
A Novel Allosteric Inhibitor Targets PLK1 in Triple Negative Breast Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Kaplan–Meier (KM) Survival Analysis
2.2. Gene Ontology
2.3. Cell Culture
2.4. Viability Assays
2.5. In Silico Docking Study
2.6. Western Blotting Analysis
2.7. Kinase Antibody Arrays
2.8. Wound Healing Assay
2.9. Mammosphere Formation Assay
2.10. Cell Cycle Analysis
2.11. Statistical Analysis
3. Results
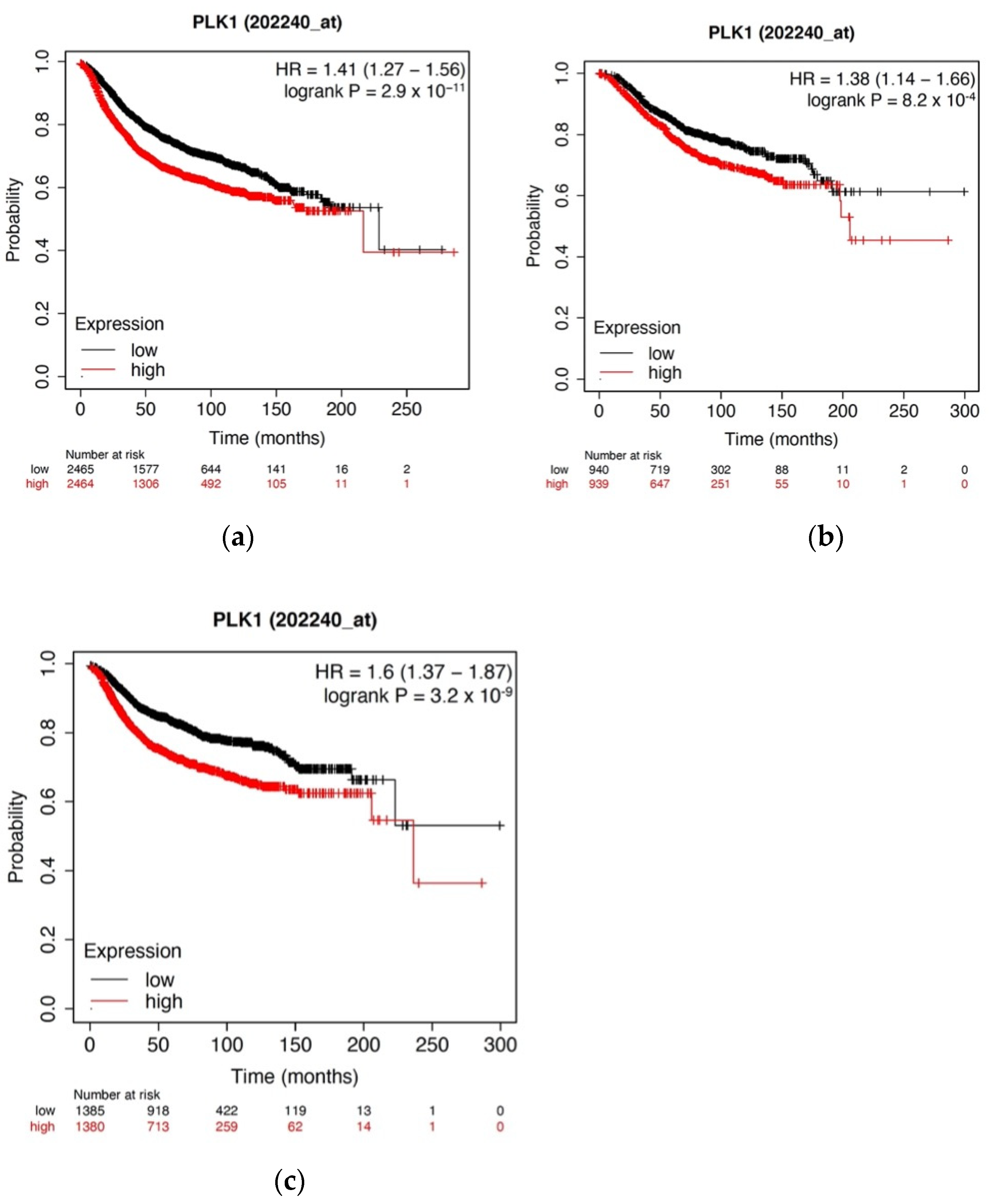
3.1. High PKL1 Expression Is Positively Correlated with Decreased Survival among Breast Cancer Patients
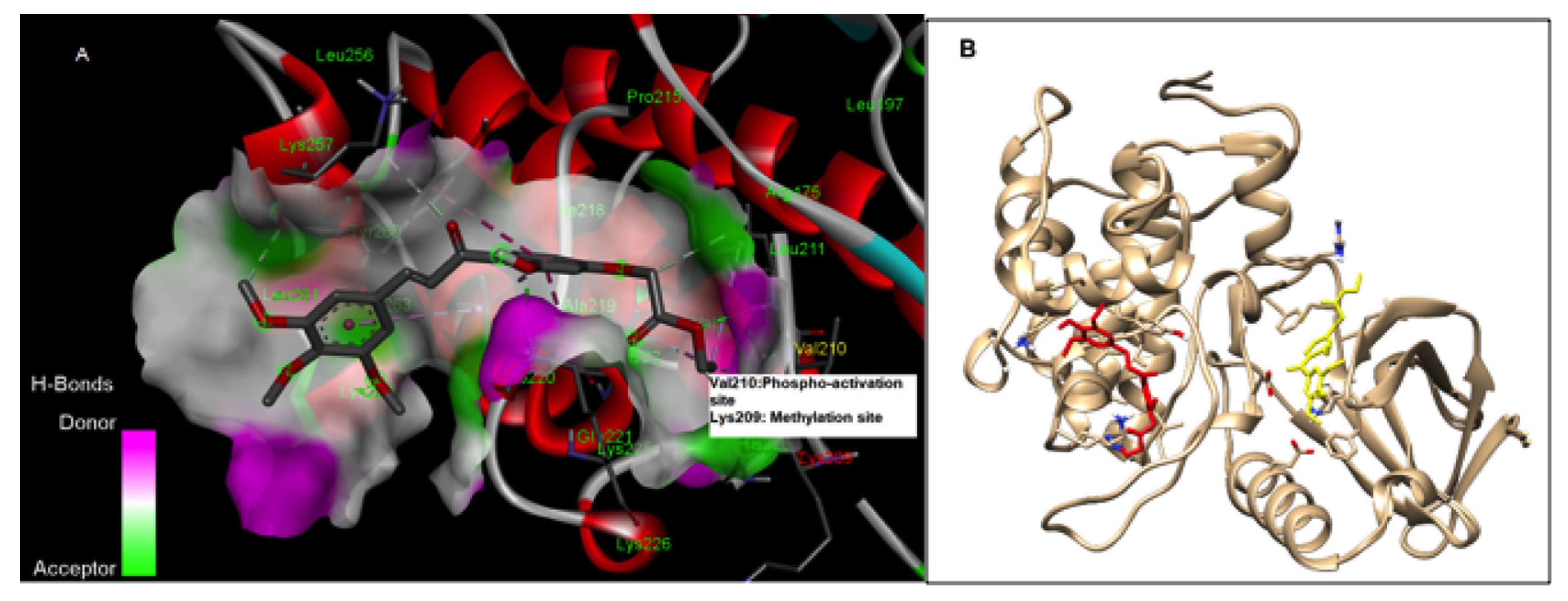
3.2. RK-10 Targets Critical Amino Acids Required for PLK1 Activation
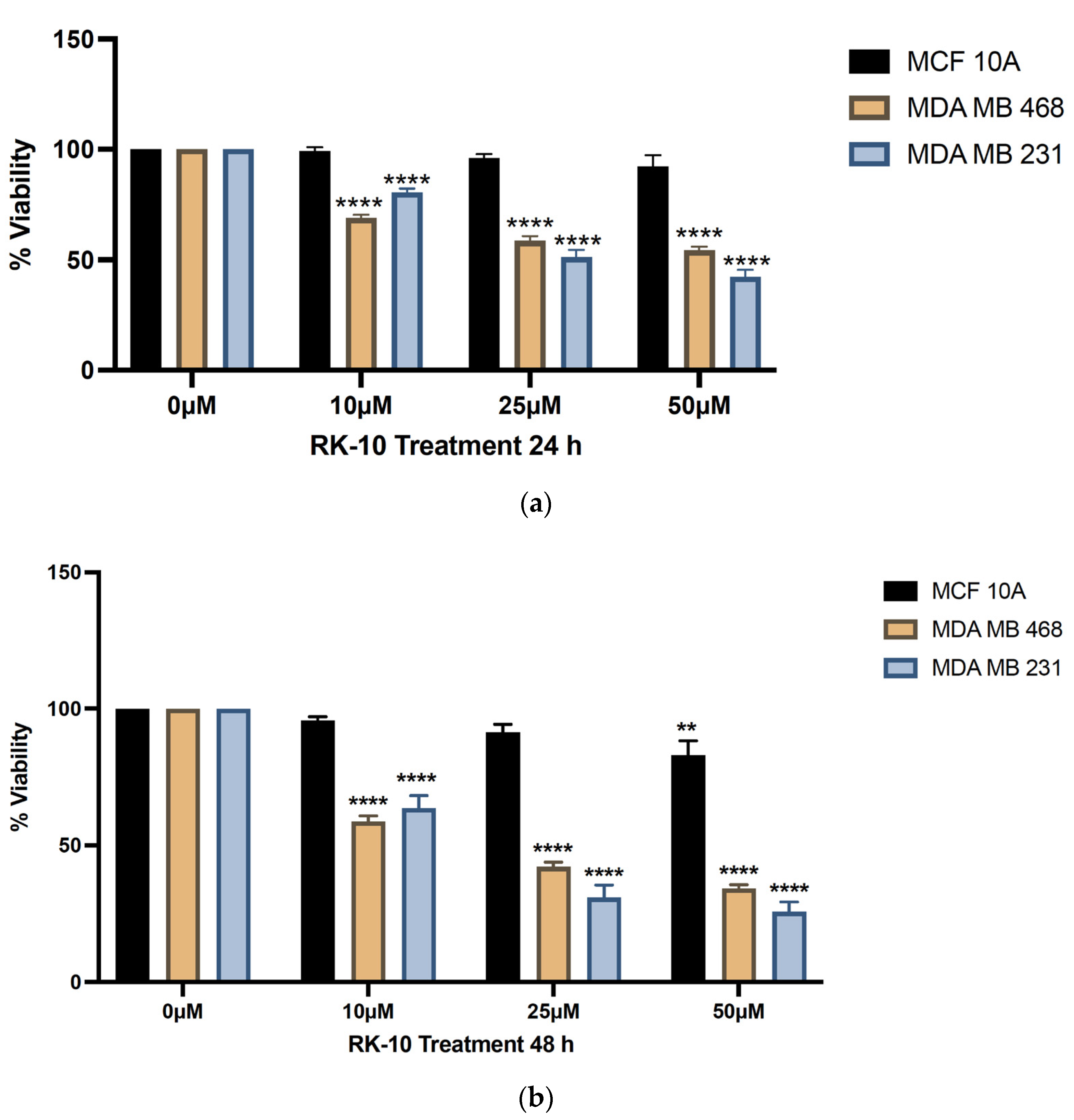
3.3. Novel Allosteric Inhibitor Decreases the Viability of TNBC Cells
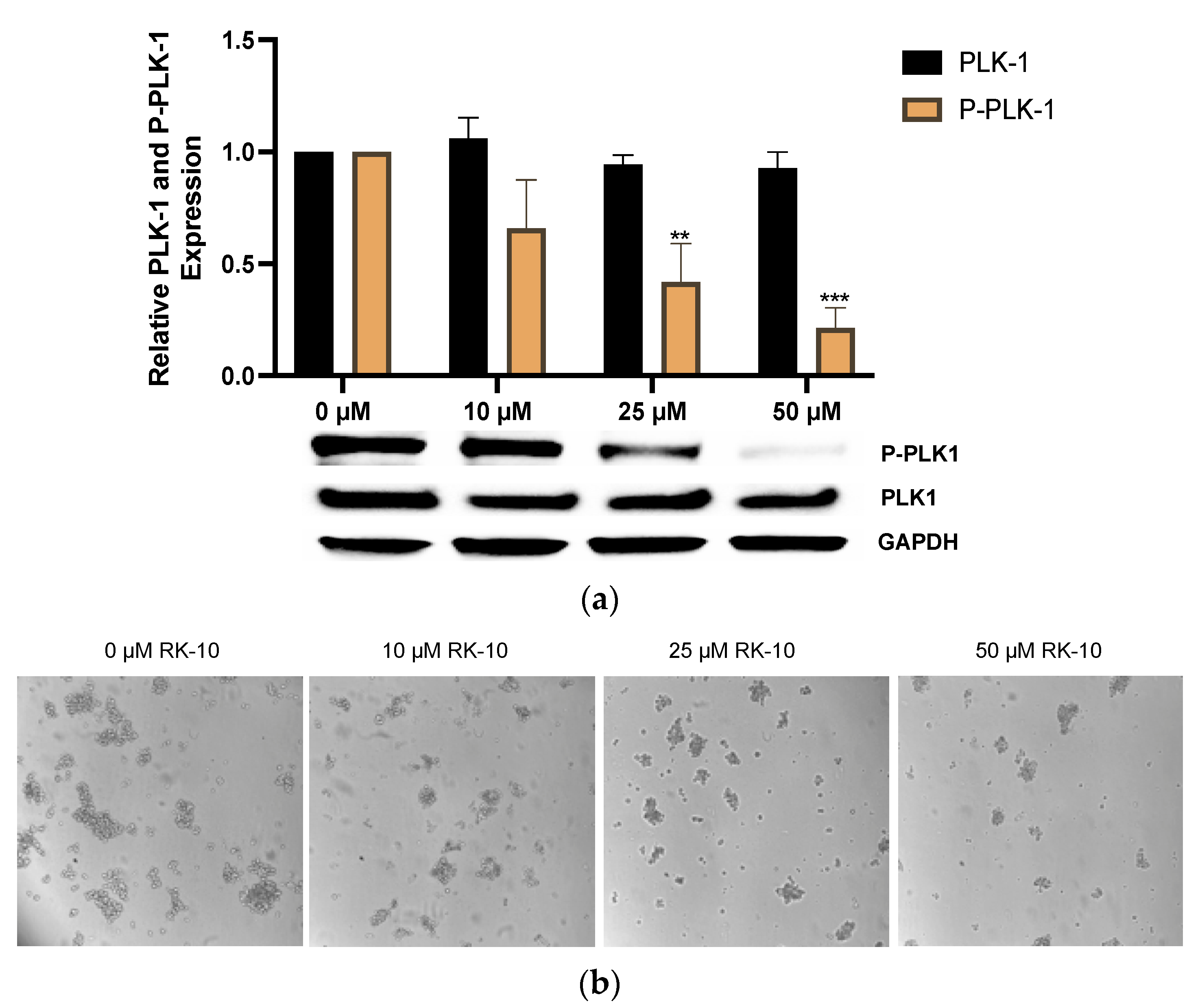
3.4. RK-10 Inhibits Phosphorylated PLK1 Protein Expression
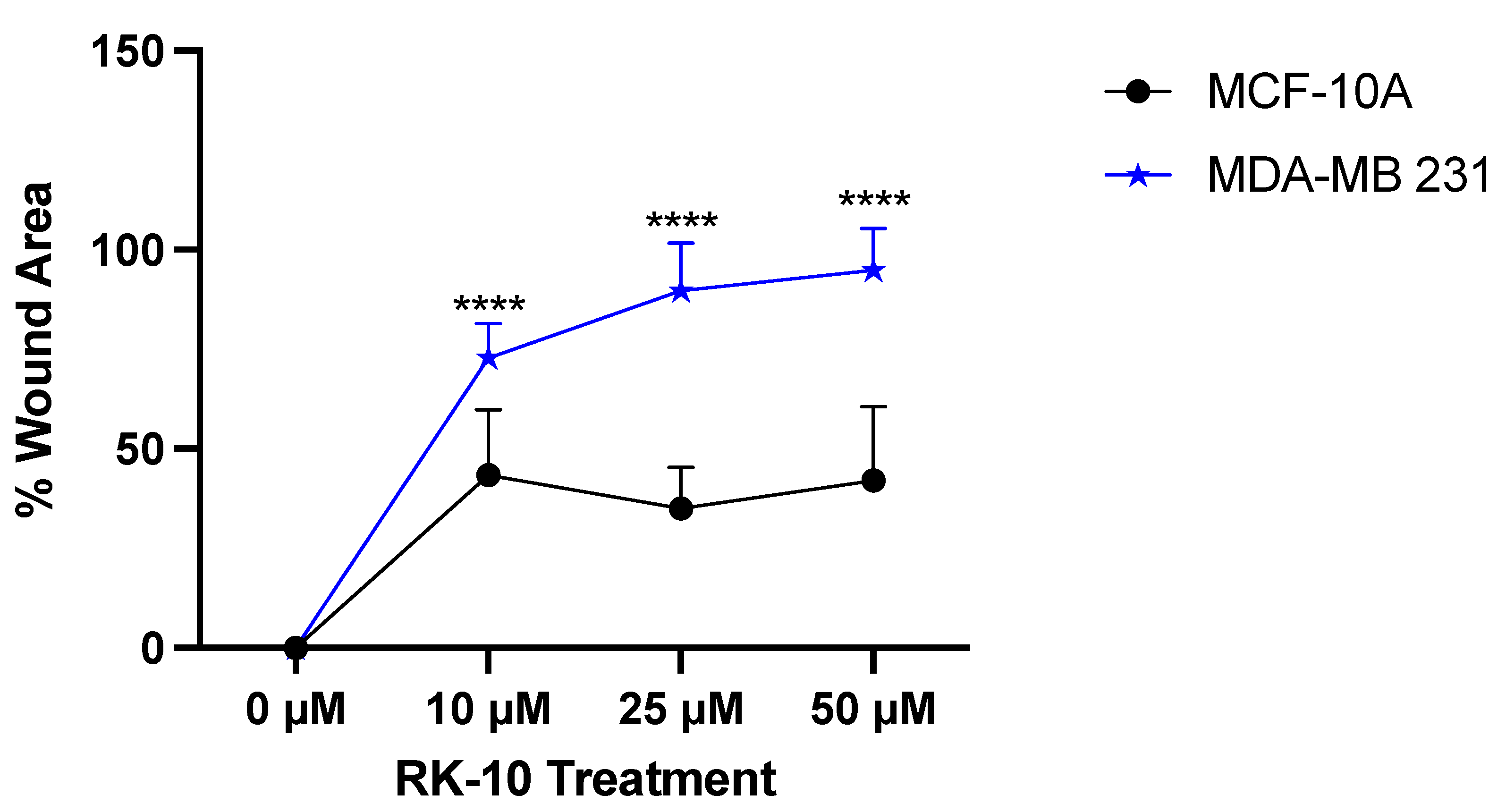
3.5. RK-10 Attenuates Cell Motility
3.6. RK-10 Selectively Retards Activation of PLK1 in TNBC Mammospheres
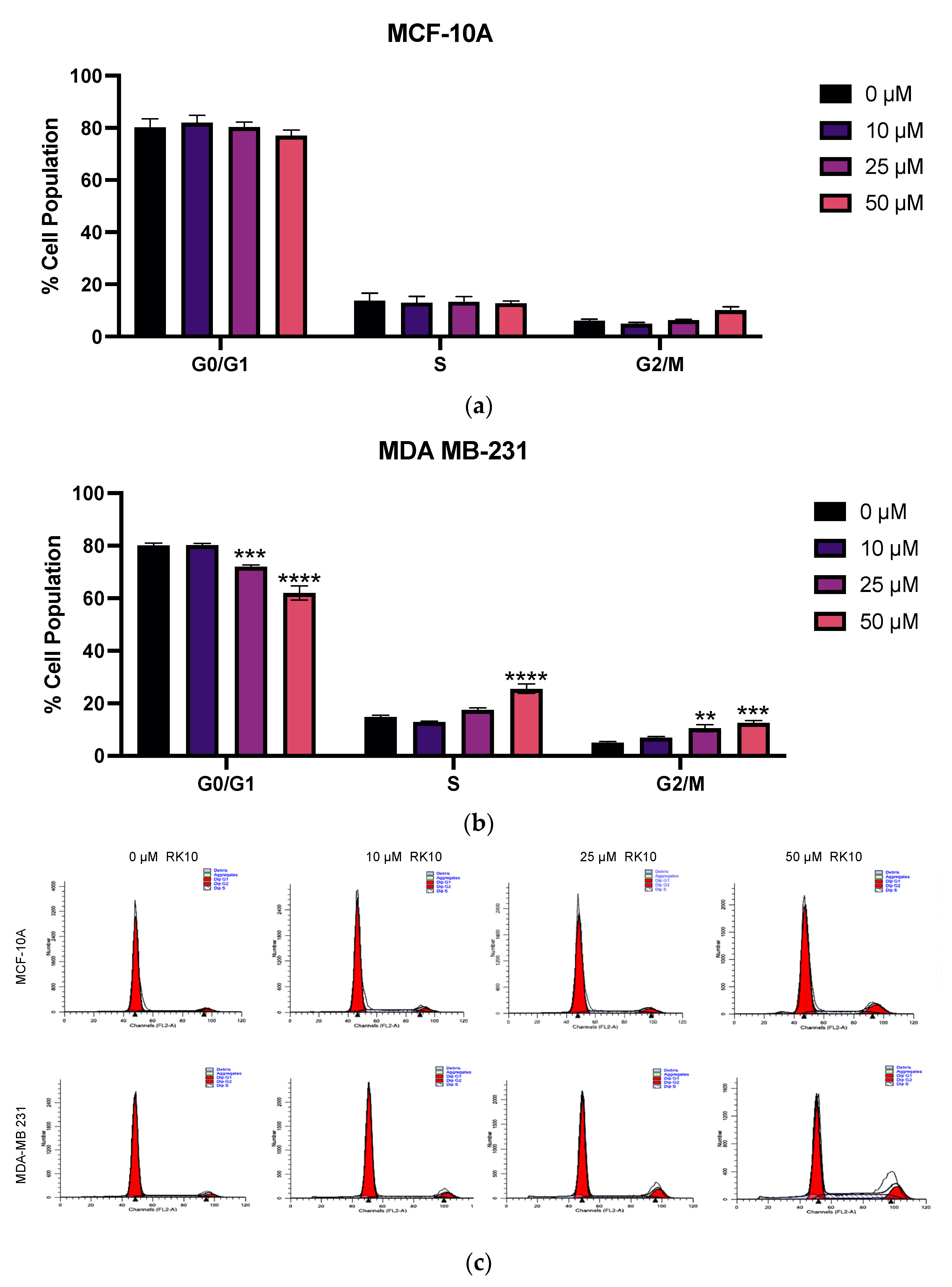
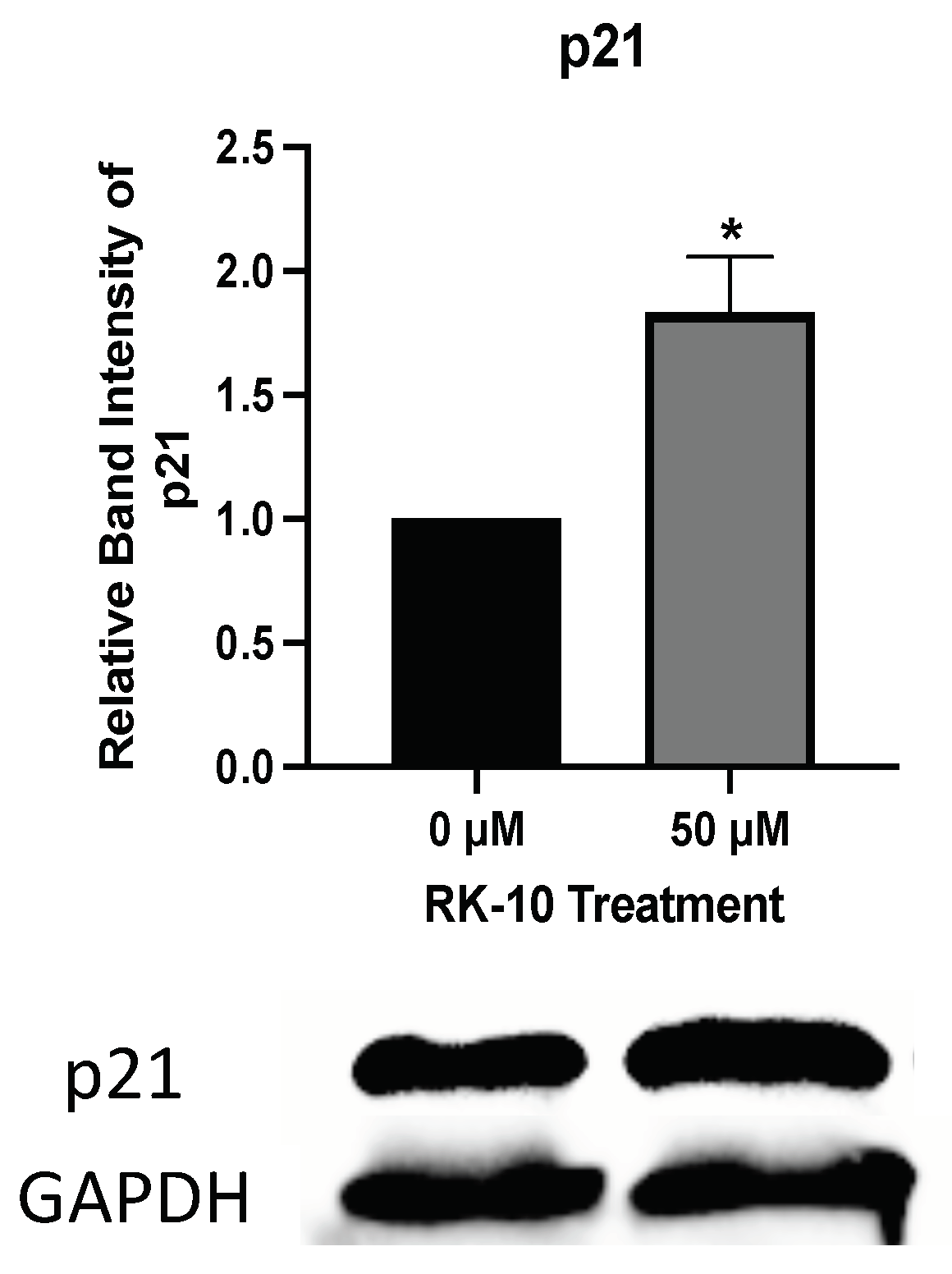
3.7. RK-10 Induces S Phase and G2/M Cell Cycle Arrest
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kappler, C.S.; Guest, S.T.; Irish, J.C.; Garrett-Mayer, E.; Kratche, Z.; Wilson, R.C.; Ethier, S.P. Oncogenic signaling in amphiregulin and EGFR-expressing PTEN-null human breast cancer. Mol. Oncol. 2015, 9, 527–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Braud, F.; Cascinu, S.; Spitaleri, G.; Pilz, K.; Clementi, L.; Liu, D.; Sikken, P.; De Pas, T. A phase I, dose-escalation study of volasertib combined with nintedanib in advanced solid tumors. Ann. Oncol. 2015, 26, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Ridinger, M.; Lin, T.L.; Becker, P.S.; Schiller, G.J.; Patel, P.A.; Spira, A.I.; Tsai, M.L.; Samuëlsz, E.; Silberman, S.L.; et al. A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 6132–6140. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.Y.; Zhao, X.; Duan, H.; Cheng, B.; Liu, Y.; Zhao, M.; Shu, W.; Mei, Y.; Wen, Z.; et al. A methylation-phosphorylation switch determines Plk1 kinase activity and function in DNA damage repair. Sci. Adv. 2019, 5, eaau7566. [Google Scholar] [CrossRef] [Green Version]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.; Lánczky, A.; Szállási, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Gallegos, K.M.; Walker, R.R.; Davidson, A.M.; Davenport, I.; Tilghman, S.L. Mammospheres of letrozole-resistant breast cancer cells enhance breast cancer aggressiveness. Oncol. Lett. 2021, 22, 620. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Jimenez, C.; Galan-Moya, E.M.; Corrales-Sanchez, V.; Noblejas-Lopez, M.D.M.; Burgos, M.; Domingo, B.; Montero, J.C.; Gomez-Juarez, M.; Picazo-Martinez, M.G.; Esparis-Ogando, A.; et al. Inhibition of the mitotic kinase PLK1 overcomes therapeutic resistance to BET inhibitors in triple negative breast cancer. Cancer Lett. 2020, 491, 50–59. [Google Scholar] [CrossRef]
- Fu, Z.; Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers 2017, 9, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.; Law, J.H.; Fotovati, A.; Dunn, S.E. Small interfering RNA library screen identified polo-like kinase-1 (PLK1) as a potential therapeutic target for breast cancer that uniquely eliminates tumor-initiating cells. Breast Cancer Res. 2012, 14, R22. [Google Scholar] [CrossRef] [Green Version]
- Maire, V.; Némati, F.; Richardson, M.; Vincent-Salomon, A.; Tesson, B.; Rigaill, G.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Lang, G.; et al. Polo-like kinase 1: A potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013, 73, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [CrossRef]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gallegos, K.M.; Patel, J.R.; Llopis, S.D.; Walker, R.R.; Davidson, A.M.; Zhang, W.; Zhang, K.; Tilghman, S.L. Quantitative Proteomic Profiling Identifies a Potential Novel Chaperone Marker in Resistant Breast Cancer. Front Oncol. 2021, 11, 540134. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Tavares, A.A.; Hagan, I.M.; Nigg, E.A.; Glover, D.M. The mitotic roles of Polo-like kinase. J. Cell Sci. 2001, 114, 2357–2358. [Google Scholar] [CrossRef]
- Murugan, R.N.; Park, J.E.; Kim, E.H.; Shin, S.Y.; Cheong, C.; Lee, K.S.; Bang, J.K. Plk1-targeted small molecule inhibitors: Molecular basis for their potency and specificity. Mol. Cells 2011, 32, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöffski, P.; Blay, J.Y.; De Greve, J.; Brain, E.; Machiels, J.P.; Soria, J.C.; Sleijfer, S.; Wolter, P.; Ray-Coquard, I.; Fontaine, C.; et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI). Eur. J. Cancer 2010, 46, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.; Vijay, G.V.; Soundararajan, R.; Yu, X.; Symmans, W.F.; Sphyris, N.; Mani, S.A. FOXC2 regulates the G2/M transition of stem cell-rich breast cancer cells and sensitizes them to PLK1 inhibition. Sci. Rep. 2016, 6, 23070. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, J.R.; Thangavelu, P.; Terrell, R.M.; Israel, B.; Sarkar, A.B.; Davidson, A.M.; Zhang, K.; Khupse, R.; Tilghman, S.L. A Novel Allosteric Inhibitor Targets PLK1 in Triple Negative Breast Cancer Cells. Biomolecules 2022, 12, 531. https://doi.org/10.3390/biom12040531
Patel JR, Thangavelu P, Terrell RM, Israel B, Sarkar AB, Davidson AM, Zhang K, Khupse R, Tilghman SL. A Novel Allosteric Inhibitor Targets PLK1 in Triple Negative Breast Cancer Cells. Biomolecules. 2022; 12(4):531. https://doi.org/10.3390/biom12040531
Chicago/Turabian StylePatel, Jankiben R., Prasad Thangavelu, Renee M. Terrell, Bridg’ette Israel, Arindam Basu Sarkar, A. Michael Davidson, Kun Zhang, Rahul Khupse, and Syreeta L. Tilghman. 2022. "A Novel Allosteric Inhibitor Targets PLK1 in Triple Negative Breast Cancer Cells" Biomolecules 12, no. 4: 531. https://doi.org/10.3390/biom12040531
APA StylePatel, J. R., Thangavelu, P., Terrell, R. M., Israel, B., Sarkar, A. B., Davidson, A. M., Zhang, K., Khupse, R., & Tilghman, S. L. (2022). A Novel Allosteric Inhibitor Targets PLK1 in Triple Negative Breast Cancer Cells. Biomolecules, 12(4), 531. https://doi.org/10.3390/biom12040531