.; Lyttle, M.H.; Nicholls, D.G.; Hudson, D. Solid Phase Synthesis of Dual Labeled Peptides: Development of Cell Permeable Calpain Specific Substrates. Int. J. Pept. Res. Ther. 2007, 13, 83–91. [Google Scholar] [CrossRef]
- Grote, J.; Laue, O.; Eiring, P.; Wehler, M. Evaluation of Brain Tissue O2 Supply Based on Results of PO2 Measurements with Needle and Surface Microelectrodes. J. Auton. Nerv. Syst. 1996, 57, 168–172. [Google Scholar] [CrossRef]
- Dourdin, N.; Bhatt, A.K.; Dutt, P.; Greer, P.A.; Arthur, J.S.; Elce, J.S.; Huttenlocher, A. Reduced Cell Migration and Disruption of the Actin Cytoskeleton in Calpain-Deficient Embryonic Fibroblasts. J. Biol. Chem. 2001, 276, 48382–48388. [Google Scholar] [CrossRef] [Green Version]
- Vesce, S.; Kirk, L.; Nicholls, D.G. Relationships between Superoxide Levels and Delayed Calcium Deregulation in Cultured Cerebellar Granule Cells Exposed Continuously to Glutamate. J. Neurochem. 2004, 90, 683–693. [Google Scholar] [CrossRef]
- Lankiewicz, S.; Marc, L.C.; Truc, B.N.; Krohn, A.J.; Poppe, M.; Cole, G.M.; Saido, T.C.; Prehn, J.H. Activation of Calpain I Converts Excitotoxic Neuron Death into a Caspase-Independent Cell Death. J. Biol. Chem. 2000, 275, 17064–17071. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.W.; Rego, A.C.; Frenguelli, B.G.; Nicholls, D.G. Mitochondrial Membrane Potential and Glutamate Excitotoxicity in Cultured Cerebellar Granule Cells. J. Neurosci. 2000, 20, 7208–7219. [Google Scholar] [CrossRef]
- Vergun, O.; Keelan, J.; Khodorov, B.I.; Duchen, M.R. Glutamate-Induced Mitochondrial Depolarisation and Perturbation of Calcium Homeostasis in Cultured Rat Hippocampal Neurones. J. Physiol. 1999, 519, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Duchen, M.R. Impaired Mitochondrial Bioenergetics Determines Glutamate-Induced Delayed Calcium Deregulation in Neurons. Biochim. Biophys. Acta 2010, 1800, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Virès, E.; Granier, C.; Prevot, P.; Lebleu, B. Structure-Activity Relationship Study of the Plasma Membrane Translocating Potential of a Short Peptide from HIV-1 Tat Protein. Lett. Pept. Sci. 1997, 4, 429–436. [Google Scholar] [CrossRef]
- Ono, Y.; Sorimachi, H. Calpains—An Elaborate Proteolytic System. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. [Google Scholar] [CrossRef] [PubMed]
- Baudry, M. Calpain-1 and Calpain-2 in the Brain: Dr. Jekill and Mr Hyde? Curr. Neuropharmacol. 2019, 17, 823–829. [Google Scholar] [CrossRef]
- Peng, T.I.; Greenamyre, J.T. Privileged Access to Mitochondria of Calcium Influx through N-Methyl-D-Aspartate Receptors. Mol. Pharm. 1998, 53, 974–980. [Google Scholar]
- Cabrera-Garcia, D.; Warm, D.; de la Fuente, P.; Fernández-Sánchez, M.T.; Novelli, A.; Villanueva-Balsera, J.M. Early Prediction of Developing Spontaneous Activity In Cultured Neuronal Networks. Sci. Rep. 2021, 11, 20407. [Google Scholar] [CrossRef]
- Rusilowicz-Jones, E.V.; Urbé, S.; Clague, M.J. Protein Degradation on the Global Scale. Mol. Cell 2022, 82, 1414–1423. [Google Scholar] [CrossRef]
- Bondada, V.; Gal, J.; Mashburn, C.; Rodgers, D.W.; Larochelle, K.E.; Croall, D.E.; Geddes, J.W. The C2 Domain of Calpain 5 Contributes to Enzyme Activation and Membrane Localization. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119019. [Google Scholar] [CrossRef]
- Caldeira, M.V.; Curcio, M.; Leal, G.; Salazar, I.L.; Mele, M.; Santos, A.R.; Melo, C.V.; Pereira, P.; Canzoniero, L.M.; Duarte, C.B. Excitotoxic Stimulation Downregulates the Ubiquitin-Proteasome System through Activation of NMDA Receptors in Cultured Hippocampal Neurons. Biochim. Biophys. Acta 2013, 1832, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.; Jones, J.W.; Hegdekar, N.; Thayer, J.A.; Kumar, A.; Faden, A.I.; Kane, M.A.; Lipinski, M.M. PLA2G4A/cPLA2-Mediated Lysosomal Membrane Damage Leads to Inhibition of Autophagy and Neurodegeneration after Brain Trauma. Autophagy 2020, 16, 466–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aits, S.; Jäättelä, M. Lysosomal Cell Death at a Glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinheckel, T.; Tholen, M. Low-Level Lysosomal Membrane Permeabilization for Limited Release and Sublethal Functions of Cathepsin Proteases in the Cytosol and Nucleus. FEBS Open Bio 2022, 12, 694–707. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Total Cell # | Non-Deregulating # | DCD # | DCD # Analyzed |
|---|---|---|---|---|
| Vehicle | 69 1 | 24 | 44 (63.8%) | 27 |
| E64d | 63 1 | 7 | 55 (87.3%) | 51 2 |
| Treatment | Mean ± SD | Lower 95% CI | Upper 95% CI | n |
|---|---|---|---|---|
| E64d vehicle | 1.19 ± 0.52 | 1.00 | 1.38 | 31 |
| E64d | 1.44 ± 0.85 | 1.20 | 1.67 | 52 |
| MG132 vehicle | 1.45 ± 1.95 | 1.09 | 1.81 | 115 |
| MG132 | 1.60 ± 1.52 | 1.32 | 1.87 | 118 |
| Rate | Non-Dereg Vehicle | Non-Dereg MG132 | Dereg Vehicle | Dereg MG132 |
|---|---|---|---|---|
| Baseline | 1.12 ± 1.34 (69) | 1.34 ± 1.13 (40) | 1.94 ± 2.56 (46) | 1.73 ± 1.68 (78) |
| Glutamate | 2.02 ± 1.52 (69) a,b | 2.05 ± 1.31 (40) a,b | 2.75 ± 2.09 (8) | 2.81 ± 2.33 (5) |
| MK-801 + NBQX | 0.69 ± 0.69 (69) a,b | 0.59 ± 0.79 (40) a,b | 1.57 ± 1.20 (7) | 1.40 (2) |
| DCD | N/A | N/A | 9.73 ± 9.99 (46) | 6.09 ± 5.46 (78) |
| Ionomycin | 6.33 ± 6.40 (66) a | 4.91 ± 3.63 (36) a | N/A | N/A |
| Treatment | Total Cell # | DCD # (Analyzed) | Non-Dereg # (Analyzed) | % DCD (Analyzed) |
|---|---|---|---|---|
| Vehicle | 174 | 50 (46) | 124 (69) | 28.7 (40.0) |
| MG132 | 205 1 | 95 (78) | 103 (40) | 45.9 (66.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polster, B.M.; Mark, K.A.; Arze, R.; Hudson, D. Calpain-Independent Intracellular Protease Activity Is Elevated in Excitotoxic Cortical Neurons Prior to Delayed Calcium Deregulation and Mitochondrial Dysfunction. Biomolecules 2022, 12, 1004. https://doi.org/10.3390/biom12071004
Polster BM, Mark KA, Arze R, Hudson D. Calpain-Independent Intracellular Protease Activity Is Elevated in Excitotoxic Cortical Neurons Prior to Delayed Calcium Deregulation and Mitochondrial Dysfunction. Biomolecules. 2022; 12(7):1004. https://doi.org/10.3390/biom12071004
Chicago/Turabian StylePolster, Brian M., Karla A. Mark, Rafael Arze, and Derek Hudson. 2022. "Calpain-Independent Intracellular Protease Activity Is Elevated in Excitotoxic Cortical Neurons Prior to Delayed Calcium Deregulation and Mitochondrial Dysfunction" Biomolecules 12, no. 7: 1004. https://doi.org/10.3390/biom12071004
APA StylePolster, B. M., Mark, K. A., Arze, R., & Hudson, D. (2022). Calpain-Independent Intracellular Protease Activity Is Elevated in Excitotoxic Cortical Neurons Prior to Delayed Calcium Deregulation and Mitochondrial Dysfunction. Biomolecules, 12(7), 1004. https://doi.org/10.3390/biom12071004