
A Structural Perspective on Calprotectin as a Ligand of Receptors Mediating Inflammation and Potential Drug Target


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
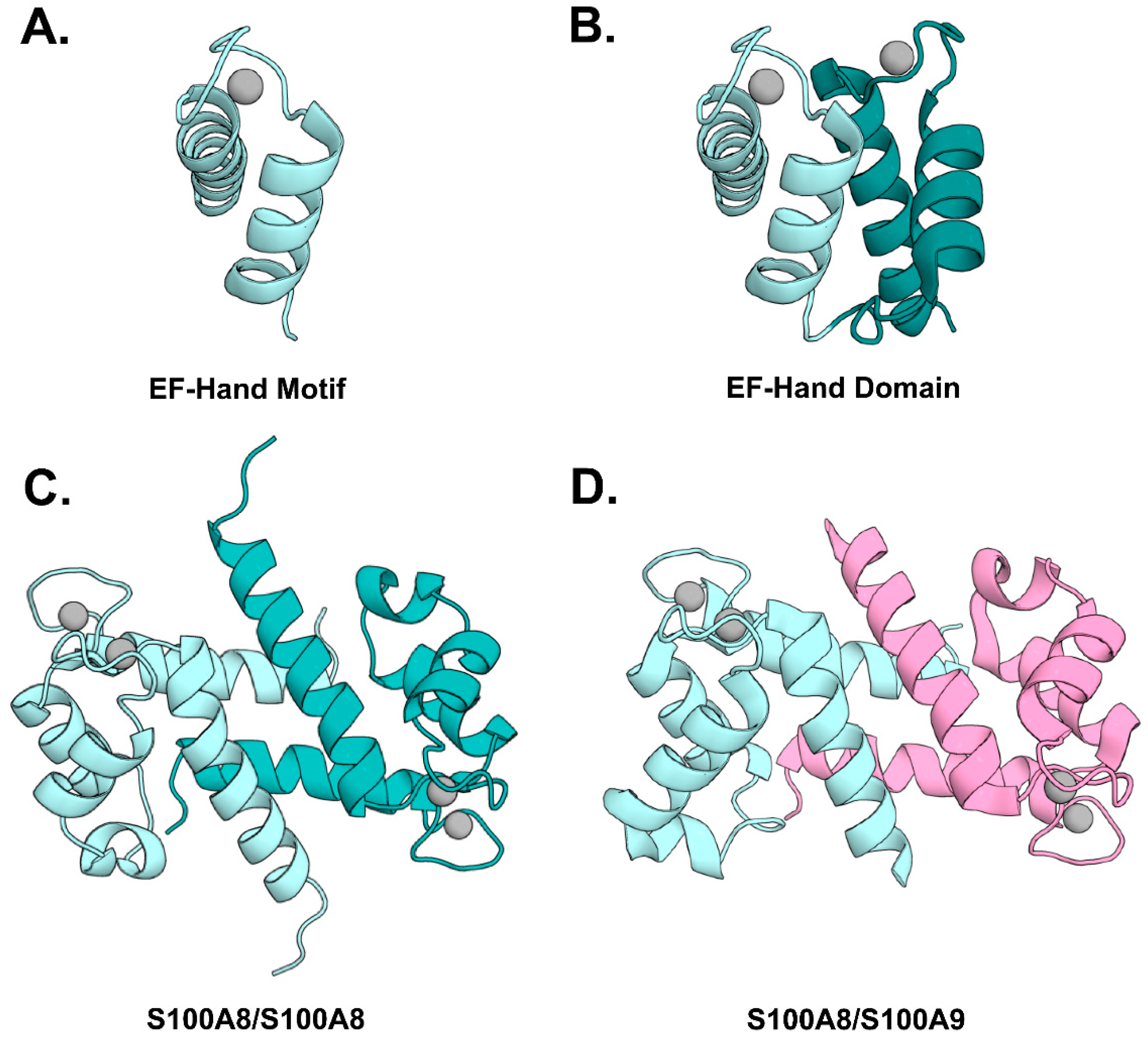
2. Biochemical and Structural Properties of CP
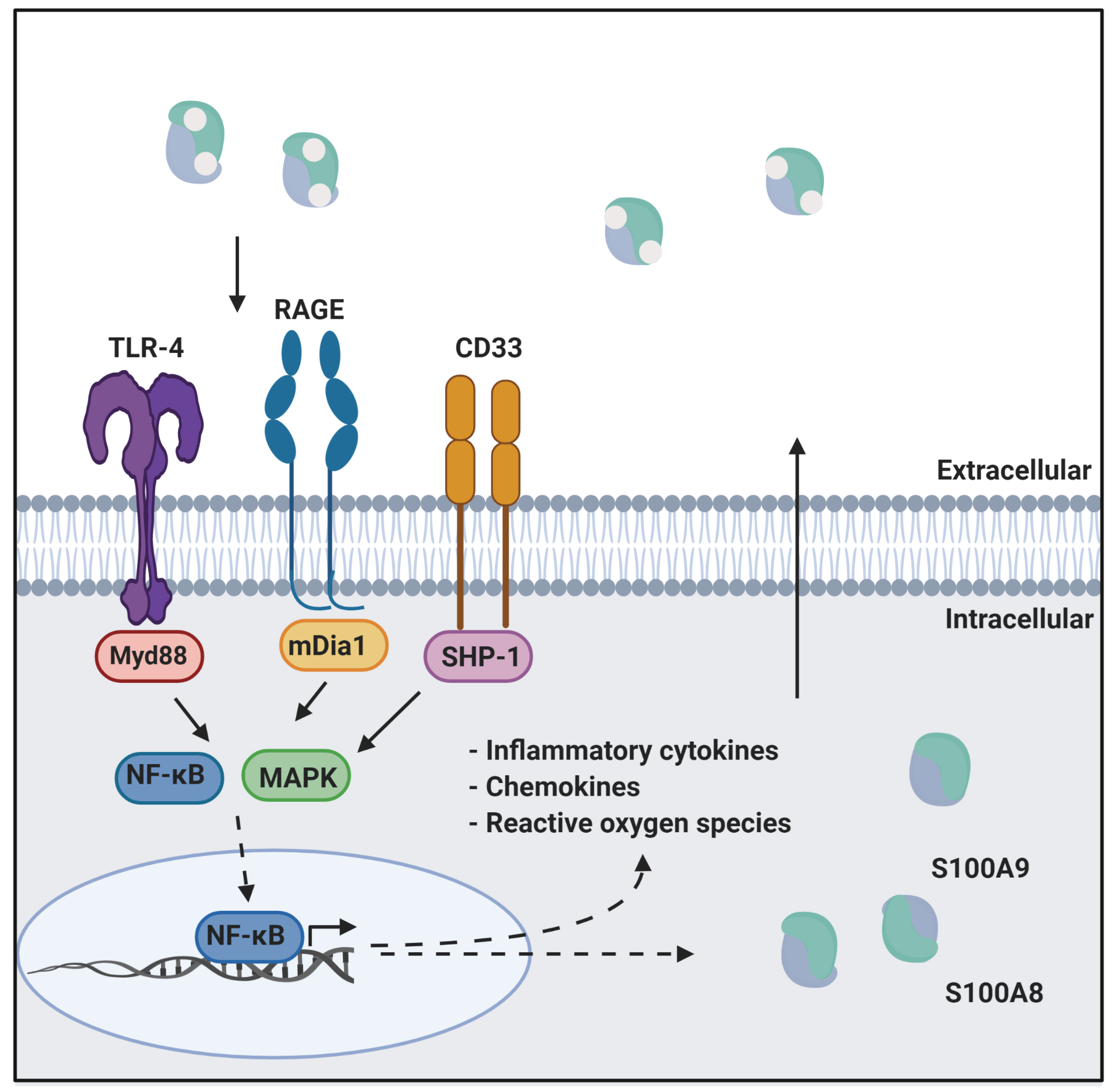
3. CP as a Ligand of Inflammatory Receptors
3.1. RAGE
3.2. TLR4
3.3. CD33
4. CP as a Drug Target
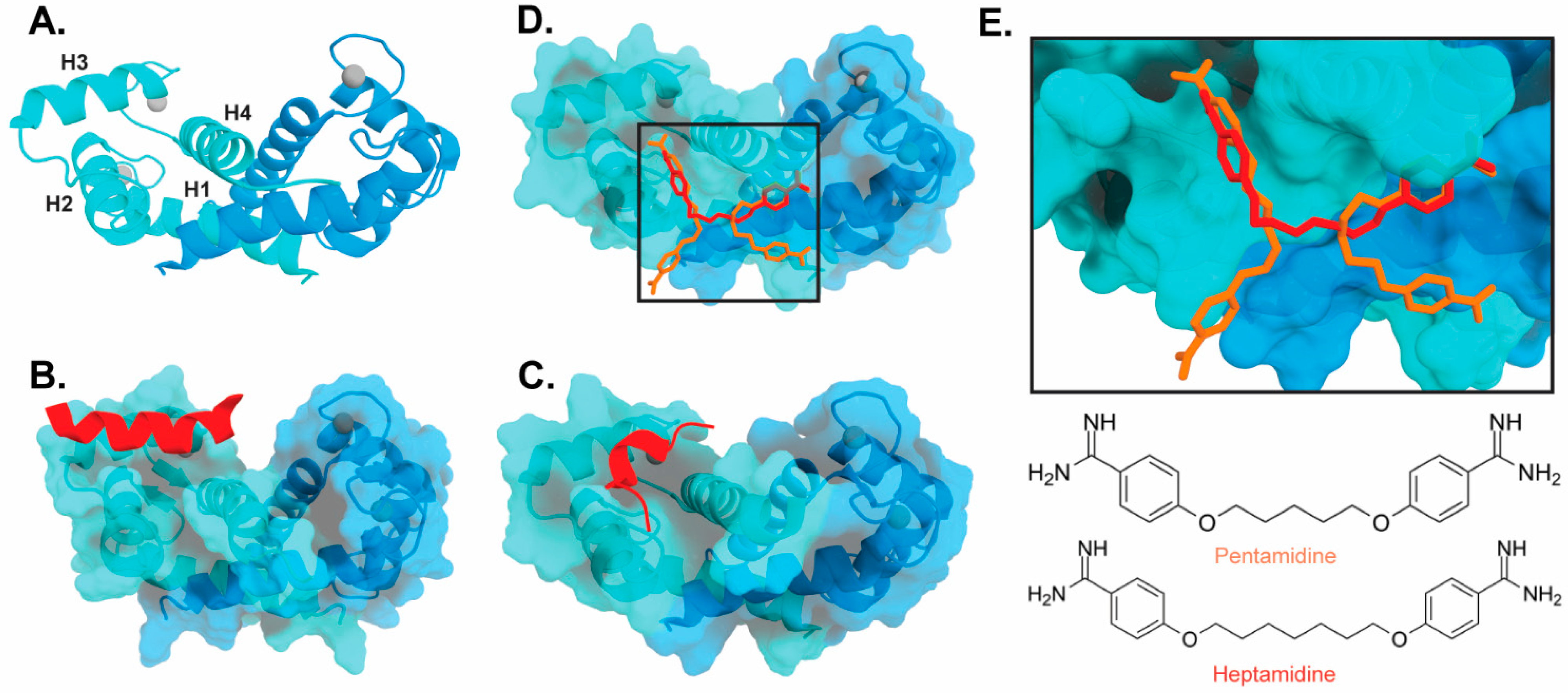
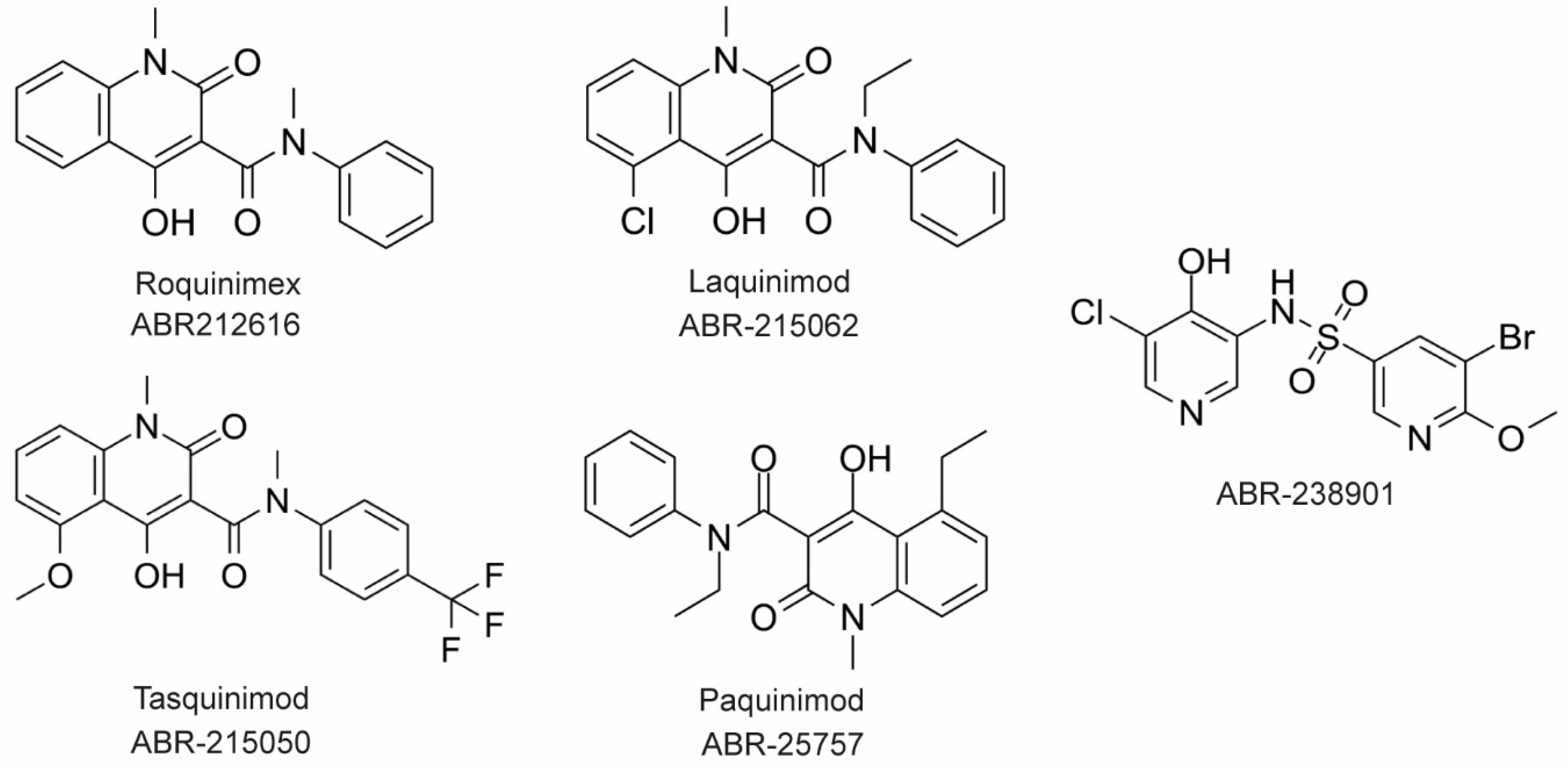
4.1. Inhibitors of S100 Proteins
4.2. Inhibitors of Calprotectin
4.3. Receptor-Targeted Drugs
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zackular, J.P.; Chazin, W.J.; Skaar, E.P. Nutritional Immunity: S100 Proteins at the Host-Pathogen Interface. J. Biol. Chem. 2015, 290, 18991–18998. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Schmidt, A.M. Characterization and Functional Analysis of the Promoter of RAGE, the Receptor for Advanced Glycation End Products. J. Biol. Chem. 1997, 272, 16498–16506. [Google Scholar] [CrossRef] [PubMed]
- Eggers, K.; Sikora, K.; Lorenz, M.; Taubert, T.; Moobed, M.; Baumann, G.; Stangl, K.; Stangl, V. RAGE-dependent regulation of calcium-binding proteins S100A8 and S100A9 in human THP-1. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2011, 119, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Roth, J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. 2004, 50, 3762–3771. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Heizmann, C.W. S100 proteins: Diagnostic and prognostic biomarkers in laboratory medicine. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 1197–1206. [Google Scholar] [CrossRef]
- Atreya, R.; Neurath, M.F. Molecular pathways controlling barrier function in IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 67–68. [Google Scholar] [CrossRef]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Nanini, H.F.; Bernardazzi, C.; Castro, F.; de Souza, H.S.P. Damage-associated molecular patterns in inflammatory bowel disease: From biomarkers to therapeutic targets. World J. Gastroenterol. 2018, 24, 4622–4634. [Google Scholar] [CrossRef] [PubMed]
- Body-Malapel, M.; Djouina, M.; Waxin, C.; Langlois, A.; Gower-Rousseau, C.; Zerbib, P.; Schmidt, A.M.; Desreumaux, P.; Boulanger, E.; Vignal, C. The RAGE signaling pathway is involved in intestinal inflammation and represents a promising therapeutic target for Inflammatory Bowel Diseases. Mucosal Immunol. 2019, 12, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, E.; Subramaniam, R.; Okada, T.; Mizoguchi, A. A Review of Selected IBD Biomarkers: From Animal Models to Bedside. Diagnostics 2021, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Harbord, M.; Annese, V.; Vavricka, S.R.; Allez, M.; Barreiro-de Acosta, M.; Boberg, K.M.; Burisch, J.; De Vos, M.; De Vries, A.-M.; Dick, A.D.; et al. The First European Evidence-based Consensus on Extra-intestinal Manifestations in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.W. A soluble protein characteristic of the nervous system. Biochem. Biophys. Res. Commun. 1965, 19, 739–744. [Google Scholar] [CrossRef]
- Schäfer, B.W.; Wicki, R.; Engelkamp, D.; Mattei, M.-G.; Heizmann, C.W. Isolation of a YAC clone covering a cluster of nine S100 genes on human chromosome 1q21: Rationale for a new nomenclature of the S100 calcium-binding protein family. Genomics 1995, 25, 638–643. [Google Scholar] [CrossRef]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef]
- Wheeler, L.C.; Donor, M.T.; Prell, J.S.; Harms, M.J. Multiple Evolutionary Origins of Ubiquitous Cu2+ and Zn2+ Binding in the S100 Protein Family. PLoS ONE 2016, 11, e0164740. [Google Scholar] [CrossRef]
- Strynadka, N.C.J.; James, M.N.G. Crystal Structures of the Helix-loop-helix Calcium-binding Proteins. Annu. Rev. Biochem. 1989, 58, 951–999. [Google Scholar] [CrossRef]
- Potts, B.C.M.; Smith, J.; Akke, M.; Macke, T.J.; Okazaki, K.; Hidaka, H.; Case, D.A.; Chazin, W.J. The structure of calcyclin reveals a novel homodimeric fold for S100 Ca2+-binding proteins. Nat. Struct. Biol. 1995, 2, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.J.; Chazin, W.J. High Level Expression and Dimer Characterization of the S100 EF-hand Proteins, Migration Inhibitory Factor-related Proteins 8 and 14. J. Biol. Chem. 1998, 273, 12427–12435. [Google Scholar] [CrossRef] [PubMed]
- Silvers, R.; Stephan, J.R.; Griffin, R.G.; Nolan, E.M. Molecular Basis of Ca(II)-Induced Tetramerization and Transition-Metal Sequestration in Human Calprotectin. J. Am. Chem. Soc. 2021, 143, 18073–18090. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.R.; Nolan, E.M. Calcium-induced tetramerization and zinc chelation shield human calprotectin from degradation by host and bacterial extracellular proteases. Chem. Sci. 2016, 7, 1962–1975. [Google Scholar] [CrossRef] [PubMed]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef] [PubMed]
- Zygiel, E.M.; Nolan, E.M. Transition Metal Sequestration by the Host-Defense Protein Calprotectin. Annu. Rev. Biochem. 2018, 87, 621–643. [Google Scholar] [CrossRef] [PubMed]
- Nakashige, T.G.; Zhang, B.; Krebs, C.; Nolan, E.M. Human calprotectin is an iron-sequestering host-defense protein. Nat. Chem. Biol. 2015, 11, 765–771. [Google Scholar] [CrossRef]
- Moroz, O.V.; Burkitt, W.; Wittkowski, H.; He, W.; Ianoul, A.; Novitskaya, V.; Xie, J.; Polyakova, O.; Lednev, I.K.; Shekhtman, A.; et al. Both Ca2+ and Zn2+ are essential for S100A12 protein oligomerization and function. BMC Biochem. 2009, 10, 11. [Google Scholar] [CrossRef]
- Hibino, T.; Sakaguchi, M.; Miyamoto, S.; Yamamoto, M.; Motoyama, A.; Hosoi, J.; Shimokata, T.; Ito, T.; Tsuboi, R.; Huh, N.-H. S100A9 Is a Novel Ligand of EMMPRIN That Promotes Melanoma Metastasis. Cancer Res. 2013, 73, 172–183. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Yamamoto, M.; Miyai, M.; Maeda, T.; Hiruma, J.; Murata, H.; Kinoshita, R.; Winarsa Ruma, I.M.; Putranto, E.W.; Inoue, Y.; et al. Identification of an S100A8 Receptor Neuroplastin-β and its Heterodimer Formation with EMMPRIN. J. Investig. Dermatol. 2016, 136, 2240–2250. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Andrassy, M.; Igwe, J.; Autschbach, F.; Volz, C.; Remppis, A.; Neurath, M.F.; Schleicher, E.; Humpert, P.M.; Wendt, T.; Liliensiek, B.; et al. Posttranslationally Modified Proteins as Mediators of Sustained Intestinal Inflammation. Am. J. Pathol. 2006, 169, 1223–1237. [Google Scholar] [CrossRef] [PubMed]
- Dattilo, B.M.; Fritz, G.; Leclerc, E.; Vander Kooi, C.W.; Heizmann, C.W.; Chazin, W.J. The Extracellular Region of the Receptor for Advanced Glycation End Products Is Composed of Two Independent Structural Units. Biochemistry 2007, 46, 6957–6970. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural basis for ligand recognition and activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef]
- Chiou, J.W.; Fu, B.; Chou, R.-H.; Yu, C. Blocking the Interactions between Calcium-Bound S100A12 Protein and the V Domain of RAGE Using Tranilast. PLoS ONE 2016, 11, e0162000. [Google Scholar] [CrossRef]
- Mohan, S.K.; Gupta, A.A.; Yu, C. Interaction of the S100A6 mutant (C3S) with the V domain of the receptor for advanced glycation end products (RAGE). Biochem. Biophys. Res. Commun. 2013, 434, 328–333. [Google Scholar] [CrossRef]
- Yatime, L.; Betzer, C.; Jensen, R.K.; Mortensen, S.; Jensen, P.H.; Andersen, G.R. The Structure of the RAGE:S100A6 Complex Reveals a Unique Mode of Homodimerization for S100 Proteins. Structure 2016, 24, 2043–2052. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Weibel, M.; Heizmann, C.W.; Galichet, A. S100B and S100A6 Differentially Modulate Cell Survival by Interacting with Distinct RAGE (Receptor for Advanced Glycation End Products) Immunoglobulin Domains. J. Biol. Chem. 2007, 282, 31317–31331. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Ostendorp, T.; Leclerc, E.; Galichet, A.; Koch, M.; Demling, N.; Weigle, B.; Heizmann, C.W.; Kroneck, P.M.H.; Fritz, G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007, 26, 3868–3878. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Laird, M.H.; Rhee, S.H.; Perkins, D.J.; Medvedev, A.E.; Piao, W.; Fenton, M.J.; Vogel, S.N. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 2009, 85, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Mineev, K.S.; Goncharuk, S.A.; Goncharuk, M.V.; Volynsky, P.E.; Novikova, E.V.; Aresinev, A.S. Spatial structure of TLR4 transmembrane domain in bicelles provides the insight into the receptor activation mechanism. Sci. Rep. 2017, 7, 6864. [Google Scholar] [CrossRef]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-like Receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef]
- Ohnishi, H.; Tochio, H.; Kato, Z.; Orii, K.E.; Li, A.; Kimura, T.; Hiroaki, H.; Kondo, N.; Shirakawa, M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 10260–10265. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Loes, A.N.; Bridgham, J.T.; Harms, M.J. Coevolution of the Toll-Like Receptor 4 Complex with Calgranulins and Lipopolysaccharide. Front. Immunol. 2018, 9, 304. [Google Scholar] [CrossRef]
- Deguchi, A.; Tomita, T.; Ohto, U.; Takemura, K.; Kitao, A.; Akashi-Takamura, S.; Miyake, K.; Maru, Y. Eritoran inhibits S100A8-mediated TLR4/MD-2 activation and tumor growth by changing the immune microenvironment. Oncogene 2016, 35, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Kessel, C.; Fuehner, S.; Zell, J.; Zimmermann, B.; Drewianka, S.; Brockmeyer, S.; Holzinger, D.; Hinze, C.; Wittkowski, H.; Foell, D. Calcium and zinc tune autoinflammatory Toll-like receptor 4 signaling by S100A12. J. Allergy Clin. Immunol. 2018, 142, 1370–1373.e8. [Google Scholar] [CrossRef]
- Vogl, T.; Stratis, A.; Wixler, V.; Völler, T.; Thurainayagam, S.; Jorch, S.K.; Zenker, S.; Dreiling, A.; Chakraborty, D.; Fröhling, M.; et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J. Clin. Investig. 2018, 128, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef]
- Alphey, M.S.; Attrill, H.; Crocker, P.R.; van Aalten, D.M.F. High Resolution Crystal Structures of Siglec-7: Insights into Ligand Specificity in the Siglec Family. J. Biol. Chem. 2003, 278, 3372–3377. [Google Scholar] [CrossRef]
- Zhao, F.; Hoechst, B.; Duffy, A.; Gamrekelashvili, J.; Fioravanti, S.; Manns, M.P.; Greten, T.F.; Korangy, F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology 2012, 136, 176–183. [Google Scholar] [CrossRef]
- Cluzeau, T.; McGraw, K.L.; Irvine, B.; Masala, E.; Ades, L.; Basiorka, A.A.; Maciejewski, J.; Auberger, P.; Wei, S.; Fenaux, P.; et al. Pro-inflammatory proteins S100A9 and tumor necrosis factor-α suppress erythropoietin elaboration in myelodysplastic syndromes. Haematologica 2017, 102, 2015–2020. [Google Scholar] [CrossRef]
- Mondet, J.; Chevalier, S.; Mossuz, P. Pathogenic Roles of S100A8 and S100A9 Proteins in Acute Myeloid and Lymphoid Leukemia: Clinical and Therapeutic Impacts. Molecules 2021, 26, 1323. [Google Scholar] [CrossRef]
- Shishibori, T.; Oyama, Y.; Matsushita, O.; Yamashita, K.; Furuichi, H.; Okabe, A.; Hata, Y.; Kobayashi, R. P–159—Three Distinct Anti-allergic Drugs, Amlexanox, Cromolyn, and Tranilast, Bind to S100A12 and A13 of S100 Family. Jpn. J. Pharmacol. 1999, 79, 161. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Logsdon, C.D. Effect of Cromolyn on S100P Interactions With RAGE and Pancreatic Cancer Growth and Invasion in Mouse Models. JNCI J. Natl. Cancer Inst. 2006, 98, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Sun, D.; Peng, Z.; Pal, A.; Maxwell, D.S.; Bornmann, W.G.; Logsdon, C.D. Designing and developing S100P inhibitor 5-methyl cromolyn for pancreatic cancer therapy. Mol. Cancer Ther. 2013, 12, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Chiang, S.-H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef]
- Marshak, D.R.; Lukas, T.J.; Watterson, D.M. Drug-protein interactions: Binding of chlorpromazine to calmodulin, calmodulin fragments, and related calcium binding proteins. Biochemistry 1985, 24, 144–150. [Google Scholar] [CrossRef]
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629. [Google Scholar] [CrossRef]
- Pingerelli, P.L.; Mizukami, H.; Wagner, A.S.; Bartnicki, D.E.; Oliver, J.P. Investigation of the Ca2+-dependent interaction of trifluoperazine with S100a: A19F NMR and circular dichroism study. J. Protein Chem. 1990, 9, 169–175. [Google Scholar] [CrossRef]
- Wilder, P.T.; Charpentier, T.H.; Liriano, M.A.; Gianni, K.; Varney, K.M.; Pozharski, E.; Coop, A.; Toth, E.A.; Mackerell, A.D.; Weber, D.J. In vitro screening and structural characterization of inhibitors of the S100B-p53 interaction. Int. J. High Throughput Screen. 2010, 2010, 109–126. [Google Scholar] [CrossRef][Green Version]
- McKnight, L.E.; Raman, E.P.; Bezawada, P.; Kudrimoti, S.; Wilder, P.T.; Hartman, K.G.; Godoy-Ruiz, R.; Toth, E.A.; Coop, A.; MacKerell, A.D.; et al. Structure-Based Discovery of a Novel Pentamidine-Related Inhibitor of the Calcium-Binding Protein S100B. ACS Med. Chem. Lett. 2012, 3, 975–979. [Google Scholar] [CrossRef]
- Young, B.D.; Yu, W.; Rodríguez, D.J.V.; Varney, K.M.; MacKerell, A.D.; Weber, D.J. Specificity of Molecular Fragments Binding to S100B versus S100A1 as Identified by NMR and Site Identification by Ligand Competitive Saturation (SILCS). Molecules 2021, 26, 381. [Google Scholar] [CrossRef]
- Garrett, S.C.; Hodgson, L.; Rybin, A.; Toutchkine, A.; Hahn, K.M.; Lawrence, D.S.; Bresnick, A.R. A Biosensor of S100A4 Metastasis Factor Activation: Inhibitor Screening and Cellular Activation Dynamics. Biochemistry 2008, 47, 986–996. [Google Scholar] [CrossRef][Green Version]
- Malashkevich, V.N.; Dulyaninova, N.G.; Ramagopal, U.A.; Liriano, M.A.; Varney, K.M.; Knight, D.; Brenowitz, M.; Weber, D.J.; Almo, S.C.; Bresnick, A.R. Phenothiazines inhibit S100A4 function by inducing protein oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 8605. [Google Scholar] [CrossRef] [PubMed]
- Agamennone, M.; Cesari, L.; Lalli, D.; Turlizzi, E.; Del Conte, R.; Turano, P.; Mangani, S.; Padova, A. Fragmenting the S100B–p53 Interaction: Combined Virtual/Biophysical Screening Approaches to Identify Ligands. ChemMedChem 2010, 5, 428–435. [Google Scholar] [CrossRef]
- Reddy, T.R.K.; Li, C.; Fischer, P.M.; Dekker, L.V. Three-Dimensional Pharmacophore Design and Biochemical Screening Identifies Substituted 1,2,4-Triazoles as Inhibitors of the Annexin A2–S100A10 Protein Interaction. ChemMedChem 2012, 7, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Camara, R.; Ogbeni, D.; Gerstmann, L.; Ostovar, M.; Hurer, E.; Scott, M.; Mahmoud, N.G.; Radon, T.; Crnogorac-Jurcevic, T.; Patel, P.; et al. Discovery of novel small molecule inhibitors of S100P with in vitro anti-metastatic effects on pancreatic cancer cells. Eur. J. Med. Chem. 2020, 203, 112621. [Google Scholar] [CrossRef]
- Zimmer, D.B.; Weber, D.J. The Calcium-Dependent Interaction of S100B with Its Protein Targets. Cardiovasc. Psychiatry Neurol. 2010, 2010, 728052. [Google Scholar] [CrossRef]
- Stålhandske, T.; Eriksoo, E.; Sandberg, B.-M. A novel quinolinecarboxamide with interesting immunomodulatory activity. Int. J. Immunopharmacol. 1982, 4, 336. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Pili, R.; Qian, D.Z.; Dalrymple, S.L.; Garrison, J.B.; Kyprianou, N.; Björk, A.; Olsson, A.; Leanderson, T. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate 2006, 66, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Schiopu, A.; Marinkovic, G.; De Camp, L.; Winkler, L.; Mares, R.; Cotoi, O.S.; Nilsson, J.; Jovinge, S. P4026Short-term blockade of the S100A8/A9 alarmin in the immediate post-myocardial infarction period inhibits acute myocardial inflammation and preserves myocardial repair. Eur. Heart J. 2017, 38. [Google Scholar] [CrossRef]
- De Veirman, K.; De Beule, N.; Maes, K.; Menu, E.; De Bruyne, E.; De Raeve, H.; Fostier, K.; Moreaux, J.; Kassambara, A.; Hose, D.; et al. Extracellular S100A9 Protein in Bone Marrow Supports Multiple Myeloma Survival by Stimulating Angiogenesis and Cytokine Secretion. Cancer Immunol. Res. 2017, 5, 839–846. [Google Scholar] [CrossRef]
- Marinković, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Al Ustwani, O.; Shen, L.; Pili, R. Mechanism of action and clinical activity of tasquinimod in castrate-resistant prostate cancer. OncoTargets Ther. 2014, 7, 223–234. [Google Scholar] [CrossRef]
- Källberg, E.; Vogl, T.; Liberg, D.; Olsson, A.; Björk, P.; Wikström, P.; Bergh, A.; Roth, J.; Ivars, F.; Leanderson, T. S100A9 interaction with TLR4 promotes tumor growth. PLoS ONE 2012, 7, e34207. [Google Scholar] [CrossRef] [PubMed]
- Björk, P.; Björk, A.; Vogl, T.; Stenström, M.; Liberg, D.; Olsson, A.; Roth, J.; Ivars, F.; Leanderson, T. Identification of Human S100A9 as a Novel Target for Treatment of Autoimmune Disease via Binding to Quinoline-3-Carboxamides. PLoS Biol. 2009, 7, e1000097. [Google Scholar] [CrossRef]
- Bierhaus, A.; Stern, D.M.; Nawroth, P.P. RAGE in inflammation: A new therapeutic target? Curr. Opin. Investig. Drugs (Lond. Engl. 2000) 2006, 7, 985–991. [Google Scholar]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-Derived RAGE Antagonistic Peptide Reduces Tumor Growth and Metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an Oral Inhibitor of Receptor for Advanced Glycation End Products (RAGE), in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef]
- Shen, C.; Ma, Y.; Zeng, Z.; Yin, Q.; Hong, Y.; Hou, X.; Liu, X. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem. Res. 2017, 42, 2902–2911. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Devarapu, S.K.; Anders, H.-J. Toll-like receptors in lupus nephritis. J. Biomed. Sci. 2018, 25, 35. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (Resatorvid), a Small-Molecule Inhibitor of Toll-Like Receptor (TLR) 4 Signaling, Binds Selectively to TLR4 and Interferes with Interactions between TLR4 and Its Adaptor Molecules. Mol. Pharmacol. 2011, 79, 34. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-R.; Liu, J.; Zhang, S.-L.; Luo, T.; Wu, F.; Dong, J.-H.; Guo, Y.-J.; Zhao, L. Corilagin ameliorates the extreme inflammatory status in sepsis through TLR4 signaling pathways. BMC Complement. Altern. Med. 2017, 17, 18. [Google Scholar] [CrossRef]
- Wang, D.; Shi, J.; Lv, S.; Xu, W.; Li, J.; Ge, W.; Xiao, C.; Geng, D.; Liu, Y. Artesunate Attenuates Lipopolysaccharide-Stimulated Proinflammatory Responses by Suppressing TLR4, MyD88 Expression, and NF-κB Activation in Microglial Cells. Inflammation 2015, 38, 1925–1932. [Google Scholar] [CrossRef]
- Stein, E.M.; Walter, R.B.; Erba, H.P.; Fathi, A.T.; Advani, A.S.; Lancet, J.E.; Ravandi, F.; Kovacsovics, T.; DeAngelo, D.J.; Bixby, D.; et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. [Google Scholar] [CrossRef]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.N.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, V.; Perera, Y.R.; Chazin, W.J. A Structural Perspective on Calprotectin as a Ligand of Receptors Mediating Inflammation and Potential Drug Target. Biomolecules 2022, 12, 519. https://doi.org/10.3390/biom12040519
Garcia V, Perera YR, Chazin WJ. A Structural Perspective on Calprotectin as a Ligand of Receptors Mediating Inflammation and Potential Drug Target. Biomolecules. 2022; 12(4):519. https://doi.org/10.3390/biom12040519
Chicago/Turabian StyleGarcia, Velia, Yasiru Randika Perera, and Walter Jacob Chazin. 2022. "A Structural Perspective on Calprotectin as a Ligand of Receptors Mediating Inflammation and Potential Drug Target" Biomolecules 12, no. 4: 519. https://doi.org/10.3390/biom12040519
APA StyleGarcia, V., Perera, Y. R., & Chazin, W. J. (2022). A Structural Perspective on Calprotectin as a Ligand of Receptors Mediating Inflammation and Potential Drug Target. Biomolecules, 12(4), 519. https://doi.org/10.3390/biom12040519