
MafA Regulation in β-Cells: From Transcriptional to Post-Translational Mechanisms



Abstract
1. Introduction
2. MafA Regulates β-Cell Function
3. MafA Target Genes
4. Regulation of MafA Transcription
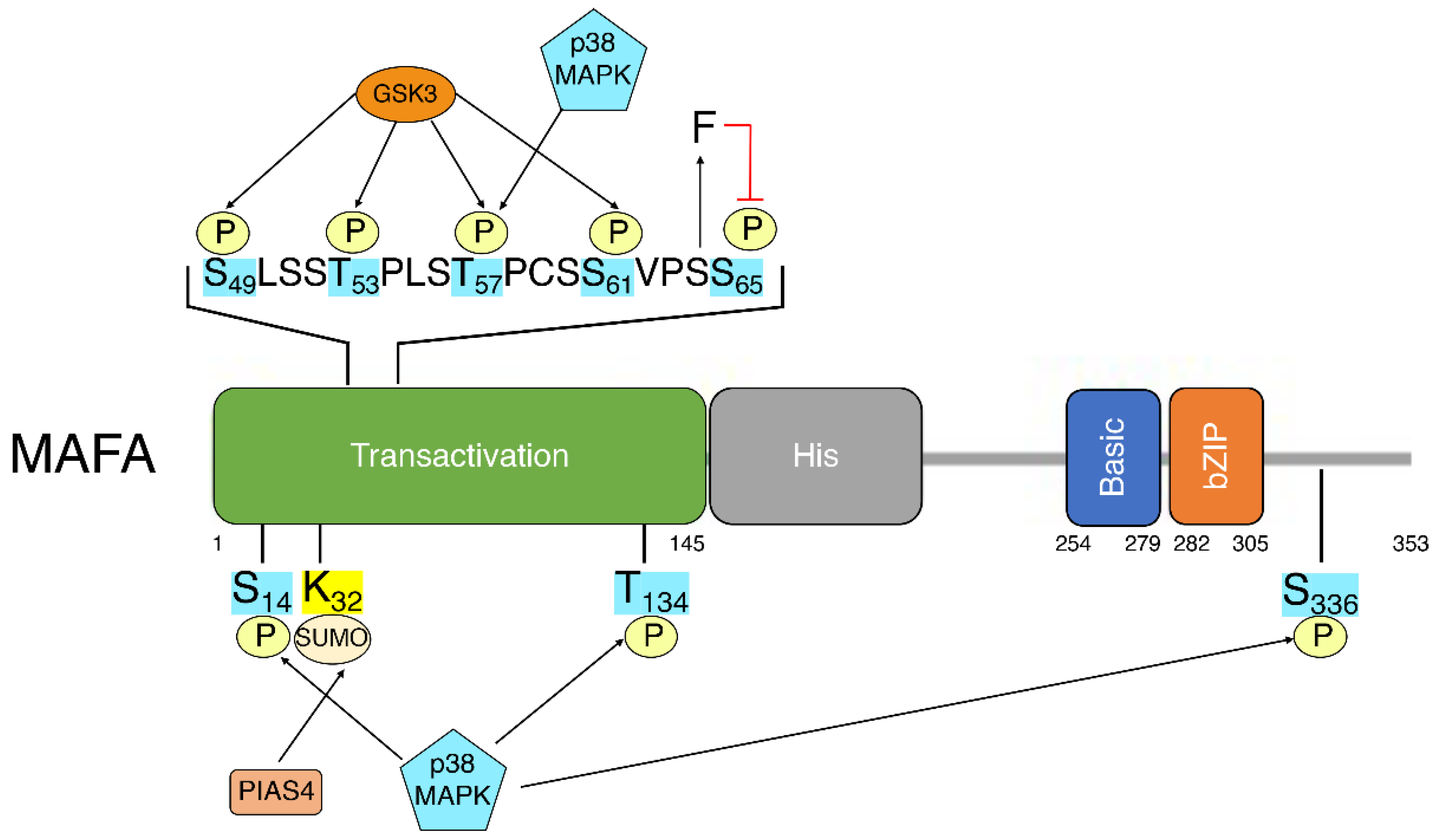
5. MafA Post-Translational Modifications
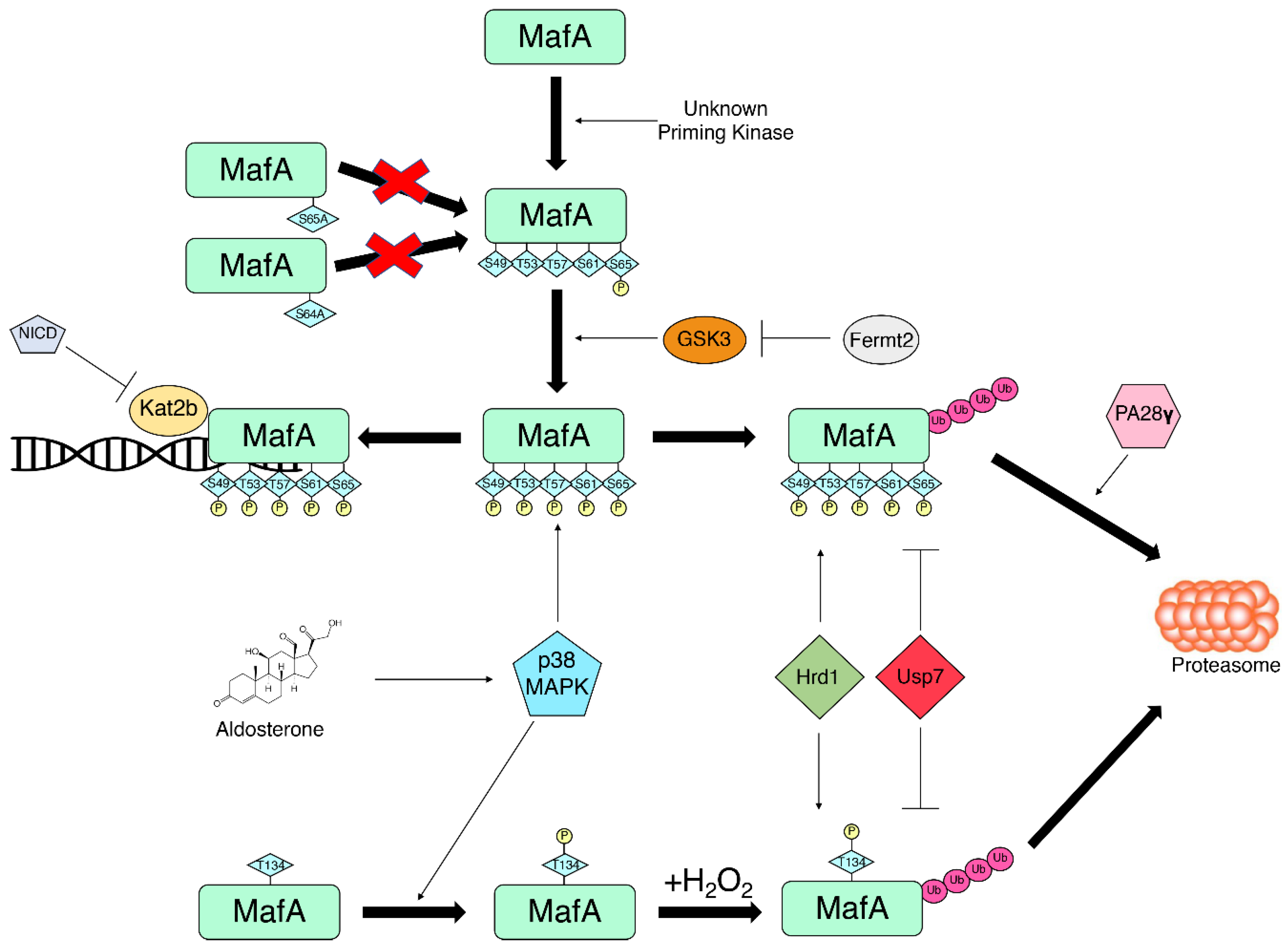
5.1. Phosphorylation
5.2. SUMOylation
5.3. Ubiquitination
5.4. Other PTMs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishizawa, M.; Kataoka, K.; Goto, N.; Fujiwara, K.T.; Kawai, S. V-maf, a viral oncogene that encodes a “leucine zipper” motif. Proc. Natl. Acad. Sci. USA 1989, 86, 7711–7715. [Google Scholar] [CrossRef] [PubMed]
- Reza, H.M.; Yasuda, K. Roles of Maf family proteins in lens development. Dev. Dyn. 2004, 229, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Guo, Z.M. Multiple functions of Maf in the regulation of cellular development and differentiation. Diabetes Metab. Res. Rev. 2015, 31, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Artner, I.; Le Lay, J.; Hang, Y.; Elghazi, L.; Schisler, J.C.; Henderson, E.; Sosa-Pineda, B.; Stein, R. MafB: An activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes 2006, 55, 297–304. [Google Scholar] [CrossRef]
- Eychene, A.; Rocques, N.; Pouponnot, C. A new MAFia in cancer. Nat. Rev. Cancer 2008, 8, 683–693. [Google Scholar] [CrossRef]
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742. [Google Scholar] [CrossRef]
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49903–49910. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol. Cell. Biol. 2003, 23, 6049–6062. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic beta-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720. [Google Scholar] [CrossRef]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, W.; Takahashi, S.; Yasuda, K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 2015, 58, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Aramata, S.; Han, S.I.; Yasuda, K.; Kataoka, K. Synergistic activation of the insulin gene promoter by the beta-cell enriched transcription factors MafA, Beta2, and Pdx1. Biochim. Biophys. Acta 2005, 1730, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Guo, M.; Matsuoka, T.A.; Hagman, D.K.; Parazzoli, S.D.; Poitout, V.; Stein, R. The islet beta cell-enriched MafA activator is a key regulator of insulin gene transcription. J. Biol. Chem. 2005, 280, 11887–11894. [Google Scholar] [CrossRef] [PubMed]
- Artner, I.; Hang, Y.; Guo, M.; Gu, G.; Stein, R. MafA is a dedicated activator of the insulin gene in vivo. J. Endocrinol. 2008, 198, 271–279. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Artner, I.; Henderson, E.; Means, A.; Sander, M.; Stein, R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 2004, 101, 2930–2933. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet beta-cell function. Mol. Endocrinol. 2007, 21, 2764–2774. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2011, 54, 583–593. [Google Scholar] [CrossRef]
- Artner, I.; Hang, Y.; Mazur, M.; Yamamoto, T.; Guo, M.; Lindner, J.; Magnuson, M.A.; Stein, R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 2010, 59, 2530–2539. [Google Scholar] [CrossRef]
- He, K.H.; Juhl, K.; Karadimos, M.; El Khattabi, I.; Fitzpatrick, C.; Bonner-Weir, S.; Sharma, A. Differentiation of pancreatic endocrine progenitors reversibly blocked by premature induction of MafA. Dev. Biol. 2014, 385, 2–12. [Google Scholar] [CrossRef]
- Nishimura, W.; Bonner-Weir, S.; Sharma, A. Expression of MafA in pancreatic progenitors is detrimental for pancreatic development. Dev. Biol. 2009, 333, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Hang, Y.; Yamamoto, T.; Benninger, R.K.; Brissova, M.; Guo, M.; Bush, W.; Piston, D.W.; Powers, A.C.; Magnuson, M.; Thurmond, D.C.; et al. The MafA transcription factor becomes essential to islet beta-cells soon after birth. Diabetes 2014, 63, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.C.; Jung, Y.; Ugboma, C.M.; Shimbo, M.; Kuno, A.; Basha, W.A.; Kudo, T.; Oishi, H.; Takahashi, S. MafB Is Critical for Glucagon Production and Secretion in Mouse Pancreatic alpha Cells In Vivo. Mol. Cell. Biol. 2018, 38, e00504-17. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, H.A.; Walker, E.M.; Hang, Y.; Dhawan, S.; Haliyur, R.; Bonatakis, L.; Avrahami, D.; Brissova, M.; Kaestner, K.H.; Bhushan, A.; et al. Examining How the MAFB Transcription Factor Affects Islet beta-Cell Function Postnatally. Diabetes 2019, 68, 337–348. [Google Scholar] [CrossRef]
- Conrad, E.; Dai, C.; Spaeth, J.; Guo, M.; Cyphert, H.A.; Scoville, D.; Carroll, J.; Yu, W.M.; Goodrich, L.V.; Harlan, D.M.; et al. The MAFB transcription factor impacts islet alpha-cell function in rodents and represents a unique signature of primate islet beta-cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E91–E102. [Google Scholar] [CrossRef] [PubMed]
- Xiafukaiti, G.; Maimaiti, S.; Ogata, K.; Kuno, A.; Kudo, T.; Shawki, H.H.; Oishi, H.; Takahashi, S. MafB Is Important for Pancreatic beta-Cell Maintenance under a MafA-Deficient Condition. Mol. Cell. Biol. 2019, 39, e00080-19. [Google Scholar] [CrossRef]
- Arda, H.E.; Li, L.; Tsai, J.; Torre, E.A.; Rosli, Y.; Peiris, H.; Spitale, R.C.; Dai, C.; Gu, X.; Qu, K.; et al. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human beta Cell Function. Cell Metab. 2016, 23, 909–920. [Google Scholar] [CrossRef]
- Russell, R.; Carnese, P.P.; Hennings, T.G.; Walker, E.M.; Russ, H.A.; Liu, J.S.; Giacometti, S.; Stein, R.; Hebrok, M. Loss of the transcription factor MAFB limits beta-cell derivation from human PSCs. Nat. Commun. 2020, 11, 2742. [Google Scholar] [CrossRef]
- Bonnavion, R.; Jaafar, R.; Kerr-Conte, J.; Assade, F.; van Stralen, E.; Leteurtre, E.; Pouponnot, C.; Gargani, S.; Pattou, F.; Bertolino, P.; et al. Both PAX4 and MAFA are expressed in a substantial proportion of normal human pancreatic alpha cells and deregulated in patients with type 2 diabetes. PLoS ONE 2013, 8, e72194. [Google Scholar] [CrossRef]
- Butler, A.E.; Robertson, R.P.; Hernandez, R.; Matveyenko, A.V.; Gurlo, T.; Butler, P.C. Beta cell nuclear musculoaponeurotic fibrosarcoma oncogene family A (MafA) is deficient in type 2 diabetes. Diabetologia 2012, 55, 2985–2988. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.A.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving Mafa expression in diabetic islet beta-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Ding, H.; Farb, T.B.; Efanov, A.M.; Sun, J.; Gore, J.L.; Syed, S.K.; Lei, Z.; Wang, Q.; Accili, D.; et al. BACH2 inhibition reverses beta cell failure in type 2 diabetes models. J. Clin. Investig. 2021, 131, e153876. [Google Scholar] [CrossRef]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef]
- Shrestha, S.; Saunders, D.C.; Walker, J.T.; Camunas-Soler, J.; Dai, X.Q.; Haliyur, R.; Aramandla, R.; Poffenberger, G.; Prasad, N.; Bottino, R.; et al. Combinatorial transcription factor profiles predict mature and functional human islet alpha and beta cells. JCI Insight 2021, 6, e151621. [Google Scholar] [CrossRef] [PubMed]
- Benkhelifa, S.; Provot, S.; Lecoq, O.; Pouponnot, C.; Calothy, G.; Felder-Schmittbuhl, M.P. mafA, a novel member of the maf proto-oncogene family, displays developmental regulation and mitogenic capacity in avian neuroretina cells. Oncogene 1998, 17, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Camunas-Soler, J.; Dai, X.Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; MacDonald, P.E. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020, 31, 1017–1031.e1014. [Google Scholar] [CrossRef]
- Nasteska, D.; Fine, N.H.F.; Ashford, F.B.; Cuozzo, F.; Viloria, K.; Smith, G.; Dahir, A.; Dawson, P.W.J.; Lai, Y.C.; Bastidas-Ponce, A.; et al. PDX1(LOW) MAFA(LOW) beta-cells contribute to islet function and insulin release. Nat. Commun. 2021, 12, 674. [Google Scholar] [CrossRef]
- Lu, X.; Guanga, G.P.; Wan, C.; Rose, R.B. A novel DNA binding mechanism for maf basic region-leucine zipper factors inferred from a MafA-DNA complex structure and binding specificities. Biochemistry 2012, 51, 9706–9717. [Google Scholar] [CrossRef]
- Kataoka, K.; Noda, M.; Nishizawa, M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell. Biol. 1994, 14, 700–712. [Google Scholar] [CrossRef]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y.; Kinjo, T.; Saitoh, I.; Watanabe, M. Mutations in the C1 element of the insulin promoter lead to diabetic phenotypes in homozygous mice. Commun. Biol. 2020, 3, 309. [Google Scholar] [CrossRef] [PubMed]
- Garin, I.; Edghill, E.L.; Akerman, I.; Rubio-Cabezas, O.; Rica, I.; Locke, J.M.; Maestro, M.A.; Alshaikh, A.; Bundak, R.; del Castillo, G.; et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3105–3110. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Maestro, M.A.; De Franco, E.; Grau, V.; Flanagan, S.; Garcia-Hurtado, J.; Mittler, G.; Ravassard, P.; Piemonti, L.; Ellard, S.; et al. Neonatal diabetes mutations disrupt a chromatin pioneering function that activates the human insulin gene. Cell Rep. 2021, 35, 108981. [Google Scholar] [CrossRef]
- ZeRuth, G.T.; Takeda, Y.; Jetten, A.M. The Kruppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol. Endocrinol. 2013, 27, 1692–1705. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.; Ye, Y.; Singh, T.; Barghouth, M.; Eliasson, L.; Artner, I.; Zhang, E.; Renstrom, E. The calcium channel subunit gamma-4 is regulated by MafA and necessary for pancreatic beta-cell specification. Commun. Biol. 2019, 2, 106. [Google Scholar] [CrossRef] [PubMed]
- Ganic, E.; Singh, T.; Luan, C.; Fadista, J.; Johansson, J.K.; Cyphert, H.A.; Bennet, H.; Storm, P.; Prost, G.; Ahlenius, H.; et al. MafA-Controlled Nicotinic Receptor Expression Is Essential for Insulin Secretion and Is Impaired in Patients with Type 2 Diabetes. Cell Rep. 2016, 14, 1991–2002. [Google Scholar] [CrossRef]
- Nagai, Y.; Matsuoka, T.A.; Shimo, N.; Miyatsuka, T.; Miyazaki, S.; Tashiro, F.; Miyazaki, J.I.; Katakami, N.; Shimomura, I. Glucotoxicity-induced suppression of Cox6a2 expression provokes beta-cell dysfunction via augmented ROS production. Biochem. Biophys. Res. Commun. 2021, 556, 134–141. [Google Scholar] [CrossRef]
- Martin, C.C.; Flemming, B.P.; Wang, Y.; Oeser, J.K.; O’Brien, R.M. Foxa2 and MafA regulate islet-specific glucose-6-phosphatase catalytic subunit-related protein gene expression. J. Mol. Endocrinol. 2008, 41, 315–328. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef]
- Ganic, E.; Johansson, J.K.; Bennet, H.; Fex, M.; Artner, I. Islet-specific monoamine oxidase A and B expression depends on MafA transcriptional activity and is compromised in type 2 diabetes. Biochem. Biophys. Res. Commun. 2015, 468, 629–635. [Google Scholar] [CrossRef]
- Aigha, I.I.; Abdelalim, E.M. NKX6.1 transcription factor: A crucial regulator of pancreatic beta cell development, identity, and proliferation. Stem Cell Res. Ther. 2020, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Vanhoose, A.M.; Samaras, S.; Artner, I.; Henderson, E.; Hang, Y.; Stein, R. MafA and MafB regulate Pdx1 transcription through the Area II control region in pancreatic beta cells. J. Biol. Chem. 2008, 283, 22612–22619. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, L.R.; Vishnu, N.; Singh, T.; Bertonnier-Brouty, L.; Bsharat, S.; Luan, C.; Renstrom, E.; Prasad, R.B.; Fex, M.; Mulder, H.; et al. The MafA-target gene PPP1R1A regulates GLP1R-mediated amplification of glucose-stimulated insulin secretion in beta-cells. Metabolism 2021, 118, 154734. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Nishimura, W.; Oishi, H.; Udagawa, H.; Kawaguchi, M.; Hiramoto, M.; Fujiwara, T.; Takahashi, S.; Yasuda, K. MafA is required for postnatal proliferation of pancreatic beta-cells. PLoS ONE 2014, 9, e104184. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Kataoka, K. MafA, NeuroD1, and HNF1beta synergistically activate the Slc2a2 (Glut2) gene in beta-cells. J. Mol. Endocrinol. 2021, 67, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.S.; Maestro, M.A.; Raum, J.C.; Guo, M.; Thompson, F.H., 3rd; Ferrer, J.; Stein, R. Hnf1alpha (MODY3) regulates beta-cell-enriched MafA transcription factor expression. Mol. Endocrinol. 2011, 25, 339–347. [Google Scholar] [CrossRef]
- Raum, J.C.; Gerrish, K.; Artner, I.; Henderson, E.; Guo, M.; Sussel, L.; Schisler, J.C.; Newgard, C.B.; Stein, R. FoxA2, Nkx2.2, and PDX-1 regulate islet beta-cell-specific mafA expression through conserved sequences located between base pairs -8118 and -7750 upstream from the transcription start site. Mol. Cell. Biol. 2006, 26, 5735–5743. [Google Scholar] [CrossRef]
- Raum, J.C.; Hunter, C.S.; Artner, I.; Henderson, E.; Guo, M.; Elghazi, L.; Sosa-Pineda, B.; Ogihara, T.; Mirmira, R.G.; Sussel, L.; et al. Islet beta-cell-specific MafA transcription requires the 5′-flanking conserved region 3 control domain. Mol. Cell. Biol. 2010, 30, 4234–4244. [Google Scholar] [CrossRef]
- Romer, A.I.; Singer, R.A.; Sui, L.; Egli, D.; Sussel, L. Murine Perinatal beta-Cell Proliferation and the Differentiation of Human Stem Cell-Derived Insulin-Expressing Cells Require NEUROD1. Diabetes 2019, 68, 2259–2271. [Google Scholar] [CrossRef]
- Hu He, K.H.; Lorenzo, P.I.; Brun, T.; Jimenez Moreno, C.M.; Aeberhard, D.; Vallejo Ortega, J.; Cornu, M.; Thorel, F.; Gjinovci, A.; Thorens, B.; et al. In vivo conditional Pax4 overexpression in mature islet beta-cells prevents stress-induced hyperglycemia in mice. Diabetes 2011, 60, 1705–1715. [Google Scholar] [CrossRef]
- Yamamoto, K.; Matsuoka, T.A.; Kawashima, S.; Takebe, S.; Kubo, F.; Miyatsuka, T.; Kaneto, H.; Shimomura, I. A novel function of Onecut1 protein as a negative regulator of MafA gene expression. J. Biol. Chem. 2013, 288, 21648–21658. [Google Scholar] [CrossRef] [PubMed]
- Du, A.; Hunter, C.S.; Murray, J.; Noble, D.; Cai, C.L.; Evans, S.M.; Stein, R.; May, C.L. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes 2009, 58, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Artner, I.; Blanchi, B.; Raum, J.C.; Guo, M.; Kaneko, T.; Cordes, S.; Sieweke, M.; Stein, R. MafB is required for islet beta cell maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 3853–3858. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Zavacki, A.M.; Marinelarena, A.; Hollister-Lock, J.; El Khattabi, I.; Marsili, A.; Weir, G.C.; Sharma, A.; Larsen, P.R.; Bonner-Weir, S. Thyroid hormone promotes postnatal rat pancreatic beta-cell development and glucose-responsive insulin secretion through MAFA. Diabetes 2013, 62, 1569–1580. [Google Scholar] [CrossRef]
- Blanchet, E.; Van de Velde, S.; Matsumura, S.; Hao, E.; LeLay, J.; Kaestner, K.; Montminy, M. Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep. 2015, 10, 1149–1157. [Google Scholar] [CrossRef]
- Accili, D.; Talchai, S.C.; Kim-Muller, J.Y.; Cinti, F.; Ishida, E.; Ordelheide, A.M.; Kuo, T.; Fan, J.; Son, J. When beta-cells fail: Lessons from dedifferentiation. Diabetes Obes. Metab. 2016, 18 (Suppl. 1), 117–122. [Google Scholar] [CrossRef]
- Xiao, X.; Chen, C.; Guo, P.; Zhang, T.; Fischbach, S.; Fusco, J.; Shiota, C.; Prasadan, K.; Dong, H.; Gittes, G.K. Forkhead Box Protein 1 (FoxO1) Inhibits Accelerated beta Cell Aging in Pancreas-specific SMAD7 Mutant Mice. J. Biol. Chem. 2017, 292, 3456–3465. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Vanderford, N.L.; Andrali, S.S.; Ozcan, S. Glucose induces MafA expression in pancreatic beta cell lines via the hexosamine biosynthetic pathway. J. Biol. Chem. 2007, 282, 1577–1584. [Google Scholar] [CrossRef]
- Leenders, F.; Groen, N.; de Graaf, N.; Engelse, M.A.; Rabelink, T.J.; de Koning, E.J.P.; Carlotti, F. Oxidative Stress Leads to beta-Cell Dysfunction Through Loss of beta-Cell Identity. Front. Immunol. 2021, 12, 690379. [Google Scholar] [CrossRef]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Long, Y.; Tong, Y.; Wan, J.; Tong, N. Activation of PPARdelta up-regulates the expression of insulin gene transcription factor MafA and ameliorates glucose-induced insulin secretion impaired by palmitate. Mol. Cell. Biochem. 2012, 366, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Vanderford, N.L.; Cantrell, J.E.; Popa, G.J.; Ozcan, S. Multiple kinases regulate mafA expression in the pancreatic beta cell line MIN6. Arch Biochem. Biophys. 2008, 480, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, J.; Wang, Y.; Wu, T.; Shan, W.; Zhu, Y.; Han, X. Aldosterone induces clonal beta-cell failure through glucocorticoid receptor. Sci. Rep. 2015, 5, 13215. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, Q.; Xu, H.; Xue, J.; Chen, C.; Yang, X.; Gao, X.; Liu, Q. miR-149 Negative Regulation of mafA Is Involved in the Arsenite-Induced Dysfunction of Insulin Synthesis and Secretion in Pancreatic Beta Cells. Toxicol. Sci. 2019, 167, 116–125. [Google Scholar] [CrossRef]
- Zhao, X.; Mohan, R.; Ozcan, S.; Tang, X. MicroRNA-30d induces insulin transcription factor MafA and insulin production by targeting mitogen-activated protein 4 kinase 4 (MAP4K4) in pancreatic beta-cells. J. Biol. Chem. 2012, 287, 31155–31164. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, Y.; Zhou, Y.; Zhang, Y.; Zhang, T.; Li, Y.; You, W.; Chang, X.; Yuan, L.; Han, X. MicroRNA-24 promotes pancreatic beta cells toward dedifferentiation to avoid endoplasmic reticulum stress-induced apoptosis. J. Mol. Cell Biol. 2019, 11, 747–760. [Google Scholar] [CrossRef]
- Marzinotto, I.; Pellegrini, S.; Brigatti, C.; Nano, R.; Melzi, R.; Mercalli, A.; Liberati, D.; Sordi, V.; Ferrari, M.; Falconi, M.; et al. miR-204 is associated with an endocrine phenotype in human pancreatic islets but does not regulate the insulin mRNA through MAFA. Sci. Rep. 2017, 7, 14051. [Google Scholar] [CrossRef]
- Arnes, L.; Sussel, L. Epigenetic modifications and long noncoding RNAs influence pancreas development and function. Trends Genet. 2015, 31, 290–299. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, Y.; Xie, M.; Wang, L.; Jin, F.; Li, Y.; Yuan, Q.; De, W. Long Noncoding RNA Meg3 Regulates Mafa Expression in Mouse Beta Cells by Inactivating Rad21, Smc3 or Sin3alpha. Cell. Physiol. Biochem. 2018, 45, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Kameswaran, V.; Golson, M.L.; Ramos-Rodriguez, M.; Ou, K.; Wang, Y.J.; Zhang, J.; Pasquali, L.; Kaestner, K.H. The Dysregulation of the DLK1-MEG3 Locus in Islets From Patients with Type 2 Diabetes Is Mimicked by Targeted Epimutation of Its Promoter With TALE-DNMT Constructs. Diabetes 2018, 67, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Kameswaran, V.; Bramswig, N.C.; McKenna, L.B.; Penn, M.; Schug, J.; Hand, N.J.; Chen, Y.; Choi, I.; Vourekas, A.; Won, K.J.; et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 2014, 19, 135–145. [Google Scholar] [CrossRef]
- Guo, S.; Vanderford, N.L.; Stein, R. Phosphorylation within the MafA N terminus regulates C-terminal dimerization and DNA binding. J. Biol. Chem. 2010, 285, 12655–12661. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cissell, M.A.; Henderson, E.; Colbran, R.; Stein, R. The RIPE3b1 activator of the insulin gene is composed of a protein(s) of approximately 43 kDa, whose DNA binding activity is inhibited by protein phosphatase treatment. J. Biol. Chem. 2000, 275, 10532–10537. [Google Scholar] [CrossRef]
- Han, S.I.; Tsunekage, Y.; Kataoka, K. Phosphorylation of MafA enhances interaction with Beta2/NeuroD1. Acta Diabetol. 2016, 53, 651–660. [Google Scholar] [CrossRef]
- Matsuoka, T.; Zhao, L.; Stein, R. The DNA binding activity of the RIPE3b1 transcription factor of insulin appears to be influenced by tyrosine phosphorylation. J. Biol. Chem. 2001, 276, 22071–22076. [Google Scholar] [CrossRef]
- Benkhelifa, S.; Provot, S.; Nabais, E.; Eychene, A.; Calothy, G.; Felder-Schmittbuhl, M.P. Phosphorylation of MafA is essential for its transcriptional and biological properties. Mol. Cell. Biol. 2001, 21, 4441–4452. [Google Scholar] [CrossRef]
- Guo, S.; Burnette, R.; Zhao, L.; Vanderford, N.L.; Poitout, V.; Hagman, D.K.; Henderson, E.; Ozcan, S.; Wadzinski, B.E.; Stein, R. The stability and transactivation potential of the mammalian MafA transcription factor are regulated by serine 65 phosphorylation. J. Biol. Chem. 2009, 284, 759–765. [Google Scholar] [CrossRef]
- Sii-Felice, K.; Pouponnot, C.; Gillet, S.; Lecoin, L.; Girault, J.A.; Eychene, A.; Felder-Schmittbuhl, M.P. MafA transcription factor is phosphorylated by p38 MAP kinase. FEBS Lett. 2005, 579, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; El Khattabi, I.; Nishimura, W.; Laybutt, D.R.; Geraldes, P.; Shah, S.; King, G.; Bonner-Weir, S.; Weir, G.; Sharma, A. p38 MAPK is a major regulator of MafA protein stability under oxidative stress. Mol. Endocrinol. 2009, 23, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- El Khattabi, I.; Sharma, A. Preventing p38 MAPK-mediated MafA degradation ameliorates beta-cell dysfunction under oxidative stress. Mol. Endocrinol. 2013, 27, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Rocques, N.; Abou Zeid, N.; Sii-Felice, K.; Lecoin, L.; Felder-Schmittbuhl, M.P.; Eychene, A.; Pouponnot, C. GSK-3-mediated phosphorylation enhances Maf-transforming activity. Mol. Cell 2007, 28, 584–597. [Google Scholar] [CrossRef]
- Han, S.I.; Aramata, S.; Yasuda, K.; Kataoka, K. MafA stability in pancreatic beta cells is regulated by glucose and is dependent on its constitutive phosphorylation at multiple sites by glycogen synthase kinase 3. Mol. Cell. Biol. 2007, 27, 6593–6605. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Zhu, K.; Lai, Y.; Cao, H.; Bai, X.; Liu, C.; Yan, Q.; Ma, L.; Chen, D.; Kanaporis, G.; Wang, J.; et al. Kindlin-2 modulates MafA and beta-catenin expression to regulate beta-cell function and mass in mice. Nat. Commun. 2020, 11, 484. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Flanagan, S.E.; Walker, E.; Quezado, R.; de Sousa Barros, F.A.; Caswell, R.; Johnson, M.B.; Wakeling, M.; Brandle, M.; Guo, M.; et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci USA 2018, 115, 1027–1032. [Google Scholar] [CrossRef]
- Walker, E.M.; Cha, J.; Tong, X.; Guo, M.; Liu, J.H.; Yu, S.; Iacovazzo, D.; Mauvais-Jarvis, F.; Flanagan, S.E.; Korbonits, M.; et al. Sex-biased islet beta cell dysfunction is caused by the MODY MAFA S64F variant by inducing premature aging and senescence in males. Cell Rep. 2021, 37, 109813. [Google Scholar] [CrossRef]
- Kanai, K.; Reza, H.M.; Kamitani, A.; Hamazaki, Y.; Han, S.I.; Yasuda, K.; Kataoka, K. SUMOylation negatively regulates transcriptional and oncogenic activities of MafA. Genes Cells 2010, 15, 971–982. [Google Scholar] [CrossRef]
- Shao, C.; Cobb, M.H. Sumoylation regulates the transcriptional activity of MafA in pancreatic beta cells. J. Biol. Chem. 2009, 284, 3117–3124. [Google Scholar] [CrossRef] [PubMed]
- Onishi, S.; Kataoka, K. PIASy is a SUMOylation-independent negative regulator of the insulin transactivator MafA. J. Mol. Endocrinol. 2019, 63, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Sarcevic, B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 2010, 5, 19. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Kanai, K.; Aramata, S.; Katakami, S.; Yasuda, K.; Kataoka, K. Proteasome activator PA28{gamma} stimulates degradation of GSK3-phosphorylated insulin transcription activator MAFA. J. Mol. Endocrinol. 2011, 47, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, A.; Zhu, C.; Sussel, L.; Pajvani, U.B. Notch signaling dynamically regulates adult beta cell proliferation and maturity. J. Clin. Investig. 2019, 129, 268–280. [Google Scholar] [CrossRef]
- He, Y.; Wang, S.; Tong, J.; Jiang, S.; Yang, Y.; Zhang, Z.; Xu, Y.; Zeng, Y.; Cao, B.; Moran, M.F.; et al. The deubiquitinase USP7 stabilizes Maf proteins to promote myeloma cell survival. J. Biol. Chem. 2020, 295, 2084–2096. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, S.; Xu, J.; Zhang, Y.; Sun, T.; Shao, Y.; Wang, J.; Tang, W.; Chen, F.; Han, X. HRD1, an Important Player in Pancreatic beta-Cell Failure and Therapeutic Target for Type 2 Diabetic Mice. Diabetes 2020, 69, 940–953. [Google Scholar] [CrossRef]
- Scoville, D.W.; Cyphert, H.A.; Liao, L.; Xu, J.; Reynolds, A.; Guo, S.; Stein, R. MLL3 and MLL4 Methyltransferases Bind to the MAFA and MAFB Transcription Factors to Regulate Islet beta-Cell Function. Diabetes 2015, 64, 3772–3783. [Google Scholar] [CrossRef]
- Hung, H.L.; Kim, A.Y.; Hong, W.; Rakowski, C.; Blobel, G.A. Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J. Biol. Chem. 2001, 276, 10715–10721. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Gene Function | Reference |
|---|---|---|---|
| Cacng4 * | Voltage-dependent calcium channel gamma-4 subunit | Enhances L-type Ca2+-mediated Ca2+ entry into β-cell | [45] |
| Chrnb4 * | Cholinergic receptor nicotinic beta 4 | Subunit of the nicotinic acetylcholine receptor | [46] |
| Cox6a2 * | Cytochrome C oxidase subunit 6A2 | One out of 13 subunits of cytochrome C oxidase complex (Complex IV), the last enzyme in the electron transport chain | [47] |
| G6pc2 * | Glucose-6-phosphate catalytic subunit related protein | Islet-specific enzyme that hydrolyzes glucose-6-phosphate, limits basal insulin secretion | [48] |
| Gck | Glucokinase | Phosphorylates glucose to glucose-6-phosphate in pancreatic islets and hepatocytes. Considered the β-cell glucose sensor | [49] |
| Glp1r | Glucagon-like peptide 1 receptor | Receptor for Glucagon-like peptide 1 (Glp1), a stimulator of insulin secretion | [49] |
| Ins1 * | Insulin I | One of two insulin genes in mouse, on chromosome 19 | [11] |
| Ins2 * | Insulin II | One of two insulin genes in mouse, on chromosome 7 | [11] |
| Maob * | Monoamine oxidase B | Metabolizes monoamine neurotransmitters, specifically benzylamine, dopamine and phenylethylamine | [50] |
| Neurod1 | Neurogenic differentiation 1 | Transactivator of genes important for β-cell maturation and function, including insulin | [49] |
| Nkx6.1 | NK6 homeobox1 | TF involved in β-cell development and regulation of genes involved in mature β-cell function | [49,51] |
| Pcsk1 | Proprotein convertase subtilisin/kexin type 1 | Proprotein convertase, which processes proinsulin in β-cells | [49] |
| Pcx | Pyruvate carboxylase | Catalyzes the conversion of pyruvate to oxaloacetate | [49] |
| Pdx1 * | Pancreatic and duodenal homeobox 1 | TF important pancreas development and for mature β-cell function | [49,52] |
| PPP1R1A | Protein phosphatase 1, regulatory inhibitor subunit 1A | Regulates cAMP/PKA signaling pathway Promotes Glp1-induced GSIS | [53] |
| Prlr | Prolactin Receptor | Involved in increasing β-cell mass during pregnancy | [54] |
| Slc2a2 * | Solute Carrier Family 2 Member 2 | Glucose transporter 2, transmembrane glucose transporter with a high Km for glucose | [49,55] |
| Slc80a8 | Solute carrier family 30 member 8 | Zinc transporter on insulin granules in β-cells | [19,46] |
| Protein Symbol | Protein Name | Mafa Transcription Mechanism | Reference |
|---|---|---|---|
| Pax6 | Paired box protein Pax-6 | Binds R1, R3 and R6 of the Mafa promoter | [58] |
| Nkx6.1 | NK6 homeobox 1 | Binds R3 of the Mafa promoter | [58] |
| Nkx2.2 | NK2 homeobox 2 | Binds R3 of the Mafa promoter | [57] |
| Pdx1 | Pancreatic and duodenal homeobox 1 | Binds R3 and R6 of the Mafa promoter | [57,58] |
| Hnf1a | Hepatocyte nuclear factor 1-alpha | Binds R3 of the Mafa promoter | [56] |
| Foxa2 | Forkhead box A2 | Binds R3 of the Mafa promoter | [57] |
| Isl1 | Insulin gene enhancer protein ISL-1 | Binds R3 of the Mafa promoter | [62] |
| Neurod1 | Neurogenic differentiation 1 | Binds R3 of the Mafa promoter | [57] |
| Pax4 | Paired box protein Pax-4 | Negative regulator of Mafa, potentially by interfering other factors from binding R3 of the Mafa promoter | [60] |
| Mafb | Transcription factor MafB | Binds R3 of the Mafa promoter | [58,63] |
| Onecut1 | One cut domain, family member 1 | Prevents Foxa2 from binding to the Mafa promoter | [61] |
| Foxo1 | Forkhead box O1 | Binds to the forkhead element of the Mafa promoter | [34] |
| Thra | Thyroid hormone receptor alpha | Binds to Thyroid hormone response element (TRE), which are located from −1927 to −1946 and from +647 to +659 (named Site 2 and Site 3, respectively) | [64] |
| CREB | cAMP responsive element binding protein | Constitutively binds to the cAMP response element (CRE), spanning from −1342 to −1346, of the Mafa promoter | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Chirikjian, M.; Pajvani, U.B.; Bartolomé, A. MafA Regulation in β-Cells: From Transcriptional to Post-Translational Mechanisms. Biomolecules 2022, 12, 535. https://doi.org/10.3390/biom12040535
Liang J, Chirikjian M, Pajvani UB, Bartolomé A. MafA Regulation in β-Cells: From Transcriptional to Post-Translational Mechanisms. Biomolecules. 2022; 12(4):535. https://doi.org/10.3390/biom12040535
Chicago/Turabian StyleLiang, Jiani, Margot Chirikjian, Utpal B. Pajvani, and Alberto Bartolomé. 2022. "MafA Regulation in β-Cells: From Transcriptional to Post-Translational Mechanisms" Biomolecules 12, no. 4: 535. https://doi.org/10.3390/biom12040535
APA StyleLiang, J., Chirikjian, M., Pajvani, U. B., & Bartolomé, A. (2022). MafA Regulation in β-Cells: From Transcriptional to Post-Translational Mechanisms. Biomolecules, 12(4), 535. https://doi.org/10.3390/biom12040535