
Nature-Inspired O-Benzyl Oxime-Based Derivatives as New Dual-Acting Agents Targeting Aldose Reductase and Oxidative Stress

 ,
,  ,
, 
 ,
, 
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
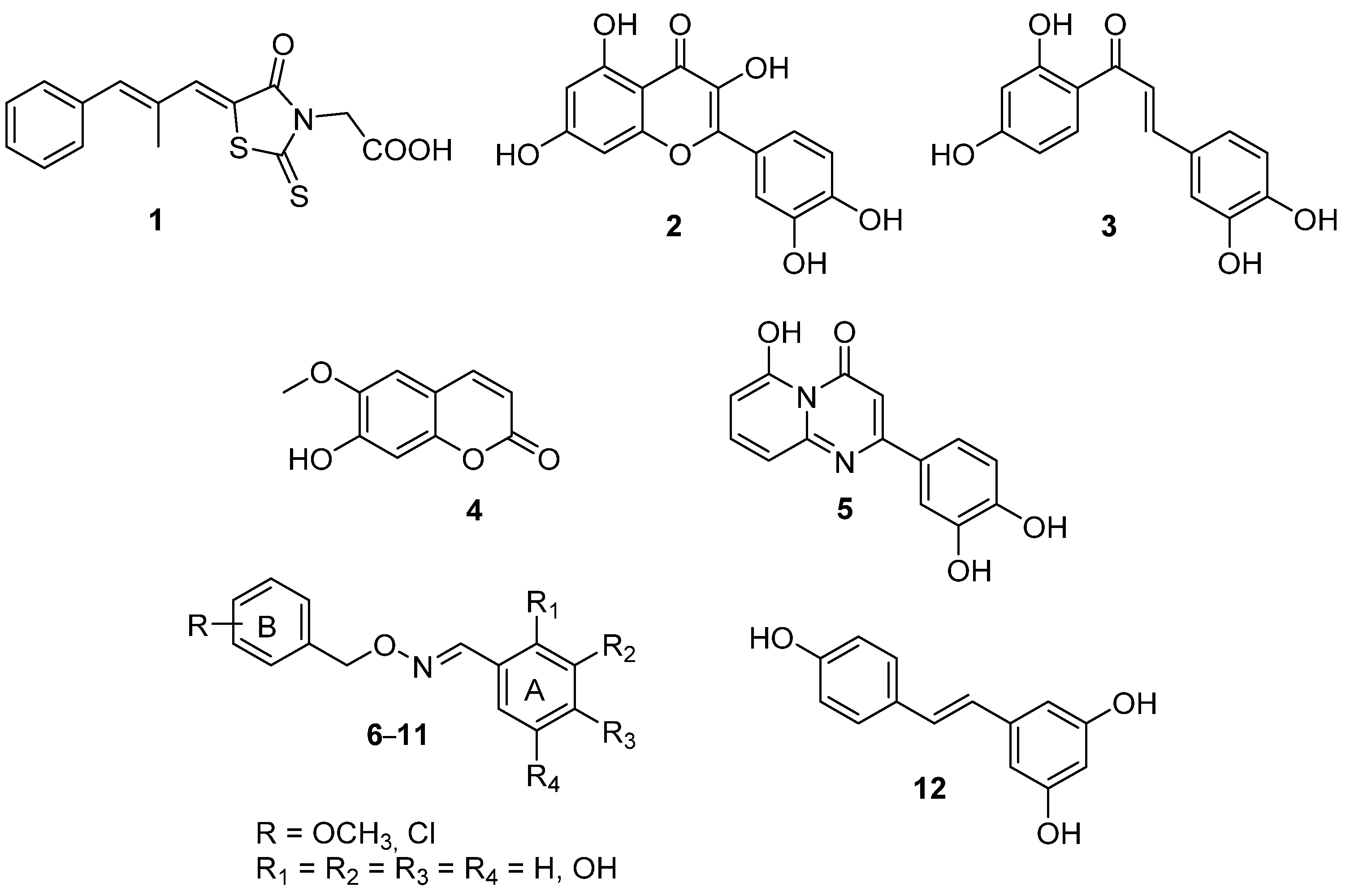
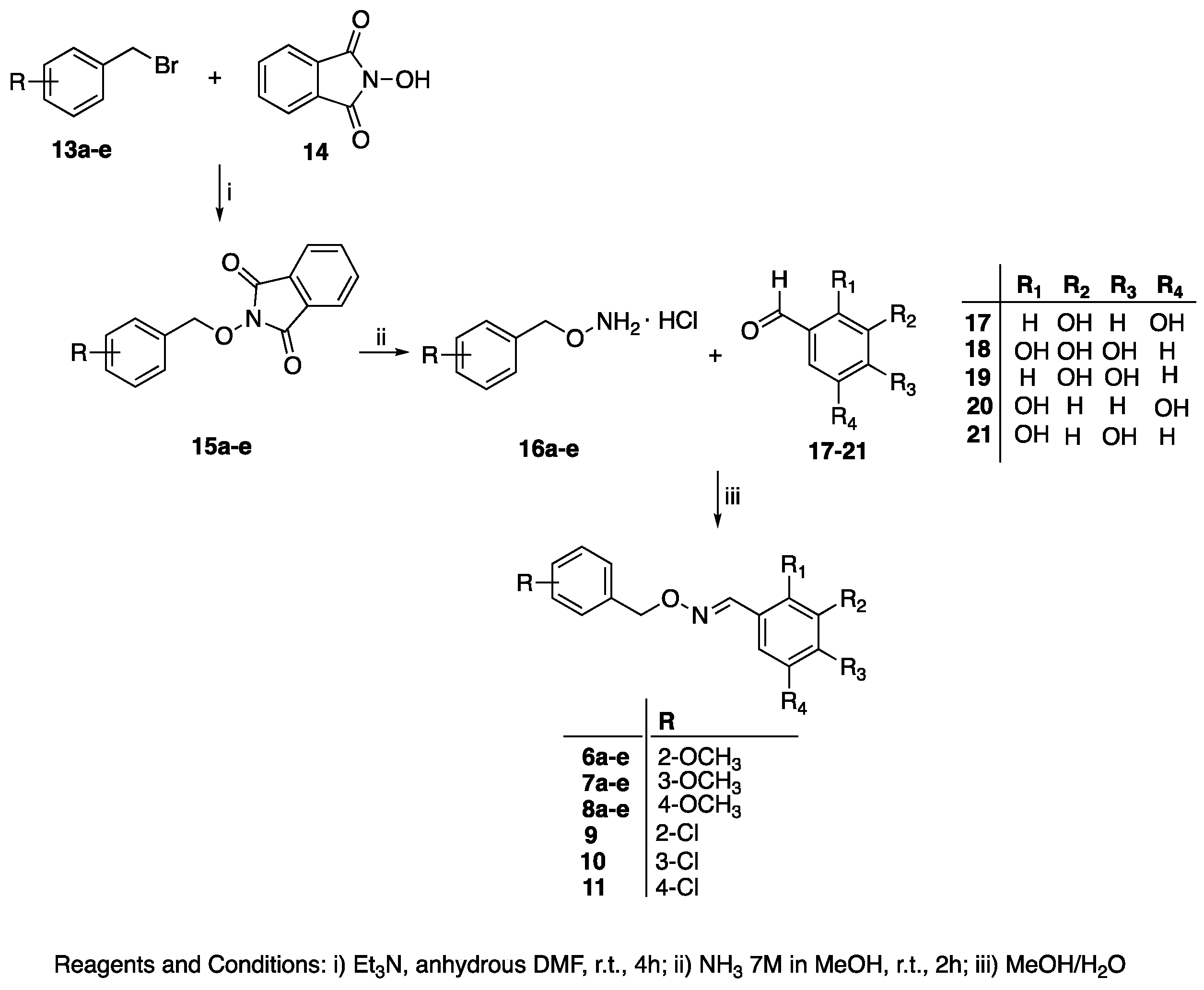
General Procedure for the Synthesis of (E)-Polyhydroxy Substituted Benzaldehyde O-(benzyl) Oximes 6a–e, 7a–e, 8a–e, and 9–11
2.2. Functional Studies
2.2.1. Aldose Reductase Inhibitory Activity
Recombinant Expression of Aldose Reductase in E. coli
- Design of the expression plasmid.
- Expression of aldose reductase.
- Purification and TEV cleavage of DSBC-aldose reductase.
- Purification of aldose reductase.
- Inhibition of aldose eductase.
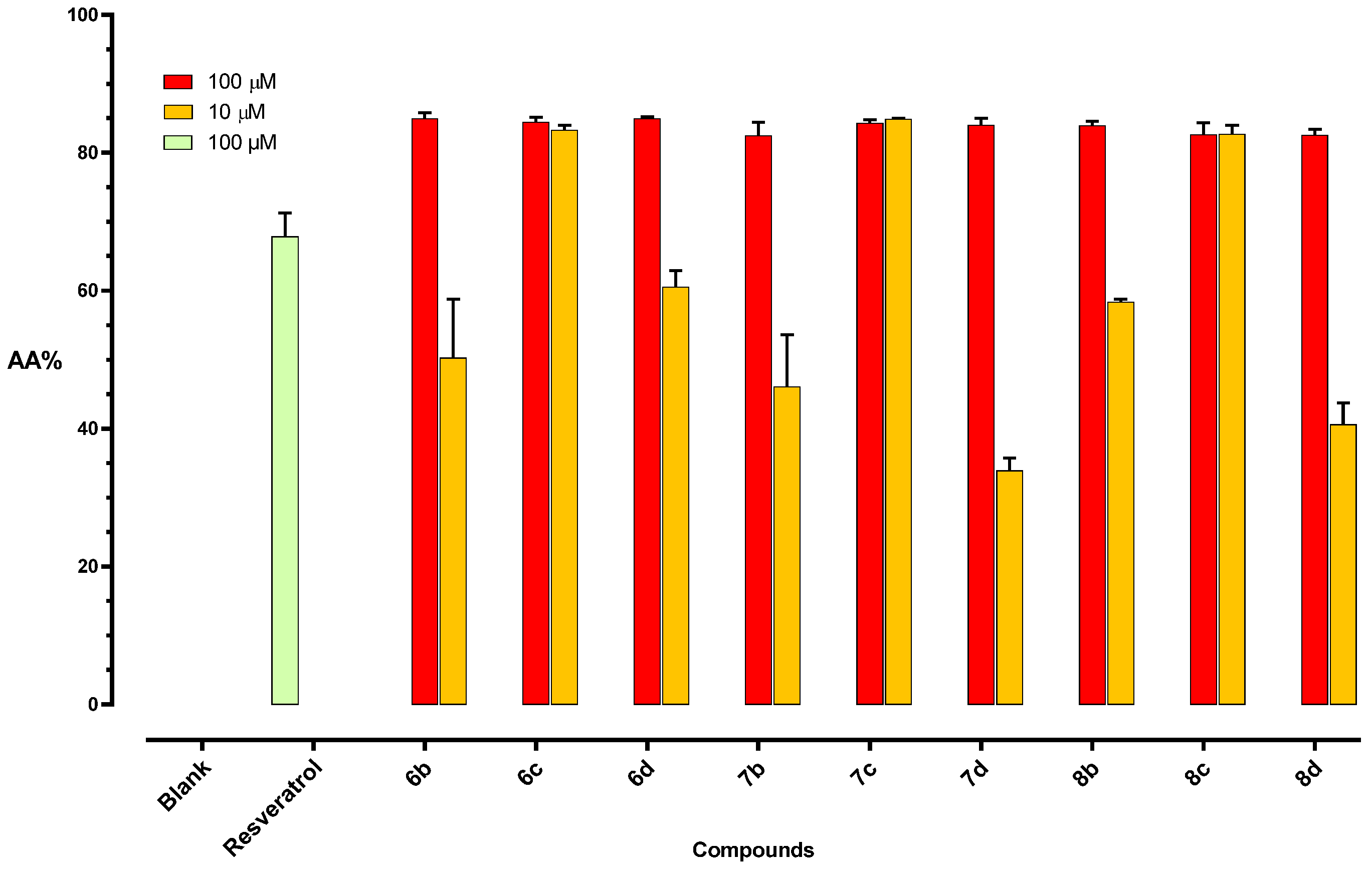
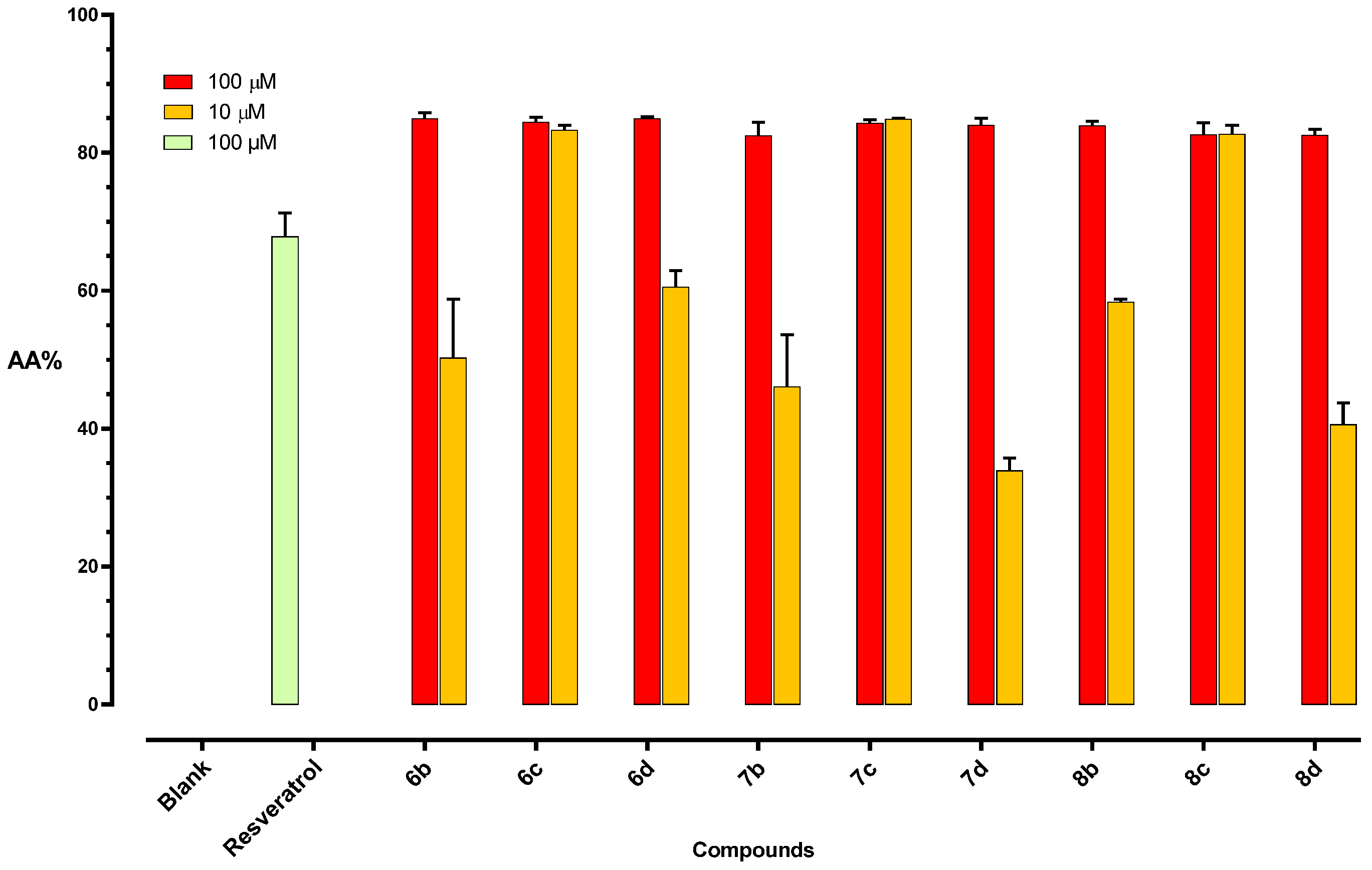
2.2.2. Anti-Oxidant Activity
Thiobarbituric Acid Reactive Substances (TBARS) Assay
DPPH Assay
2.3. Computational Investigations
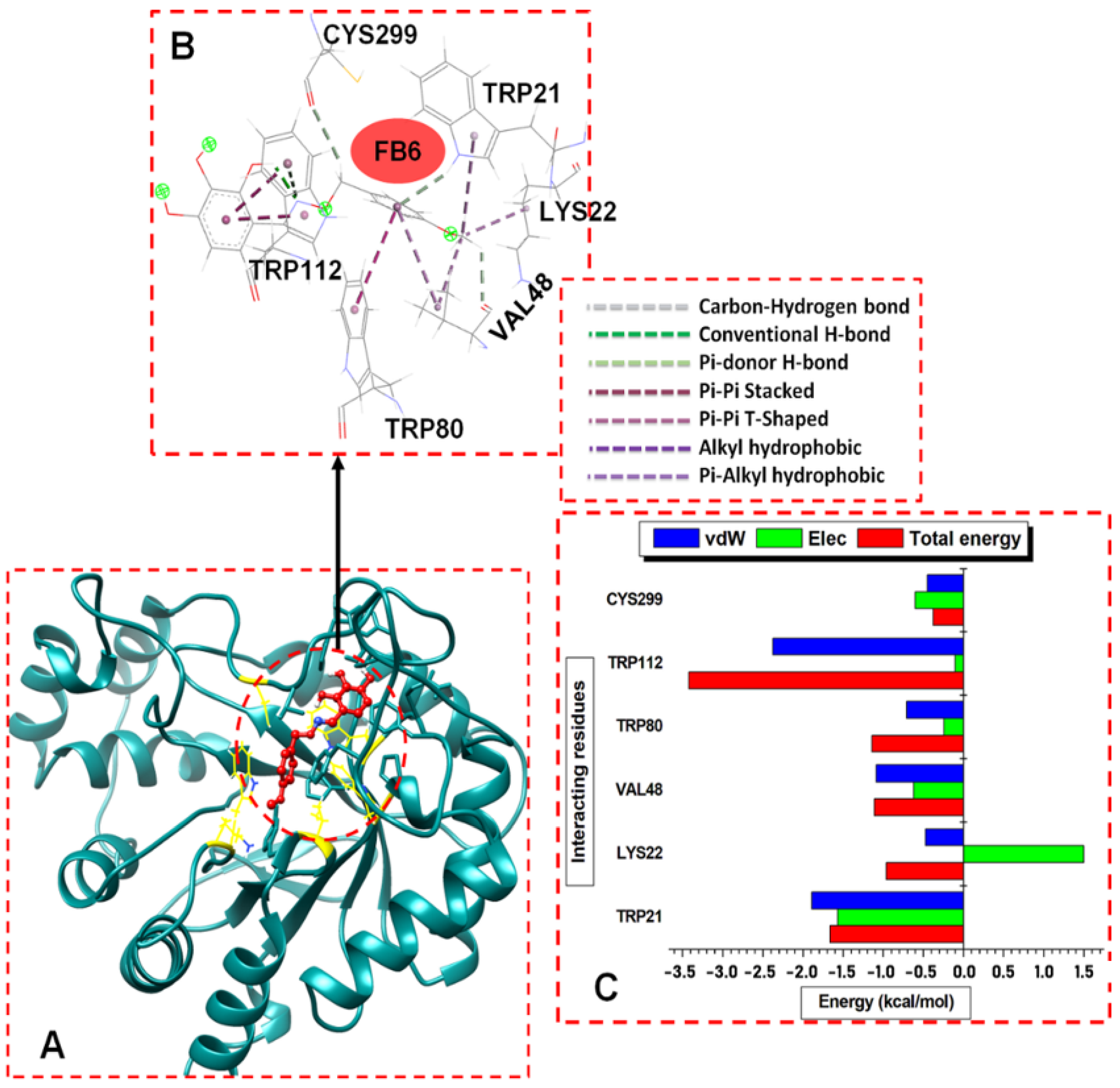
2.3.1. Docking Studies
System Preparation
Molecular Docking Calculations
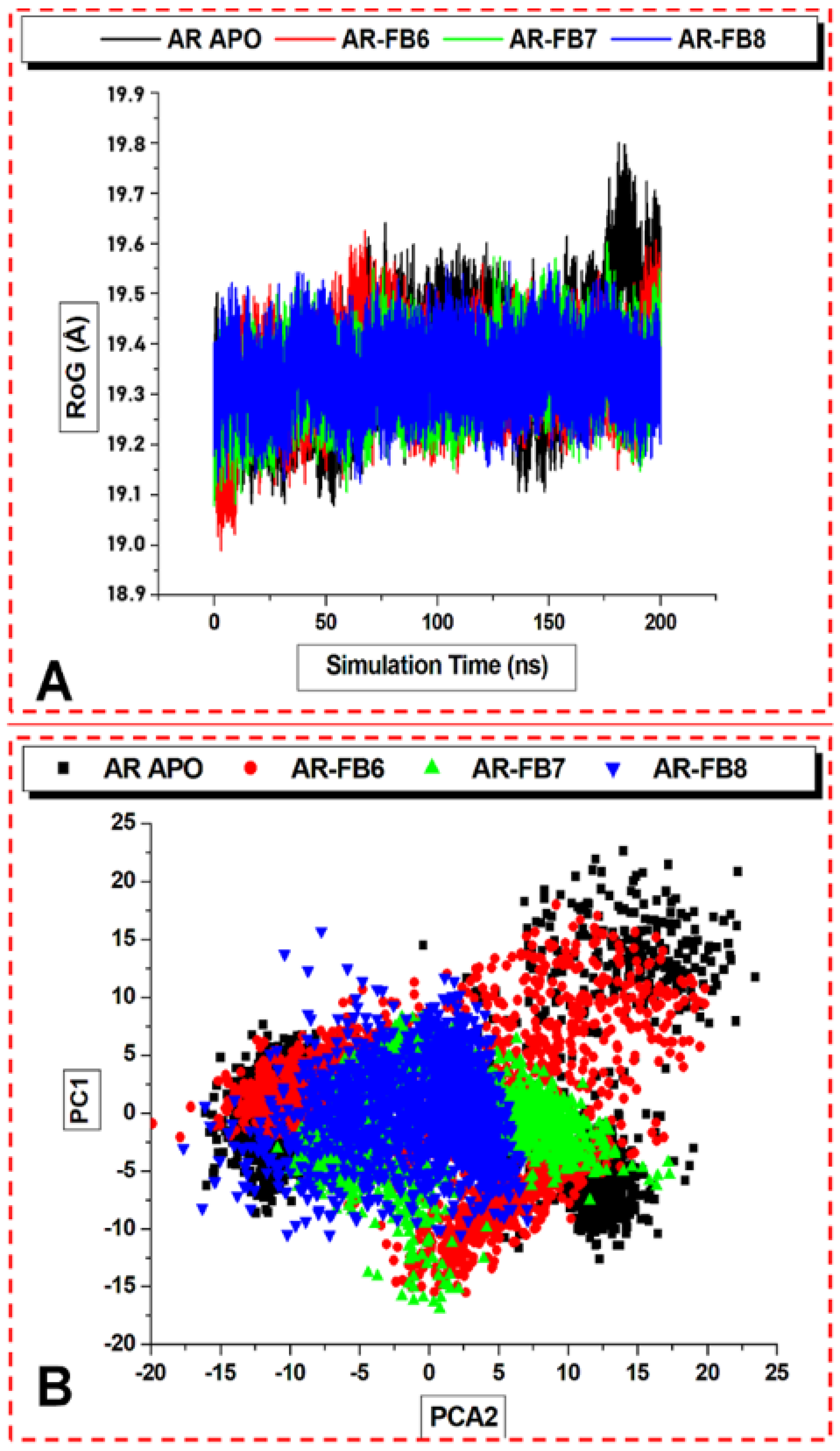
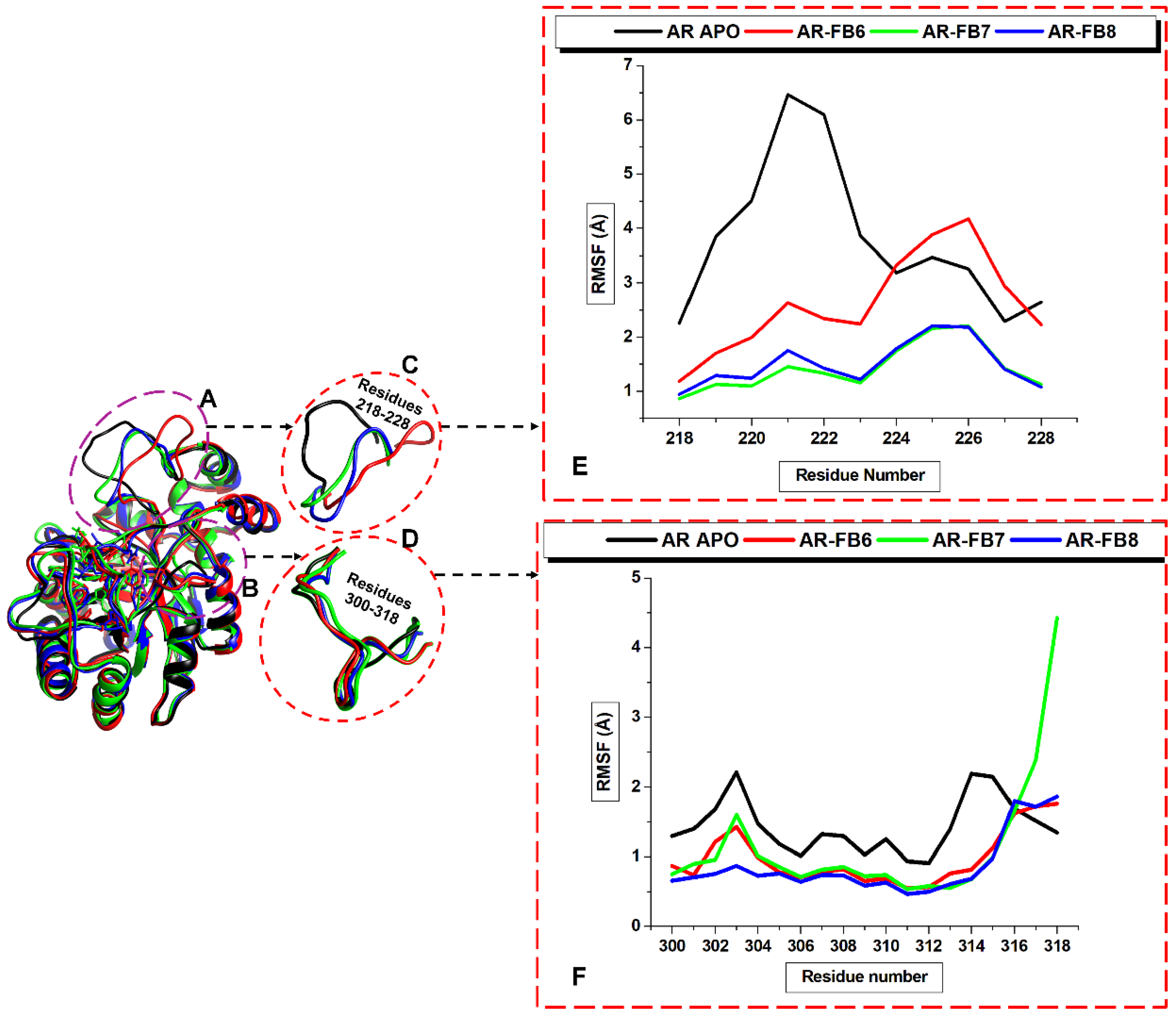
2.3.2. Molecular Dynamics Studies
Retrieval and Preparation of Aldose Reductase
Molecular Dynamics (MD) Simulations
Binding Free Energy Computations
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jannapureddy, S.; Sharma, M.; Yepuri, G.; Schmidt, A.M.; Ramasamy, R. Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 2021, 12, 636267. [Google Scholar] [CrossRef] [PubMed]
- Quattrini, L.; La Motta, C. Aldose reductase inhibitors: 2013–present. Expert Opin. Ther. Pat. 2019, 29, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Traditional Medicine Strategy: 2014–2023; WHO Press: Geneva, Switzerland, 2013. [Google Scholar]
- Del Corso, A.; Barsacchi, D.; Riannessi, M.; Tozzi, M.G.; Camici, M.; Mura, U. Change in stereospecificity of bovine lens aldose reductase modified by oxidative stress. J. Biol. Chem. 1989, 264, 17653–17655. [Google Scholar] [CrossRef]
- Grimshaw, C.E.; Lai, C.J. Oxidized aldose reductase: In Vivo factor not in vitro artifact. Arch. Biochem. Biophys. 1996, 327, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, F.; Sartini, S.; Piano, I.; Franceschi, M.; Quattrini, L.; Guazzelli, L.; Ciccone, L.; Orlandini, E.; Gargini, C.; La Motta, C.; et al. Oxy-imino saccharidic derivatives as a new structural class of aldose reductase inhibitors endowed with anti-oxidant activity. J. Enzyme Inhib. Med. Chem. 2020, 35, 1194–1205. [Google Scholar] [CrossRef]
- Nencetti, S.; La Motta, C.; Rossello, A.; Sartini, S.; Nuti, E.; Ciccone, L.; Orlandini, E. N-(Aroyl)-N-(arylmethyloxy)-α-alanines: Selective inhibitors of aldose reductase. Bioorg. Med. Chem. 2017, 25, 3068–3076. [Google Scholar] [CrossRef]
- Del Corso, A.; Balestri, F.; Di Bugno, E.; Moschini, R.; Cappiello, M.; Sartini, S.; La Motta, C.; Da Settimo, F.; Mura, U. A new approach to control the enigmatic activity of aldose reductase. PLoS ONE 2013, 8, e74076. [Google Scholar] [CrossRef] [Green Version]
- Ramunno, A.; Cosconati, S.; Sartini, S.; Maglio, V.; Angiuoli, S.; La Pietra, V.; Di Maro, S.; Giustiniano, M.; La Motta, C.; Da Settimo, F.; et al. Progresses in the pursuit of aldose reductase inhibitors: The structure-based lead optimization step. Eur. J. Med. Chem. 2012, 51, 16–26. [Google Scholar] [CrossRef]
- Da Settimo, F.; Primofiore, G.; La Motta, C.; Salerno, S.; Novellino, E.; Greco, G.; Lavecchia, A.; Laneri, S.; Boldrini, E. Spirohydantoin derivatives of thiopyrano[2,3-b]pyridin-4(4H)-one as potent in vitro and in vivo aldose reductase inhibitors. Bioorg. Med. Chem. 2005, 13, 491–499. [Google Scholar] [CrossRef]
- Da Settimo, F.; Primofiore, G.; Da Settimo, A.; La Motta, C.; Simorini, F.; Novellino, E.; Greco, G.; Lavecchia, A.; Boldrini, E. Novel, highly potent aldose reductase inhibitors: Cyano(2-oxo-2,3-dihydroindol-3-yl)acetic acid derivatives. J. Med. Chem. 2003, 46, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- La Motta, C.; Sartini, S.; Mugnaini, L.; Simorini, F.; Taliani, S.; Salerno, S.; Marini, A.M.; Da Settimo, F.; Lavecchia, A.; Novellino, E.; et al. Pyrido[1,2-a]pyrimidin-4-one derivatives as a novel class of selective aldose reductase inhibitors exhibiting antioxidant activity. J. Med. Chem. 2007, 50, 4917–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousido-Siah, A.; Ruiz, F.X.; Mitschler, A.; Porté, S.; de Lera, Á.R.; Martín, M.J.; Manzanaro, S.; de la Fuente, J.A.; Terwesten, F.; Betz, M.; et al. Identification of a novel polyfluorinated compound as a lead to inhibit the human enzymes aldose reductase and AKR1B10: Structure determination of both ternary complexes and implications for drug design. Acta Cryst. 2014, D70, 889–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza Bastos, A.; Graves, D.T.; de Melo Loureiro, A.P.; Rossa Júnior, C.; Tfaile Corbi, S.C.; Frizzera, F.; Scarel-Caminaga, R.M.; Câmara, N.O.; Andriankaja, O.M.; Hiyane, M.I.; et al. Diabetes and increased lipid peroxidation are associated with systemic inflammation even in well-controlled patients. J. Diabetes Complicat. 2016, 30, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niki, E.; Noguchi, N. Evaluation of Antioxidant Capacity. What Capacity is Being Measured by Which Method? IUBMB Life 2000, 50, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, A.; Belfiore, M.; Macchia, M.; Martini, C.; Nencetti, S.; Orlandini, E.; Rossello, A. Synthesis and aldose reductase inhibitory activity of N-(arylsulfonyl)- and N-(aroyl)-N-(arylmethyloxy)glycines. Eur. J. Med. Chem. 1994, 29, 787–794. [Google Scholar] [CrossRef]
- Balsamo, A.; Breschi, M.C.; Chiellini, G.; Lapucci, A.; Lazzeri, N.; Machia, M.; Martinelli, A.; Micali, E.; Nencetti, S.; Rossello, A. Synthesis and β-adrenergic properties of (Z)-N-[3-(alkylamino)-2-hydroxypropylidene](aryl-methyloxy)amines: Effects of the configuration around the methyloxyiminomethyl (MOIM) double bond on the biopharmacological properties of MOIM-type β-blocking agents. Bioorg. Med. Chem. 1998, 6, 2151–2160. [Google Scholar] [CrossRef]
- Gentili, D.; Macchia, M.; Menchini, E.; Nencetti, S.; Orlandini, E.; Rossello, A.; Broccali, G.; Limonta, D. Synthesis and antimicrobial properties of cephalosporin derivatives substituted on the C(7) nitrogen with arylmethyloxyimino or arylmethyloxyamino alkanoyl groups. Il Farm. 1999, 54, 224–231. [Google Scholar] [CrossRef]
- Nozach, H.; Fruchart-Gaillard, C.; Fenaille, F.; Beau, F.; Ramos, O.H.; Douzi, B.; Saez, N.J.; Moutiez, M.; Servent, D.; Gondry, M.; et al. High throughput screening identifies disulfide isomerase DsbC as a very efficient partner for recombinant expression of small disulfide-rich proteins in E. coli. Microb. Cell Fact. 2013, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Cantore, M.; Siano, S.; Coronnello, M.; Mazzetti, L.; Franchi-Micheli, S.; Boldrini, E.; Ciuffi, M.; Failli, P. Pirenoxine prevents oxidative effects of argon fluoride excimer laser irradiation in rabbit corneas: Biochemical, histological and cytofluorimetric evaluations. J. Photochem. Photobiol. B 2005, 78, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.K.; Shibamoto, T. Antioxidant assays for plant and food components. J. Agric. Food Chem. 2009, 57, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Steuber, H.; Zentgraf, M.; Gerlach, C.; Sotriffer, C.A.; Heine, A.; Klebe, G. Expect the unexpected or caveat for drug designers: Multiple structure determinations using aldose reductase crystals treated under varying soaking and co-crystallization conditions. J. Mol. Biol. 2006, 363, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, H.; Zhao, Y.; Li, Z.; Chen, S.; Zhai, J.; Chen, Y.; Xie, W.; Wang, Z.; Li, Q.; et al. Inhibitor selectivity between Aldo-keto reductase superfamily members AKR1B10 and AKR1B1: Role of Trp112 (Trp111). FEBS Lett. 2013, 587, 3681–3686. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Cough, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adewumi, A.T.; Elrashedy, A.; Soremekun, O.S.; Ajadi, M.B.; Soliman, M.E.S. Weak spots inhibition in the Mycobacterium tuberculosis antigen 85C target for antitubercular drug design through selective irreversible covalent inhibitor-SER124. J. Biomol. Struct. Dyn. 2020, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.chemaxon.com (accessed on 26 January 2022).
- Available online: https://chemicalize.com (accessed on 26 January 2022).
- Available online: http://molexus.io/molegro-molecular-viewer (accessed on 26 January 2022).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Chemoinform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://avogadro.cc/ (accessed on 26 January 2022).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Adewumi, A.T.; Ajadi, M.B.; Soremekun, O.S.; Soliman, M.E.S. Thompson loop: Opportunities for antitubercular demethylmenaquinone methyltransferase protein. RSC Adv. 2020, 10, 23466–23483. [Google Scholar] [CrossRef]
- Silva, N.S.R.; Gonçalves, L.K.S.; Duarte, J.L.; Silva, J.S.; Santos, C.S.; Braga, F.S.; Silva, R.C.; Costa, J.S.; Hage-Melim, L.I.S.; dos Santos, C.B.R. Computational analysis of physicochemical, pharmacokinetic and toxicological properties of deoxyhypusine synthase inhibitors with antimalarial activity. Comput. Mol. Biosci. 2014, 4, 47–57. [Google Scholar] [CrossRef]
- Available online: http:www.accelrys.com (accessed on 26 January 2022).
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins Struct. Funct. Genet. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.ambermd.org (accessed on 26 January 2022).
- Lee, T.S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Zheng, S.; Tang, Q.; He, J.; Du, S.; Xu, S.; Wang, C.; Xu, Y.; Lin, F. VFFDT: A new software for preparing AMBER force field parameters for metal-containing molecular systems. J. Chem. Inf. Model. 2016, 56, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Adewumi, A.T.; Ramharack, P.; Soremekun, O.S.; Soliman, M.E.S. Delving into the characteristic features of “Menace” Mycobacterium tuberculosis homologs: A structural dynamics and proteomics perspectives. Protein J. 2020, 39, 118–132. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Oluyemi, W.M.; Samuel, B.B.; Adewumi, A.T.; Adekunle, Y.A.; Soliman, M.E.S.; Krenn, L. An allosteric inhibitory potential of triterpenes from Combretum racemosum on the structural and functional dynamics of Plasmodium falciparum lactate dehydrogenase binding landscape. Chem. Biodivers. 2022, 19, 1–20. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Post, M.; Wolf, S.; Stock, G. Principal component analysis of nonequilibrium molecular dynamics simulations. J. Chem. Phys. 2019, 150, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hou, T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf. Model. 2012, 52, 1199–1212. [Google Scholar] [CrossRef] [Green Version]
- Gapsys, V.; Michielssens, S.; Peters, J.H.; de Groot, B.L.; Leonov, H. Calculation of Binding Free Energies. Methods Mol. Biol. 2015, 1215, 173–209. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM_PBSA and MM_GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Ciccone, L.; Nencetti, S.; Tonali, N.; Fruchart-Gaillard, C.; Shepard, W.; Nuti, E.; Camodeca, C.; Rossello, A.; Orlandini, E. Monoaryl derivatives as transthyretin fibril formation inhibitors: Design, synthesis, biological evaluation and structural analysis. Bioorg. Med. Chem. 2020, 28, 115673. [Google Scholar] [CrossRef] [PubMed]
- Bargagna, B.; Ciccone, L.; Nencetti, S.; Santos, M.A.; Chaves, S.; Camodeca, C.; Orlandini, E. Multifunctional Small Molecules as Potential Anti-Alzheimer’s Disease Agents. Molecules 2021, 26, 6015. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular Docking: A powerful approach for structure-based drug drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Cosconati, S.; Marinelli, L.; La Motta, C.; Sartini, S.; Da Settimo, F.; Olson, A.J.; Novellino, E. Pursuing aldose reductase inhibitors through in situ cross-docking and similarity-based virtual screening. J. Med. Chem. 2009, 52, 5578–5581. [Google Scholar] [CrossRef]
- Steuber, H.; Zentgraf, M.; La Motta, C.; Sartini, S.; Heine, A.; Klebe, G. Evidence for a novel binding site conformer of aldose reductase in ligand-bound state. J. Mol. Biol. 2007, 369, 186–197. [Google Scholar] [CrossRef]
- Zentgraf, M.; Steuber, H.; Koch, C.; La Motta, C.; Sartini, S.; Sotriffer, C.A.; Klebe, G. How reliable are current docking approaches for structure-based drug design? Lessons from aldose reductase. Angew. Chem. Int. Ed. Engl. 2007, 46, 3575–3578. [Google Scholar] [CrossRef]
- Olanlokun, J.O.; Olotu, A.F.; David, O.M.; Idowu, T.O.; Soliman, E.M.; Olorunsogo, O.O. A novel compound purified from Alstonia boonei inhibits Plasmodium falciparum lactate dehydrogenase and plasmepsin II. J. Biomol. Struct. Dyn. 2019, 37, 2193–2200. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.Q. Insights into protein-ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.M.; Archontis, G. MM-GB(PB)SA Calculations of Protein-Ligand Binding Free Energies. In Molecular Dynamics-Studies of Synthetic and Biological Macromolecules; InTech Europe: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef] [Green Version]
- Priya, R.; Sumitha, R.; Doss, C.G.P.; Rajasekaran, C.; Babu, S.; Seenivasan, R.; Siva, R. Molecular docking and molecular dynamics to identify a novel human immunodeficiency virus inhibitor from alkaloids of Toddalia asiatica. Pharmacogn. Mag. 2015, 11, S414–S422. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef] [PubMed]
- Durham, E.; Dorr, B.; Woetzel, N.; Staritzbichler, R.; Meiler, J. Solvent accessible surface area approximations for rapid and accurate protein structure prediction. J. Mol. Model. 2009, 15, 1093–1108. [Google Scholar] [CrossRef] [Green Version]
- Kijewska, M.; Sharfalddin, A.A.; Jaremko, Ł.; Cal, M.; Setner, B.; Siczek, M.; Stefanowicz, P.; Hussien, M.A.; Emwas, A.-H.; Jaremko, M. Lossen Rearrangement of p-Toluenesulfonates of N-Oxyimides in Basic Condition, Theoretical Study, and Molecular Docking. Front. Chem. 2021, 9, 662533. [Google Scholar] [CrossRef]
- Stefanowicz, P.; Jaremko, L.; Jaremko, M.; Lis, T. Crystal-state studies on p-toluenesulfonates of N-oxyimides—A possible structural basis of serine proteases inhibition. New J. Chem. 2006, 30, 258–265. [Google Scholar] [CrossRef]
- Türkkan, B.; Ozyürek, M.; Bener, M.; Güçlü, K.; Apak, R. Synthesis, characterization and antioxidant capacity of naringenin-oxime. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 85, 235–240. [Google Scholar] [CrossRef]
- Potaniec, B.; Grabarczyk, M.; Stompor, M.; Szumny, A.; Zieliński, P.; Żołnierczyk, A.K.; Anio, M. Antioxidant activity and spectroscopic data of isoxanthohomol oxime and related compounds. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 118, 716–723. [Google Scholar] [CrossRef]

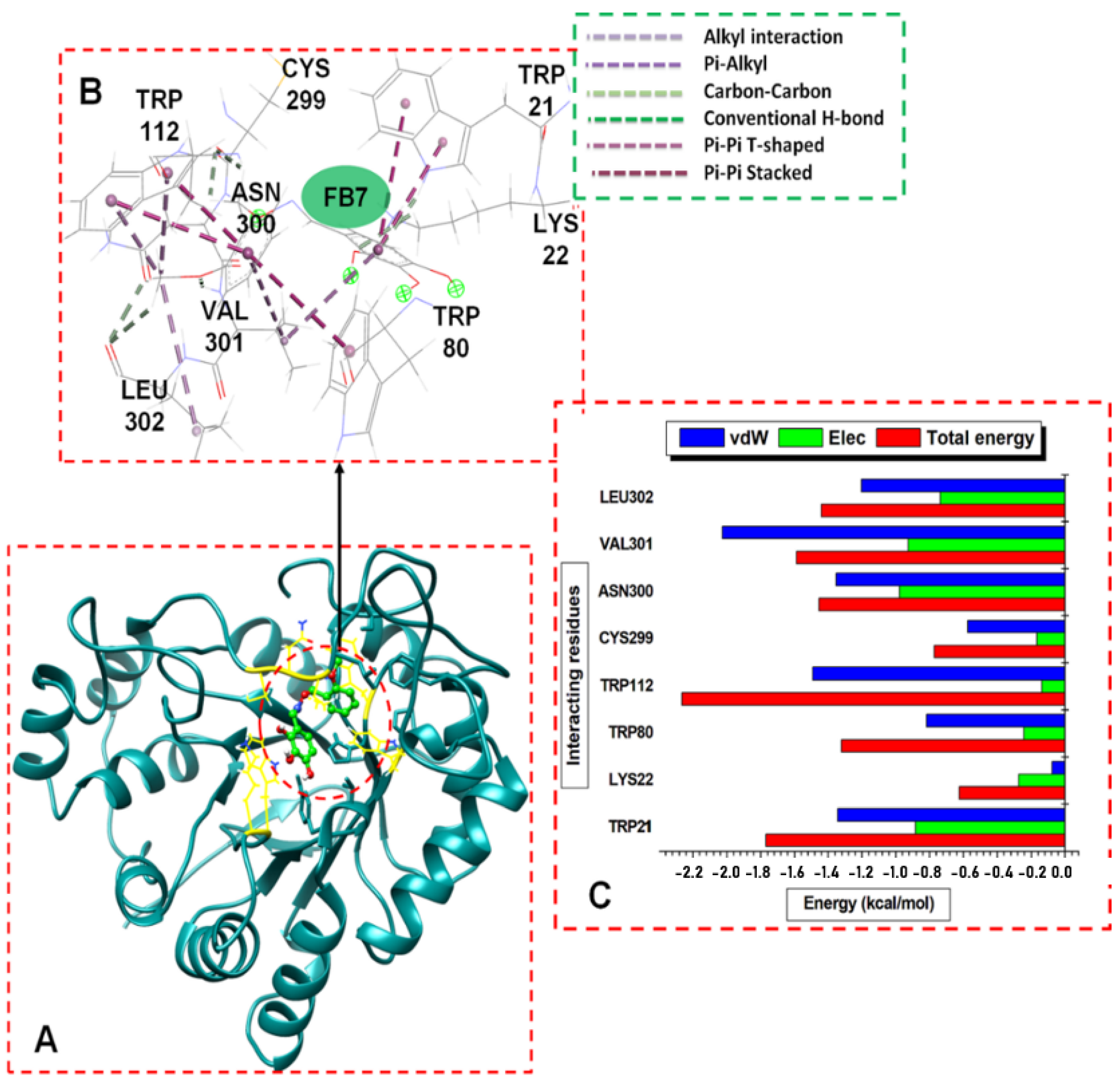
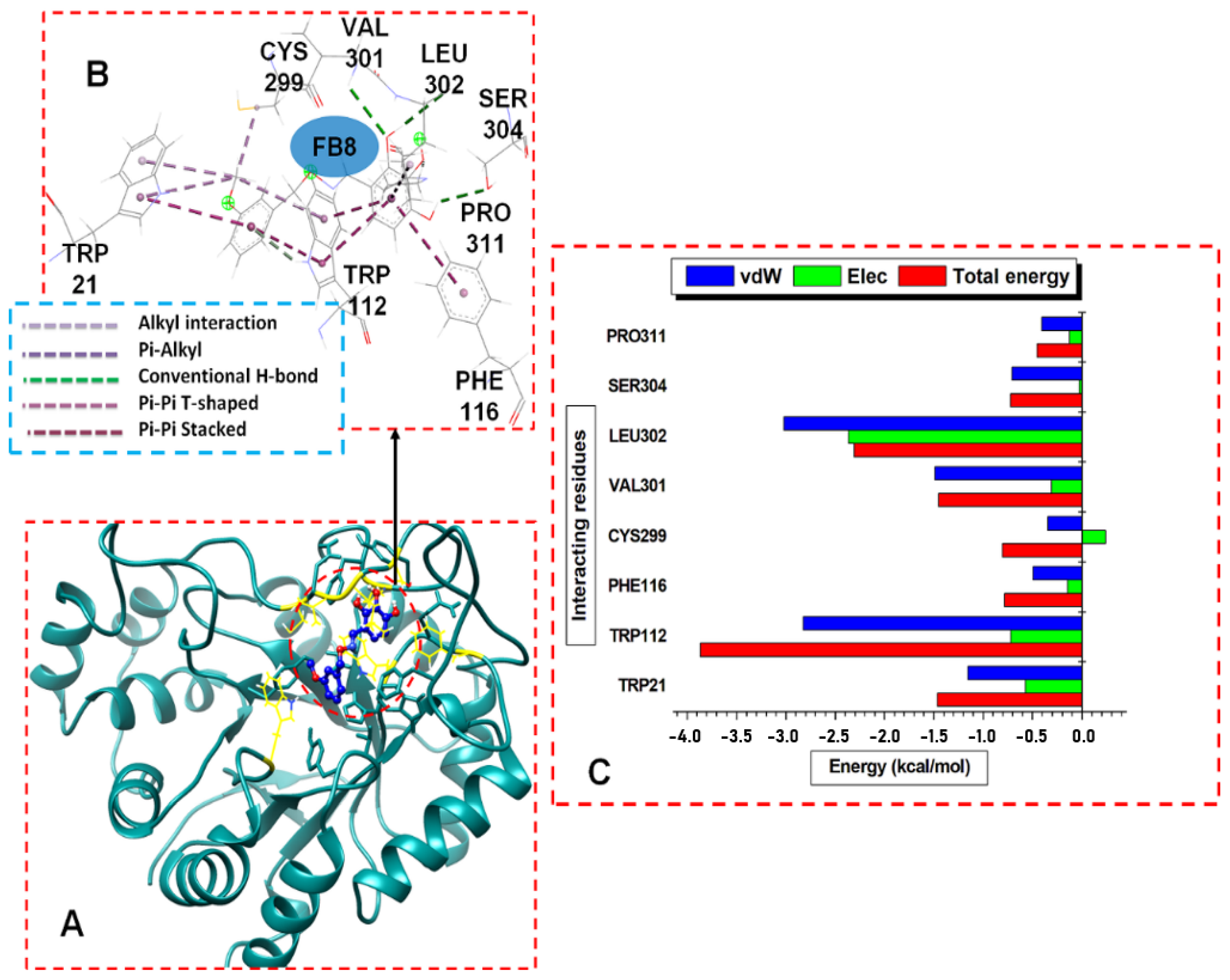



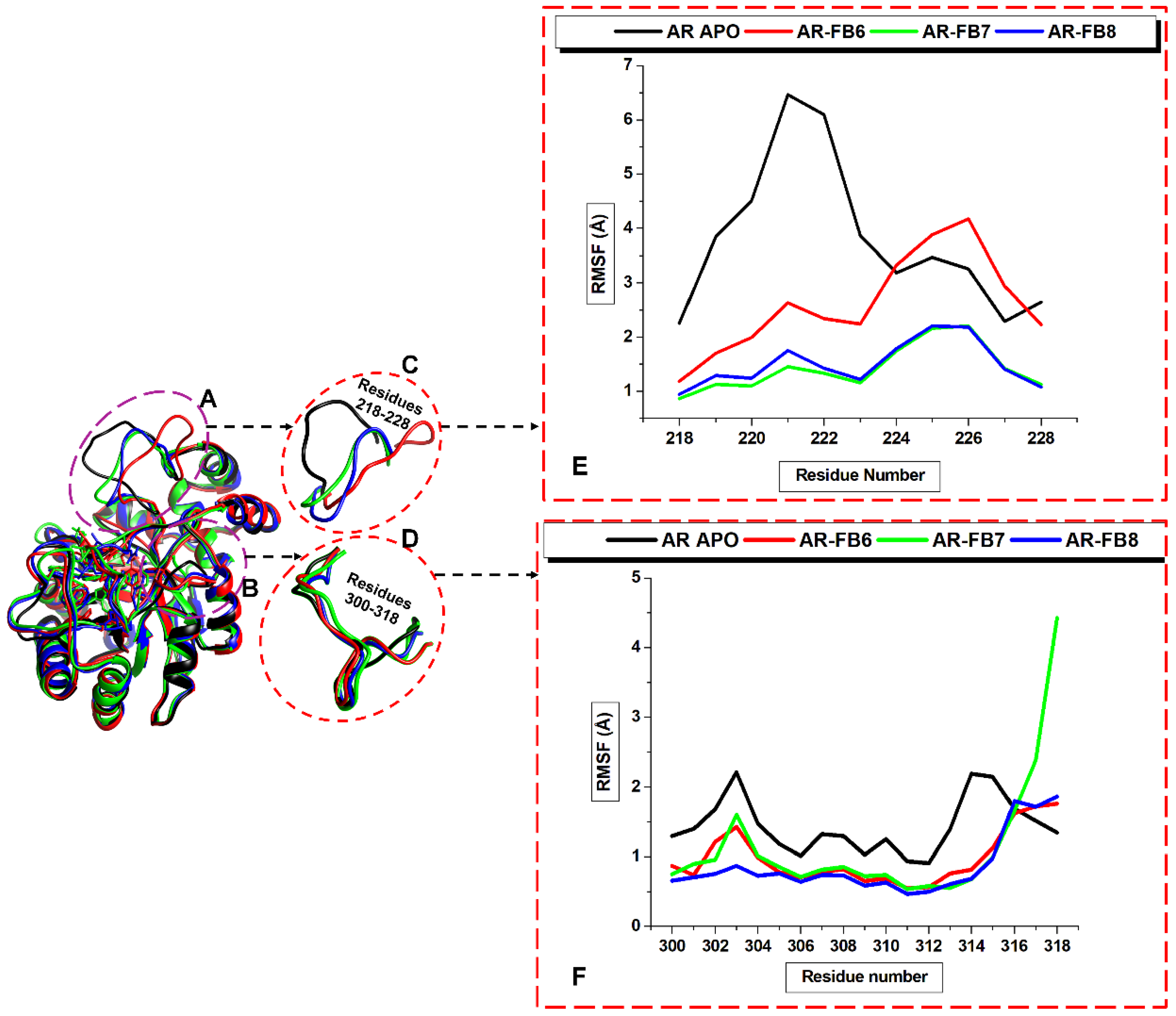
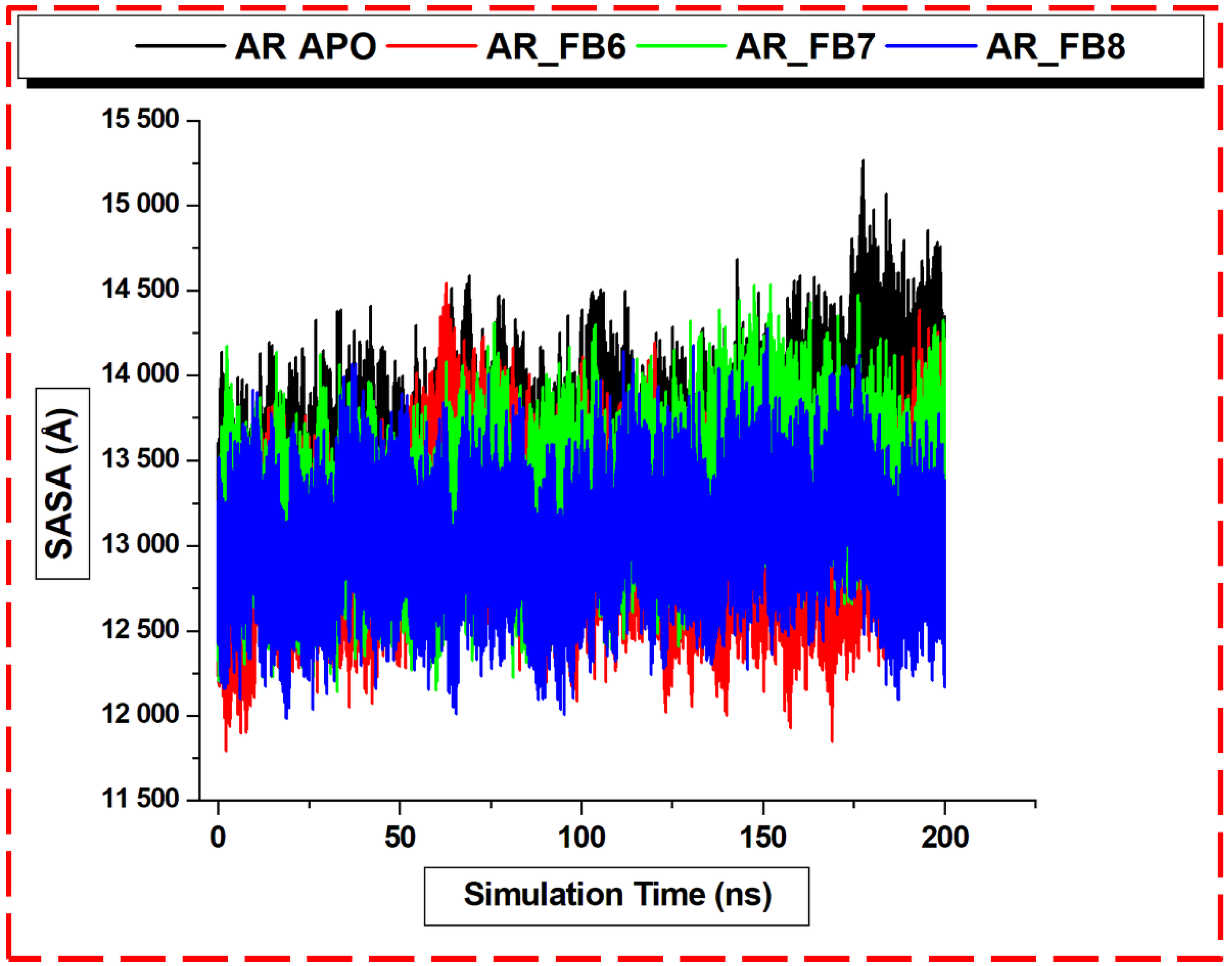

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|
| 6a | 2-OCH3 | H | OH | H | OH |
| 6b | 2-OCH3 | OH | OH | OH | H |
| 6c | 2-OCH3 | H | OH | OH | H |
| 6d | 2-OCH3 | OH | H | H | OH |
| 6e | 2-OCH3 | OH | H | OH | H |
| 7a | 3-OCH3 | H | OH | H | OH |
| 7b | 3-OCH3 | OH | OH | OH | H |
| 7c | 3-OCH3 | H | OH | OH | H |
| 7d | 3-OCH3 | OH | H | H | OH |
| 7e | 3-OCH3 | OH | H | OH | H |
| 8a | 4-OCH3 | H | OH | H | OH |
| 8b | 4-OCH3 | OH | OH | OH | H |
| 8c | 4-OCH3 | H | OH | OH | H |
| 8d | 4-OCH3 | OH | H | H | OH |
| 8e | 4-OCH3 | OH | H | OH | H |
| 9 | 2-Cl | H | OH | H | OH |
| 10 | 3-Cl | H | OH | H | OH |
| 11 | 4-Cl | H | OH | H | OH |
| Compound | % Inhibition a | Compound | % Inhibition a |
|---|---|---|---|
| 6a | n.a. b | 8a | n.a. |
| 6b | 44 | 8b | 67 |
| 6c | 19 | 8c | 25 |
| 6d | 28 | 8d | n.a. |
| 6e | 23 | 8e | 22 |
| 7a | n.a. | 9 | 10 |
| 7b | 63 | 10 | 44 |
| 7c | 20 | 11 | n.a. |
| 7d | n.a. | ||
| 7e | 23 |
| Binding Energies (kcal/mol) | |||
|---|---|---|---|
| PDB Code | ALR2-6b | ALR2-7b | ALR2-8b |
| 2F2B | −4.9 | −7.6 | −4.8 |
| 4JIR | −7.3 | −8.1 | −7.8 |
| 4IGS | −8.0 | −8.4 | −8.0 |
| 4JII | −9.0 | −9.4 | −8.2 |
| Complex | Energy Components (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ΔEvdW | ΔEelec | ΔGgas | EGGB | ESA | ΔGsolv | ΔGbind | |
| ALR2-6b | −39.29 (±3.22) | −11.81 (±6.19) | −51.10 (±7.66) | 30.94 (±4.71) | −5.24 (±0.23) | 25.70 (±4.69) | −25.40 (±4.66) |
| ALR2-7b | −40.99 (±3.93) | −21.88 (±6.55) | −62.88 (±6.65) | 33.87 (±3.42) | −5.50 (±0.37) | 28.38 (±3.30) | −34.51 (±4.81) |
| ALR2-8b | −42.22 (±1.73) | −7.50 (±4.27) | −49.72 (±4.63) | 23.89 (±2.76) | −5.65 (±0.15) | 18.24 (±2.76) | −31.48 (±2.92) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccone, L.; Petrarolo, G.; Barsuglia, F.; Fruchart-Gaillard, C.; Cassar Lajeunesse, E.; Adewumi, A.T.; Soliman, M.E.S.; La Motta, C.; Orlandini, E.; Nencetti, S. Nature-Inspired O-Benzyl Oxime-Based Derivatives as New Dual-Acting Agents Targeting Aldose Reductase and Oxidative Stress. Biomolecules 2022, 12, 448. https://doi.org/10.3390/biom12030448
Ciccone L, Petrarolo G, Barsuglia F, Fruchart-Gaillard C, Cassar Lajeunesse E, Adewumi AT, Soliman MES, La Motta C, Orlandini E, Nencetti S. Nature-Inspired O-Benzyl Oxime-Based Derivatives as New Dual-Acting Agents Targeting Aldose Reductase and Oxidative Stress. Biomolecules. 2022; 12(3):448. https://doi.org/10.3390/biom12030448
Chicago/Turabian StyleCiccone, Lidia, Giovanni Petrarolo, Francesca Barsuglia, Carole Fruchart-Gaillard, Evelyne Cassar Lajeunesse, Adeniyi T. Adewumi, Mahmoud E. S. Soliman, Concettina La Motta, Elisabetta Orlandini, and Susanna Nencetti. 2022. "Nature-Inspired O-Benzyl Oxime-Based Derivatives as New Dual-Acting Agents Targeting Aldose Reductase and Oxidative Stress" Biomolecules 12, no. 3: 448. https://doi.org/10.3390/biom12030448
APA StyleCiccone, L., Petrarolo, G., Barsuglia, F., Fruchart-Gaillard, C., Cassar Lajeunesse, E., Adewumi, A. T., Soliman, M. E. S., La Motta, C., Orlandini, E., & Nencetti, S. (2022). Nature-Inspired O-Benzyl Oxime-Based Derivatives as New Dual-Acting Agents Targeting Aldose Reductase and Oxidative Stress. Biomolecules, 12(3), 448. https://doi.org/10.3390/biom12030448