
Sulphoraphane Affinity-Based Chromatography for the Purification of Myrosinase from Lepidium sativum Seeds


Abstract
:1. Introduction
2. Materials and Methods
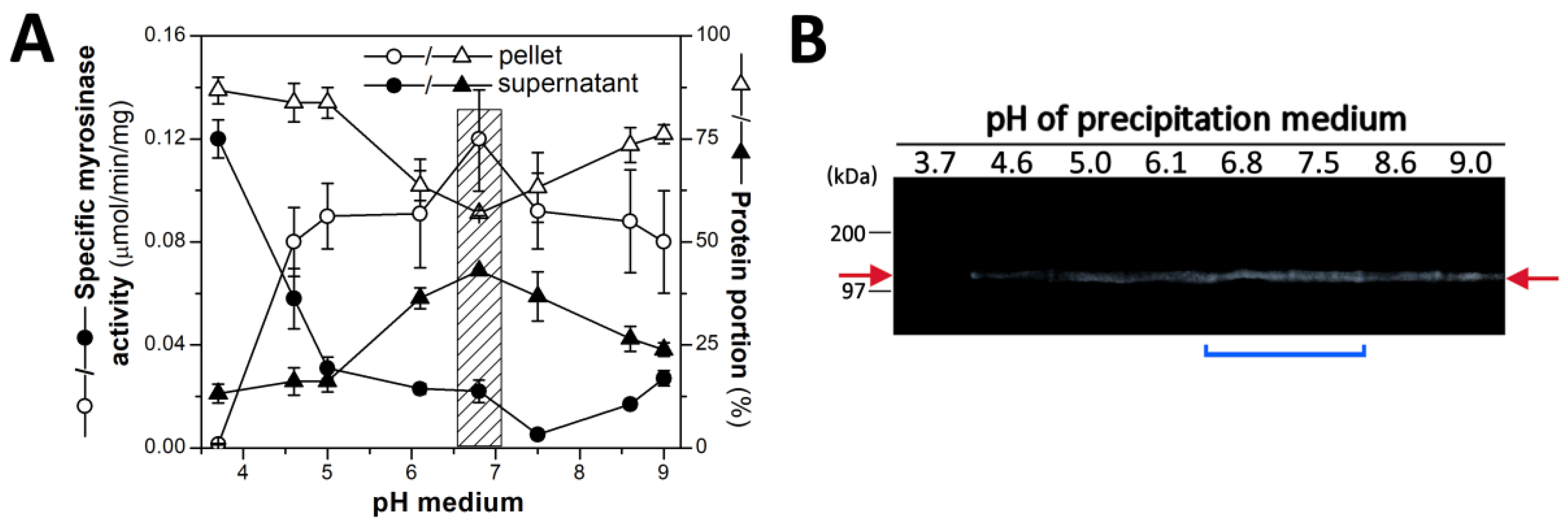
2.1. Plant Material, Its Homogenization and Preparation the Myrosinase Fraction by Isoelectric Precipitation by AS
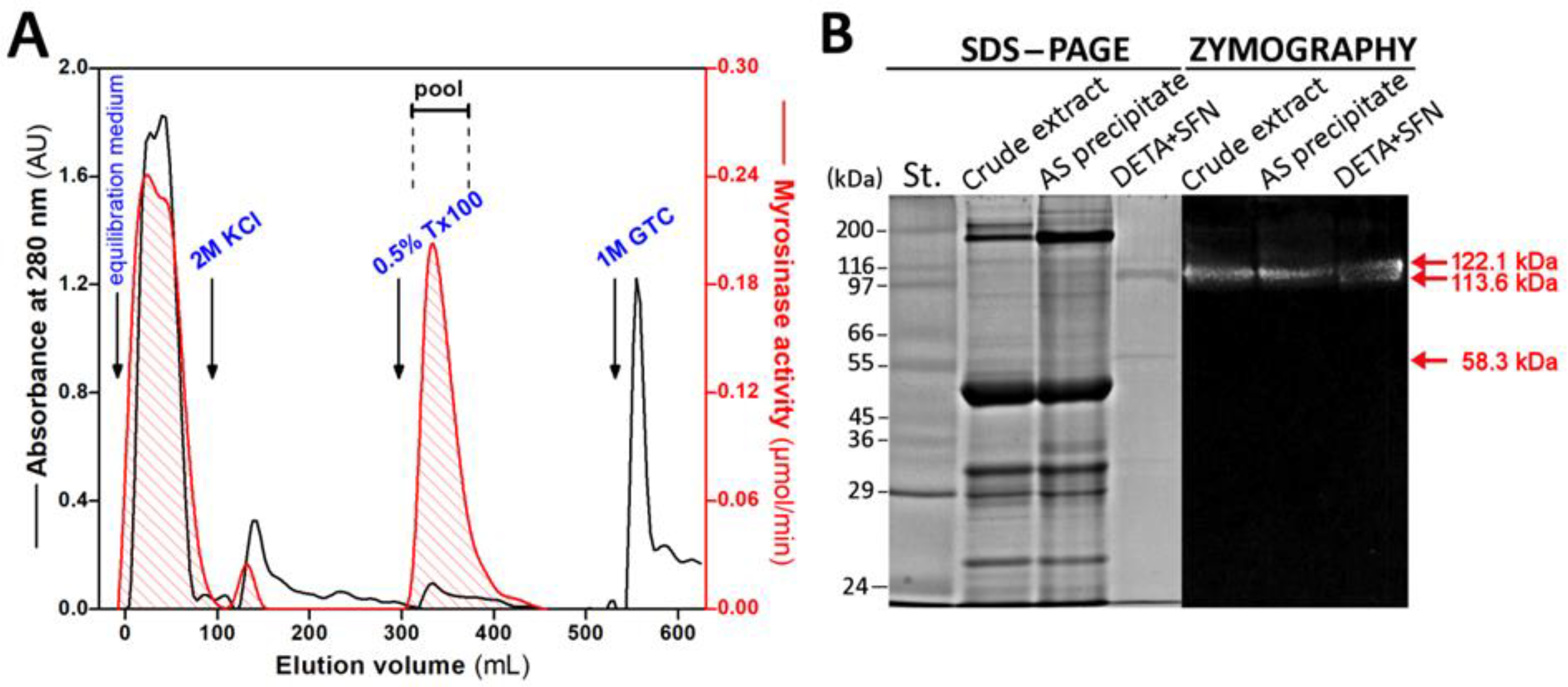
2.2. Purification of Myrosinase on Affinity Chromatography Column with SFN Ligand
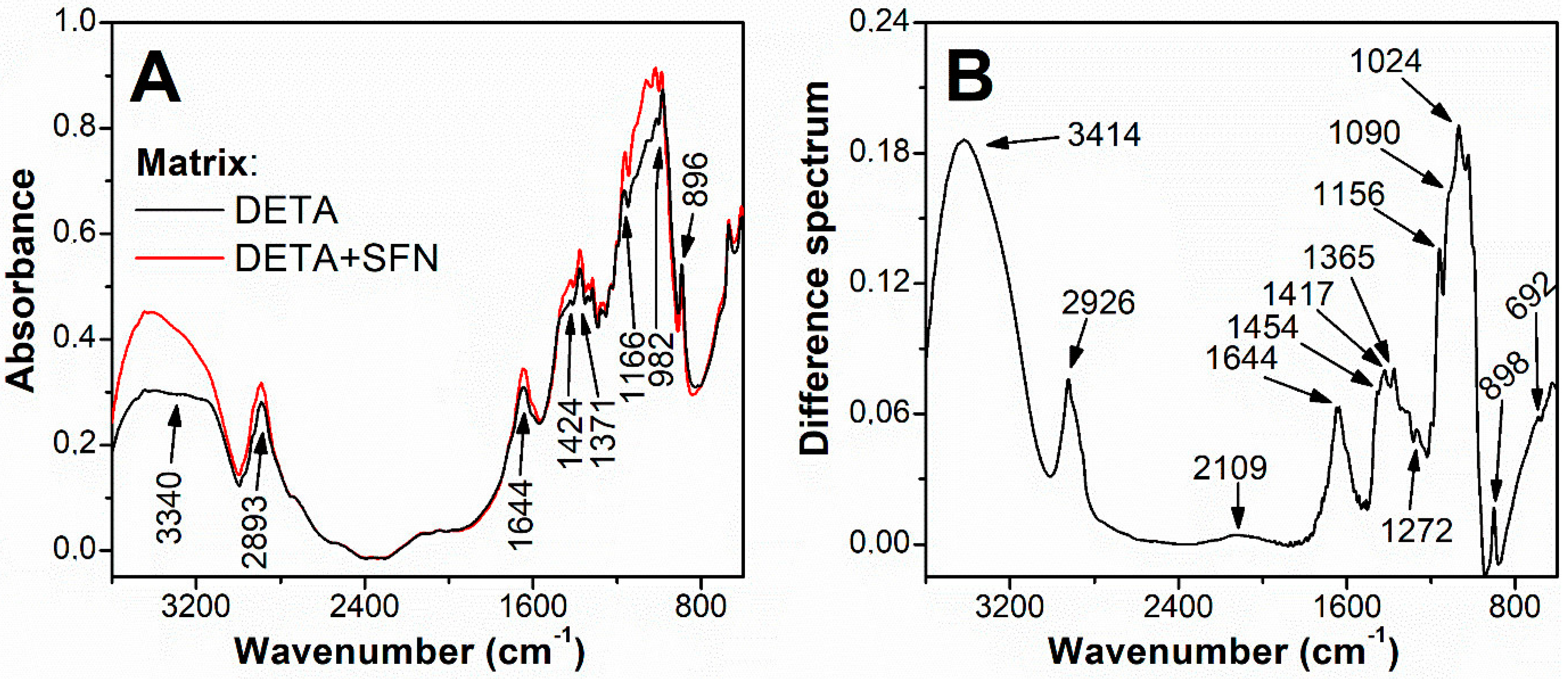
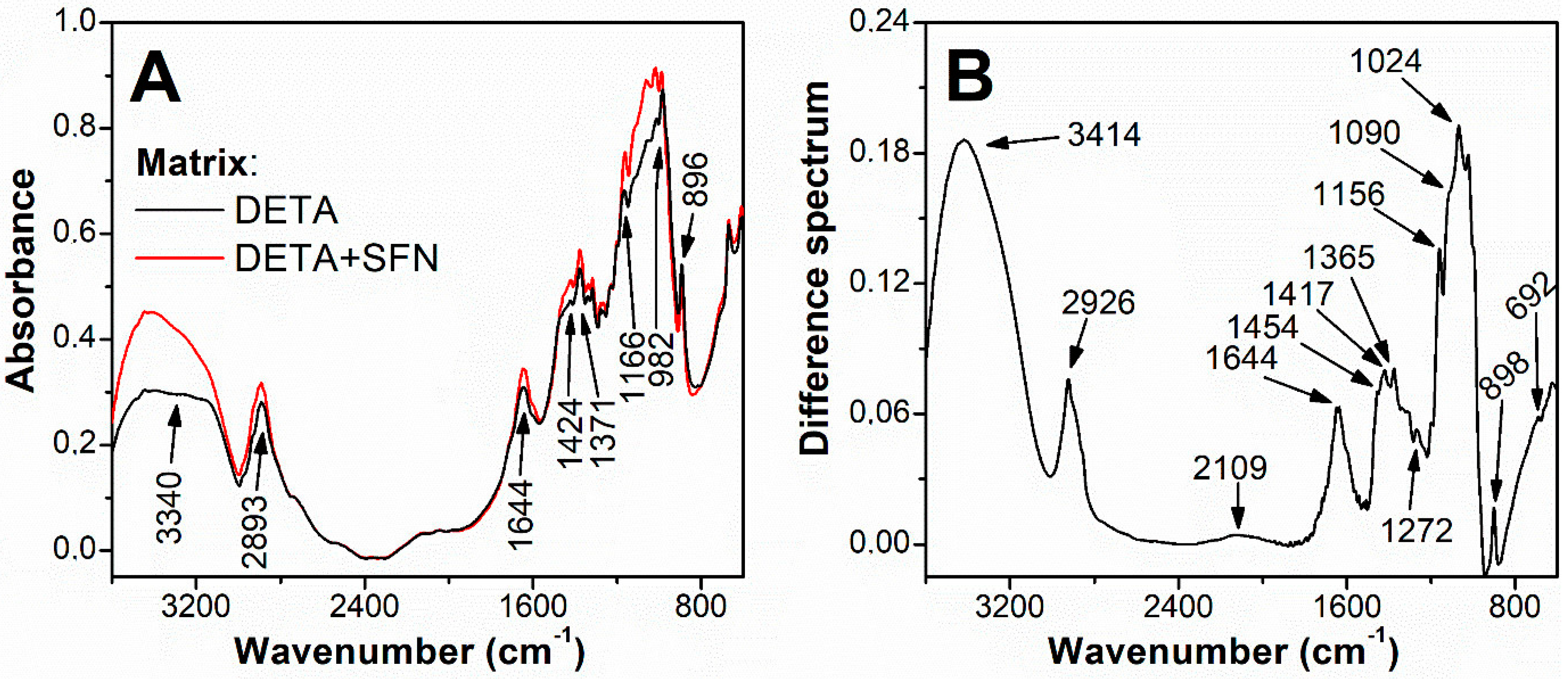
2.3. Preparation of Affinity Chromatography Column with SFN as Ligand
2.4. Protein Concentration Determination
2.5. Assay for Myrosinase Activity and Characterization of Myrosinase Properties
2.6. Electrophoresis of Proteins and Zymography Detection of Myrosinase Activity under Non-Denaturated Conditions
2.7. Identification of Proteins by MALDI-MS
2.8. Chemicals
3. Results and Discussion
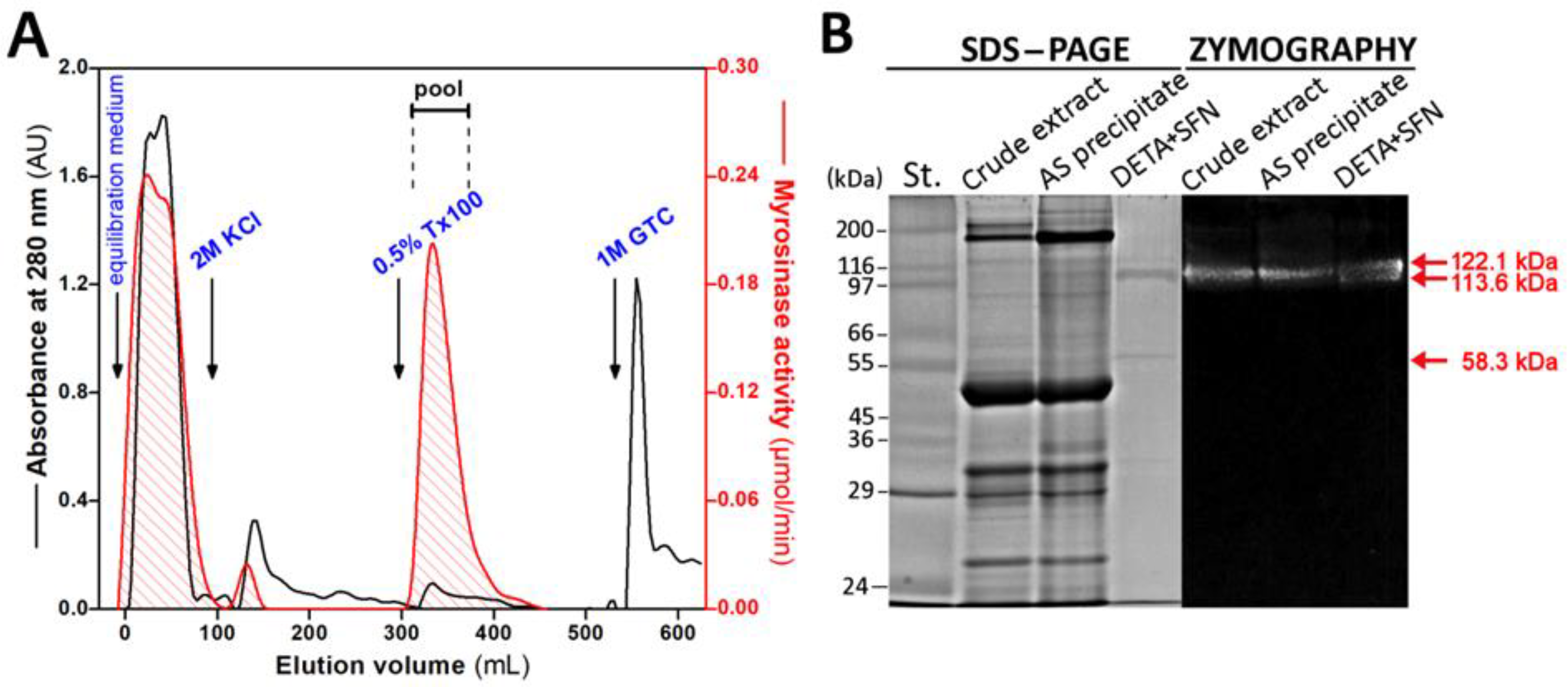
3.1. Purification of Myrosinase from L. sativum Seeds
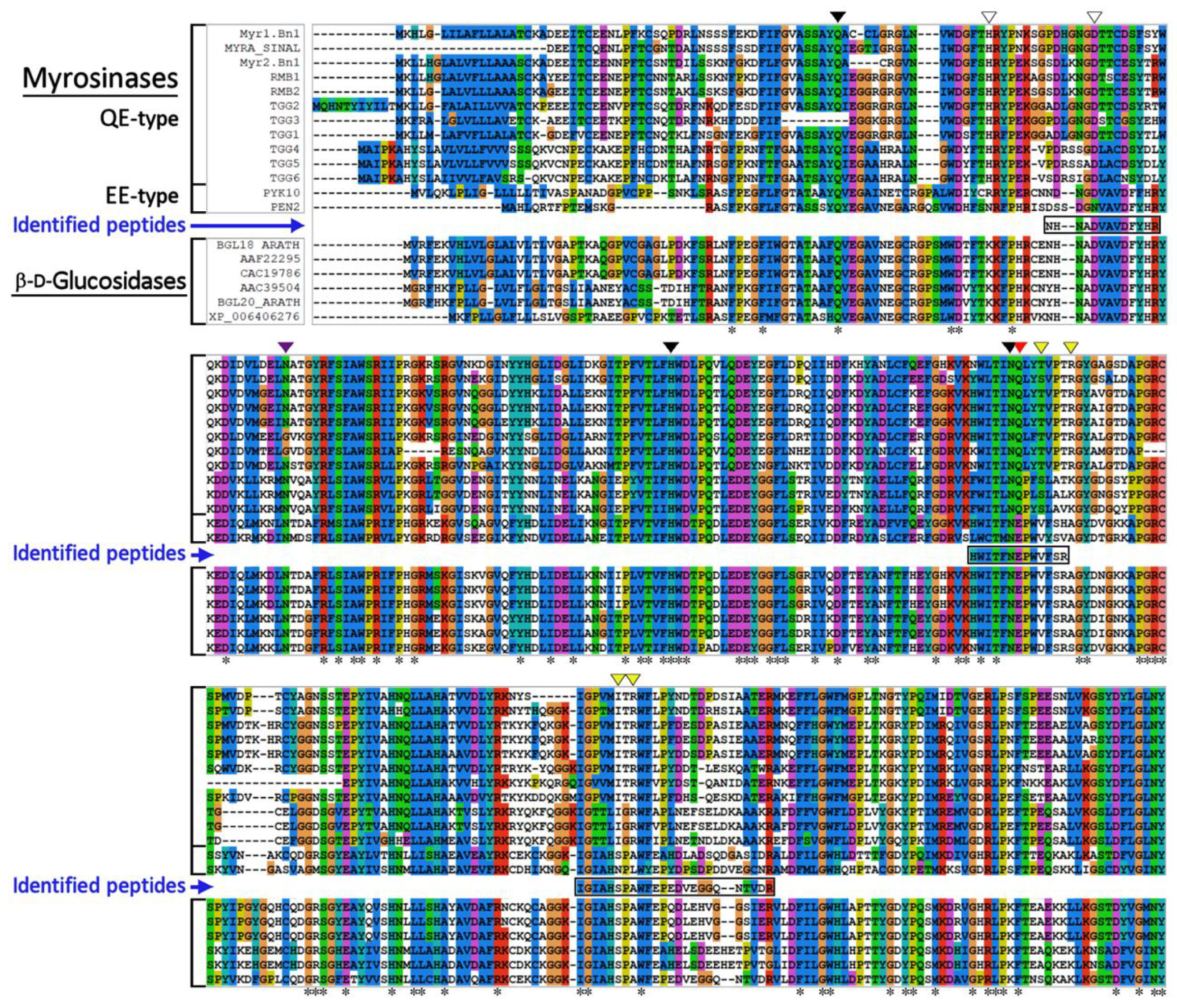
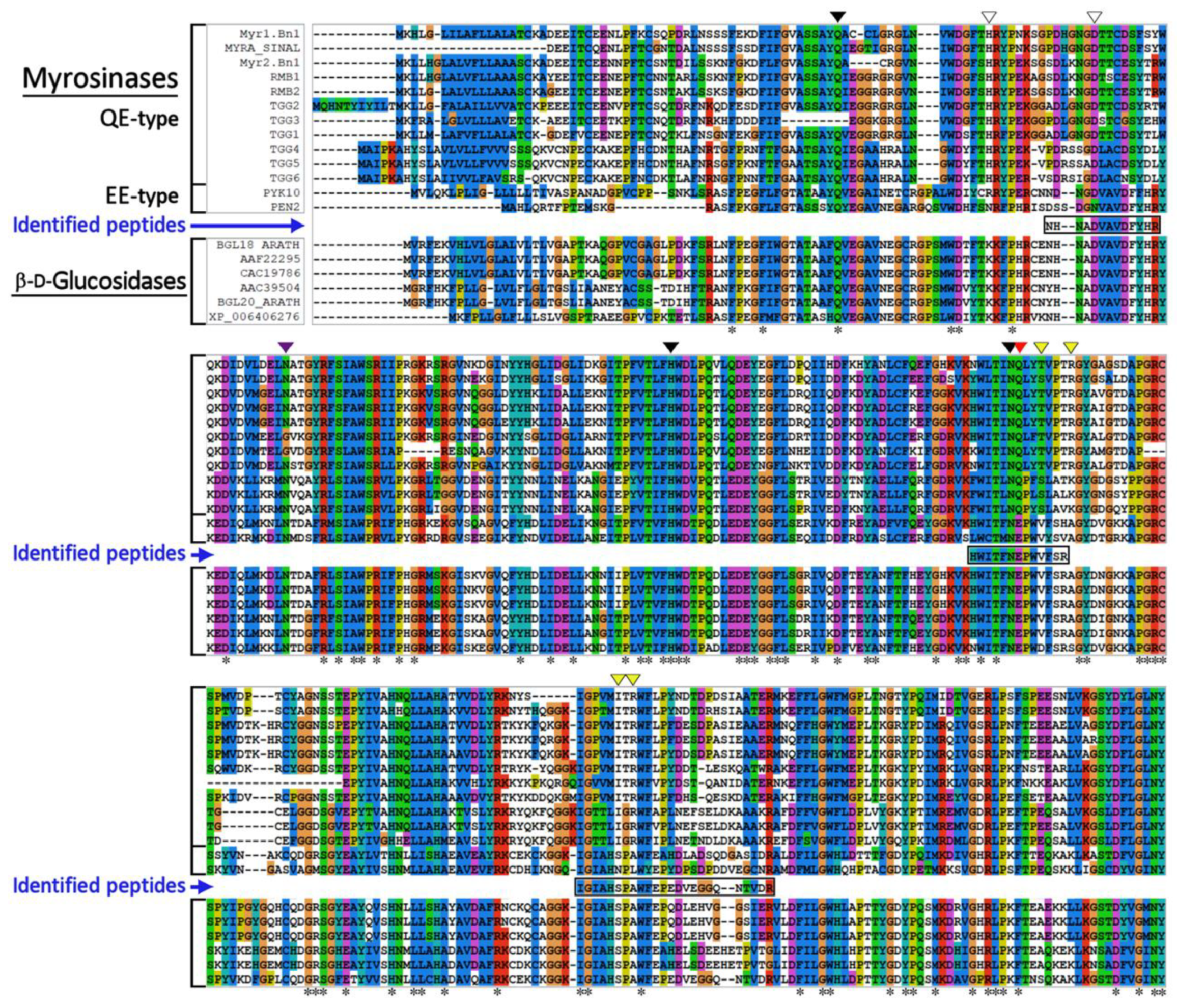
3.2. Identification of Myrosinase by Mass Spectrometry Analysis
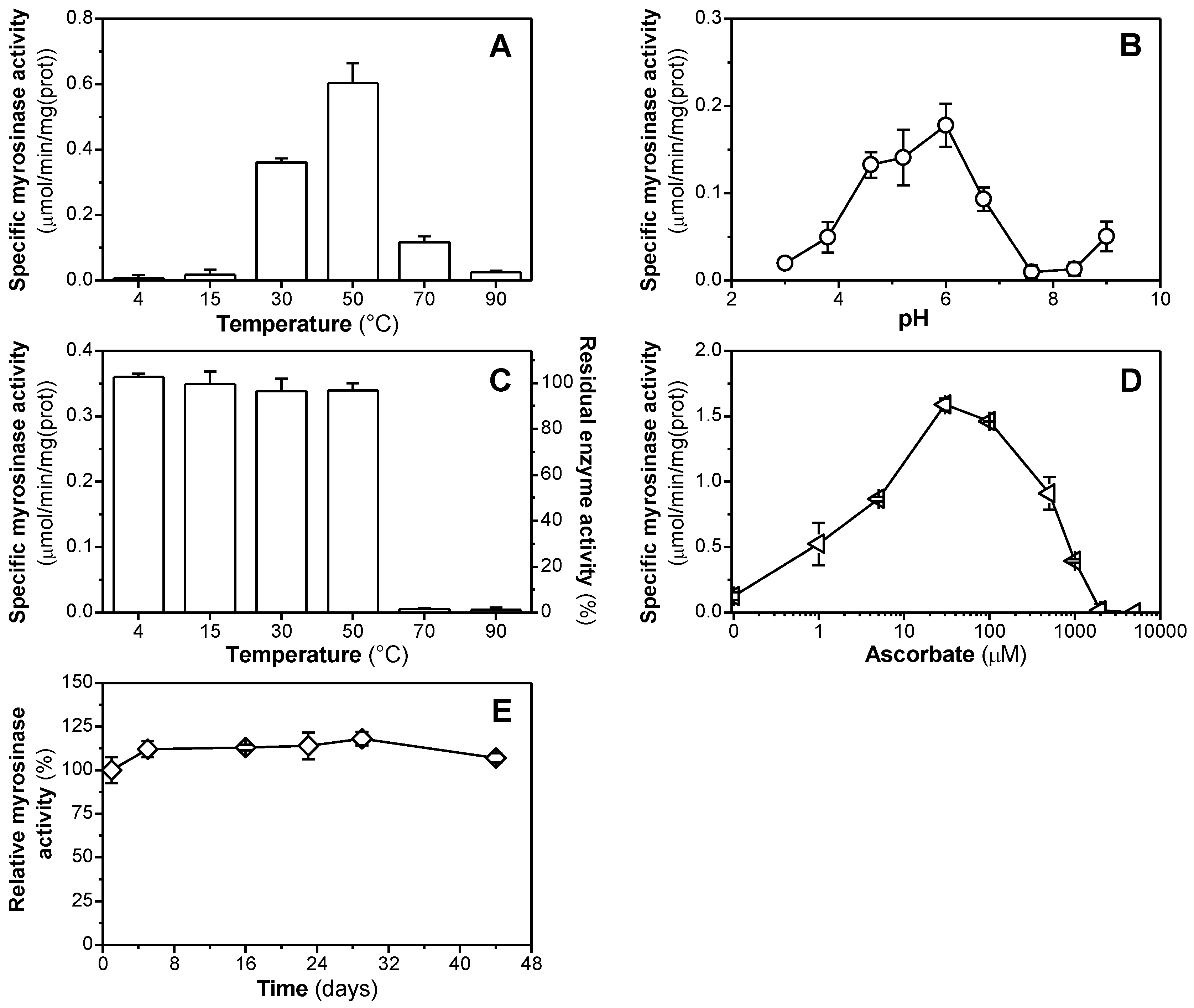
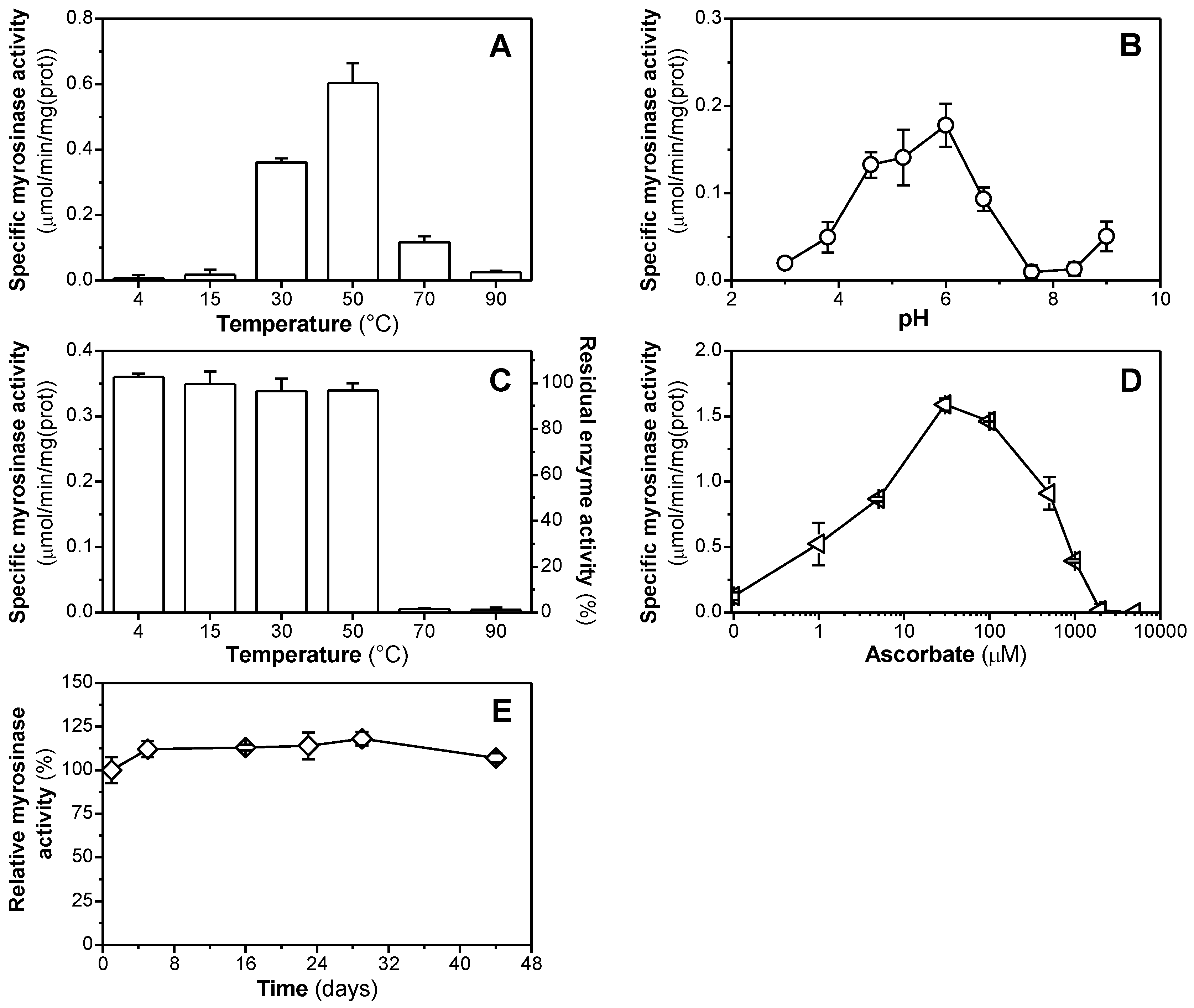
3.3. Enzymatic Properties of Purified Myrosinase
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AS | ammonium sulfate |
| 1,2-BDT | 1,2-benzenedithiol |
| CHCA | α-cyano-4-hydroxycinnamic acid |
| DETA | diethyltriamine |
| DTT | DL-dithiothreitol |
| EQM | equilibration buffer/medium |
| GOP | glucose oxidase-peroxidase |
| MES | 2-(N-morpholino)ethanesulfonic acid |
| pNPG | p-nitrophenyl-O-β-glucopyranoside |
| SFN | sulforaphane |
| TFA | trifluoroacetic acid |
| Tris | Tris(hydroxymethyl)aminomethane |
References
- Bhat, R.; Vyas, D. Myrosinase: Insights on structural, catalytic, regulatory, and environmental interactions. Crit. Rev. Biotechnol. 2019, 39, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bones, A.M.; Rossiter, J.T. The myrosinase-glucosinolate system, its organisation and biochemistry. Physiol. Plant. 1996, 97, 194–208. [Google Scholar] [CrossRef]
- Kelly, P.J.; Bones, A.; Rossiter, J.T. Sub-cellular immunolocalization of the glucosinolate sinigrin in seedlings of Brassica juncea. Planta 1998, 206, 370–377. [Google Scholar] [CrossRef]
- Textor, S.; Gershenzon, J. Herbivore induction of the glucosinolate–myrosinase defense system: Major trends, biochemical bases and ecological significance. Phytochem. Rev. 2009, 8, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Drakopoulos, D.; Kägi, A.; Gimeno, A.; Six, J.; Jenny, E.; Forrer, H.-R.; Musa, T.; Meca, G.; Vogelgsang, S. Prevention of Fusarium head blight infection and mycotoxins in wheat with cut-and-carry biofumigation and botanicals. Field Crops Res. 2020, 246, 107681. [Google Scholar] [CrossRef]
- Gimsing, A.L.; Kirkegaard, J.A. Glucosinolate and isothiocyanate concentration in soil following incorporation of Brassica biofumigants. Soil Biol. Biochem. 2006, 38, 2255–2264. [Google Scholar] [CrossRef]
- Horáková, K.; Drobnica, L.; Nemec, P.; Antos, A.; Kristián, P. Cytotoxic and cancerostatis activity of isothiocyanates and related compounds. I. Activity of some naturally occurring isothiocyanates and their synthetic analogues on HeLa-cells. Neoplasma 1968, 15, 169–176. [Google Scholar]
- Maina, S.; Misinzo, G.; Bakari, G.; Kim, H.-Y. Human, Animal and Plant Health Benefits of Glucosinolates and Strategies for Enhanced Bioactivity: A Systematic Review. Molecules 2020, 25, 3682. [Google Scholar] [CrossRef]
- Esteve, M. Mechanisms Underlying Biological Effects of Cruciferous Glucosinolate-Derived Isothiocyanates/Indoles: A Focus on Metabolic Syndrome. Front. Nutr. 2020, 7, 111. [Google Scholar] [CrossRef]
- Rakariyatham, K.; Yang, X.; Gao, Z.; Song, M.; Han, Y.; Chen, X.; Xiao, H. Synergistic chemopreventive effect of allyl isothiocyanate and sulforaphane on non-small cell lung carcinoma cells. Food Funct. 2019, 10, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Kontar, S.; Imrichova, D.; Bertova, A.; Mackova, K.; Poturnayova, A.; Sulova, Z.; Breier, A. Cell Death Effects Induced by Sulforaphane and Allyl Isothiocyanate on P-Glycoprotein Positive and Negative Variants in L1210 Cells. Molecules 2020, 25, 2093. [Google Scholar] [CrossRef] [PubMed]
- Drobnica, Ľ.; Kristián, P.; Augustín, J. The chemistry of the—NCS group. In Cyanates and Their Thio Derivatives (1977); John Wiley & Sons: Hoboken, NJ, USA, 1977; pp. 1003–1221. [Google Scholar] [CrossRef]
- Melrose, J. The Glucosinolates: A Sulphur Glucoside Family of Mustard Anti-Tumour and Antimicrobial Phytochemicals of Potential Therapeutic Application. Biomedicines 2019, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Carmen Martinez-Ballesta, M.; Carvajal, M. Myrosinase in Brassicaceae: The most important issue for glucosinolate turnover and food quality. Phytochem. Rev. 2015, 14, 1045–1051. [Google Scholar] [CrossRef]
- Lim, S.; Lee, E.J.; Kim, J. Decreased sulforaphene concentration and reduced myrosinase activity of radish (Raphanus sativus L.) root during cold storage. Postharvest Biol. Technol. 2015, 100, 219–225. [Google Scholar] [CrossRef]
- Bellostas, N.; Petersen, I.L.; Sørensen, J.C.; Sørensen, H. A fast and gentle method for the isolation of myrosinase complexes from Brassicaceous seeds. J. Biochem. Biophys. Methods 2008, 70, 918–925. [Google Scholar] [CrossRef]
- Li, X.; Kushad, M.M. Purification and characterization of myrosinase from horseradish (Armoracia rusticana) roots. Plant Physiol. Biochem. 2005, 43, 503–511. [Google Scholar] [CrossRef]
- Härtel, F.V.; Brandt, A. Characterization of a Brassica napus myrosinase expressed and secreted by Pichia pastoris. Protein Expr. Purif. 2002, 24, 221–226. [Google Scholar] [CrossRef]
- Ohtsuru, M.; Hata, T. The interaction of L-ascorbic acid with the active center of myrosinase. Biochim. Biophys. Acta 1979, 567, 384–391. [Google Scholar] [CrossRef]
- Wade, K.L.; Ito, Y.; Ramarathnam, A.; Holtzclaw, W.D.; Fahey, J.W. Purification of active myrosinase from plants by aqueous two-phase counter-current chromatography. Phytochem. Anal. PCA 2015, 26, 47–53. [Google Scholar] [CrossRef]
- Pihakaski, K.; Iversen, T.-H. Myrosinase in Brassicaceae: I. Localization of Myrosinase in Cell Fractions of Roots of Sinapis alba L. J. Exp. Bot. 1976, 27, 242–258. [Google Scholar] [CrossRef]
- Zhang, Y. The 1,2-benzenedithiole-based cyclocondensation assay: A valuable tool for the measurement of chemopreventive isothiocyanates. Crit. Rev. Food Sci. Nutr. 2012, 52, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wade, K.L.; Prestera, T.; Talalay, P. Quantitative determination of isothiocyanates, dithiocarbamates, carbon disulfide, and related thiocarbonyl compounds by cyclocondensation with 1,2-benzenedithiol. Anal. Biochem. 1996, 239, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Wang, C.-S.; Smith, R.L. Lowry determination of protein in the presence of Triton X-100. Anal. Biochem. 1975, 63, 414–417. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Bones, A.; Slupphaug, G. Purification, Characterization and Partial Amino Acid Sequencing of β-thioglucosidase from Brassica napus L. J. Plant Physiol. 1989, 134, 722–729. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Burmeister, W.P.; Cottaz, S.; Driguez, H.; Iori, R.; Palmieri, S.; Henrissat, B. The crystal structures of Sinapis alba myrosinase and a covalent glycosyl–enzyme intermediate provide insights into the substrate recognition and active-site machinery of an S-glycosidase. Structure 1997, 5, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Roman, J.; Castillo, A.; Cottet, L.; Mahn, A. Molecular Modelling of Broccoli Myrosinase and Its Interaction with Different Ligands. Chem. Eng. Trans. 2018, 64, 373–378. [Google Scholar] [CrossRef]
- Román, J.; Castillo, A.; Mahn, A. Molecular Docking of Potential Inhibitors of Broccoli Myrosinase. Molecules 2018, 23, 1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarem, M.; Lee, C.M.; Kafle, K.; Huang, S.; Chae, I.; Yang, H.; Kubicki, J.D.; Kim, S.H. Probing cellulose structures with vibrational spectroscopy. Cellulose 2019, 26, 35–79. [Google Scholar] [CrossRef]
- Wu, H.; Liang, H.; Yuan, Q.; Wang, T.; Yan, X. Preparation and stability investigation of the inclusion complex of sulforaphane with hydroxypropyl-β-cyclodextrin. Carbohydr. Polym. 2010, 82, 613–617. [Google Scholar] [CrossRef]
- Wu, Y.; Zou, L.; Mao, J.; Huang, J.; Liu, S. Stability and encapsulation efficiency of sulforaphane microencapsulated by spray drying. Carbohydr. Polym. 2014, 102, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Madhurambal, G.; Mariappan, M.; Ravindran, B.; Mojumdar, S.C. Thermal and FTIR spectral studies in various proportions of urea thiourea mixed crystal. J. Therm. Anal. Calorim. 2011, 104, 885–891. [Google Scholar] [CrossRef]
- Bernardi, R.; Finiguerra, M.G.; Rossi, A.A.; Palmieri, S. Isolation and biochemical characterization of a basic myrosinase from ripe Crambe abyssinica seeds, highly specific for epi-progoitrin. J. Agric. Food Chem. 2003, 51, 2737–2744. [Google Scholar] [CrossRef]
- Mahn, A.; Angulo, A.; Cabanas, F. Purification and characterization of broccoli (Brassica oleracea var. italica) myrosinase (beta-thioglucosidase glucohydrolase). J. Agric. Food Chem. 2014, 62, 11666–11671. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Cottaz, S.; Rollin, P.; Vasella, A.; Henrissat, B. High resolution X-ray crystallography shows that ascorbate is a cofactor for myrosinase and substitutes for the function of the catalytic base. J. Biol. Chem. 2000, 275, 39385–39393. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, S.; Andréasson, E.; Ekbom, B.; Granér, G.; Pontoppidan, B.; Taipalensuu, J.; Zhang, J.; Rask, L.; Meijer, J. Complex Formation of Myrosinase Isoenzymes in Oilseed Rape Seeds Are Dependent on the Presence of Myrosinase-Binding Proteins. Plant Physiol. 2002, 129, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Geshi, N.; Brandt, A. Two jasmonate-inducible myrosinase-binding proteins from Brassica napus L. seedlings with homology to jacalin. Planta 1998, 204, 295–304. [Google Scholar] [CrossRef]
- Taipalensuu, J.; Falk, A.; Ek, B.; Rask, L. Myrosinase-binding proteins are derived from a large wound-inducible and repetitive transcript. Eur. J. Biochem. 1997, 243, 605–611. [Google Scholar] [CrossRef]
- Rosenbergová, Z.; Kántorová, K.; Šimkovič, M.; Breier, A.; Rebroš, M. Optimisation of Recombinant Myrosinase Production in Pichia pastoris. Int. J. Mol. Sci. 2021, 22, 3677. [Google Scholar] [CrossRef]
- Kötzler, M.P.; Hancock, S.M.; Withers, S.G. Glycosidases: Functions, Families and Folds; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 1–14. [Google Scholar] [CrossRef]
- Nong, H.; Zhang, J.M.; Li, D.Q.; Wang, M.; Sun, X.P.; Zhu, Y.J.; Meijer, J.; Wang, Q.H. Characterization of a novel β-thioglucosidase CpTGG1 in Carica papaya and its substrate-dependent and ascorbic acid-independent O-β-glucosidase activity. J. Integr. Plant Biol. 2010, 52, 879–890. [Google Scholar] [CrossRef]
- Tsuruo, I.; Hata, T. Studies on Myrosinase in Mustard Seed Part V on the β-Glucosidase Activity of Myrosinase and the Interaction of Ascorbate with Myrosinase. Agric. Biol. Chem. 1968, 32, 1425–1431. [Google Scholar] [CrossRef]
- Durham, P.L.; Poulton, J.E. Enzymic Properties of Purified Myrosinase from Lepidium sativum Seedlings. Z. Für Nat. C 1990, 45, 173–178. [Google Scholar] [CrossRef]
- Andersson, D.; Chakrabarty, R.; Bejai, S.; Zhang, J.; Rask, L.; Meijer, J. Myrosinases from root and leaves of Arabidopsis thaliana have different catalytic properties. Phytochemistry 2009, 70, 1345–1354. [Google Scholar] [CrossRef]
- Durham, P.L.; Poulton, J.E. Effect of Castanospermine and Related Polyhydroxyalkaloids on Purified Myrosinase from Lepidium sativum Seedlings. Plant Physiol. 1989, 90, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Ludikhuyze, L.; Ooms, V.; Weemaes, C.; Hendrickx, M. Kinetic study of the irreversible thermal and pressure inactivation of myrosinase from broccoli (Brassica oleracea L. Cv. italica). J. Agric. Food Chem. 1999, 47, 1794–1800. [Google Scholar] [CrossRef]
- Oliviero, T.; Verkerk, R.; Van Boekel, M.A.; Dekker, M. Effect of water content and temperature on inactivation kinetics of myrosinase in broccoli (Brassica oleracea var. italica). Food Chem. 2014, 163, 197–201. [Google Scholar] [CrossRef]
- Verkerk, R.; Dekker, M. Glucosinolates and myrosinase activity in red cabbage (Brassica oleracea L. var. Capitata f. rubra DC.) after various microwave treatments. J. Agric. Food Chem. 2004, 52, 7318–7323. [Google Scholar] [CrossRef]
- Yen, G.-C.; Wei, Q.-K. Myrosinase activity and total glucosinolate content of cruciferous vegetables, and some properties of cabbage myrosinase in Taiwan. J. Sci. Food Agric. 1993, 61, 471–475. [Google Scholar] [CrossRef]
- Ghawi, S.K.; Methven, L.; Rastall, R.R.; Iranjan, K. Thermal and high hydrostatic pressure inactivation of myrosinase from green cabbage: A kinetic study. Food Chem. 2012, 131, 1240–1247. [Google Scholar] [CrossRef]
- Sangkret, S.; Pongmalai, P.; Devahastin, S.; Chiewchan, N. Enhanced production of sulforaphane by exogenous glucoraphanin hydrolysis catalyzed by myrosinase extracted from Chinese flowering cabbage (Brassica rapa var. parachinensis). Sci. Rep. 2019, 9, 9882. [Google Scholar] [CrossRef]
- Hochkoeppler, A.; Palmieri, S. Kinetic Properties of Myrosinase in Hydrated Reverse Micelles. Biotechnol. Prog. 1992, 8, 91–96. [Google Scholar] [CrossRef]
- Okunade, O.A.; Ghawi, S.K.; Methven, L.; Niranjan, K. Thermal and pressure stability of myrosinase enzymes from black mustard (Brassica nigra L. W.D.J. Koch. var. nigra), brown mustard (Brassica juncea L. Czern. var. juncea) and yellow mustard (Sinapsis alba L. subsp. maire) seeds. Food Chem. 2015, 187, 485–490. [Google Scholar] [CrossRef]
- Curiqueo, C.; Mahn, A.; Castillo, A. Broccoli Myrosinase cDNA Expression in Escherichia coli and Saccharomyces cerevisiae. Biomolecules 2022, 12, 233. [Google Scholar] [CrossRef]
- Rosenbergova, Z.; Hegyi, Z.; Ferko, M.; Andelova, N.; Rebros, M. Improved Production of Recombinant Myrosinase in Pichia pastoris. Int. J. Mol. Sci. 2021, 22, 11889. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, H.; Liang, X.; Zhou, W.; Qiu, Y.; Xue, C.; Sun, J.; Mao, X. Preparation of Sulforaphene from Radish Seed Extracts with Recombinant Food-Grade Yarrowia lipolytica Harboring High Myrosinase Activity. J. Agric. Food Chem. 2021, 69, 5363–5371. [Google Scholar] [CrossRef]
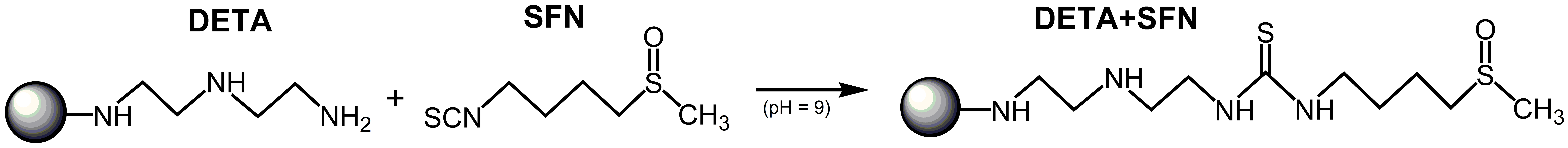

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
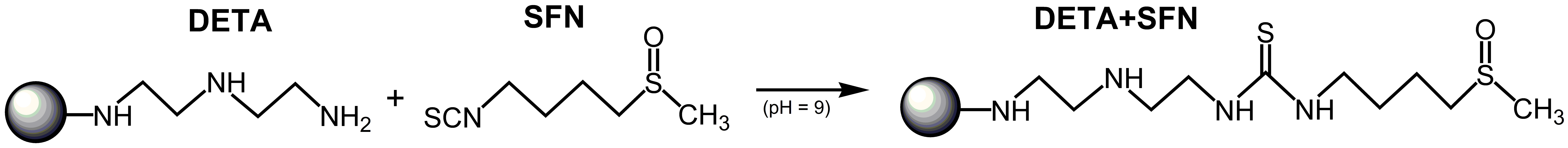{kind=link}
| Fraction | Protein (mg) | Activity (μmol/min) | Specific Activity (μmol/min/mg) | Recovery (%) | Purif. Factor (n-fold) |
|---|---|---|---|---|---|
| Crude extract | 203 ± 30 | 22.4 ± 3.3 | 0.11 ± 0.02 | 100 ± 15 | 1 ± 0.14 |
| AS (45%) | 61 ± 7 | 11.6 ± 1.3 | 0.2 ± 0.02 | 51.7 ± 4.1 | 1.8 ± 0.2 |
| DETA + SFN (Triton X-100 pool) | 0.44 ± 0.16 | 8.2 ± 2.8 | 18.6 ± 6.3 | 36.6 ± 8.9 | 169 ± 18 |
| Substrate | KM (×10−3 mol/L) | Vmax (×10−6 mol/L/s) | kcat (s−1) | kcat/KM (s−1·M−1) |
|---|---|---|---|---|
| Sinigrin | 0.574 ± 0.237 | 1.293 ± 0.147 | 7.9 ± 0.9 | 13,763 |
| Glucoraphanin | 1.134 ± 0.256 | 1.378 ± 0.102 | 8.3 ± 0.6 | 7319 |
| Sinalbin | 0.632 ± 0.235 | 0.846 ± 0.091 | 5.1 ± 0.5 | 8070 |
| Substrate | Specific Enzyme Activity (µmol/min/mg) | Relative Activity (% of Control) |
|---|---|---|
| Sinigrin (control) | 0.125 ± 0.014 | 100 |
| p-NP-β-D-Glc | 0.017 ± 0.001 | 13 |
| p-NP-α-D-Glc | 0.001 ± 0.001 | <1 |
| p-NP-β-D-Gal | 0.001 ± 0.001 | <1 |
| o-NP-β-D-Gal | 0.001 ± 0.001 | <1 |
| p-NP-α-D-Man | 0.001 ± 0.001 | <1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galádová, H.; Polozsányi, Z.; Breier, A.; Šimkovič, M. Sulphoraphane Affinity-Based Chromatography for the Purification of Myrosinase from Lepidium sativum Seeds. Biomolecules 2022, 12, 406. https://doi.org/10.3390/biom12030406
Galádová H, Polozsányi Z, Breier A, Šimkovič M. Sulphoraphane Affinity-Based Chromatography for the Purification of Myrosinase from Lepidium sativum Seeds. Biomolecules. 2022; 12(3):406. https://doi.org/10.3390/biom12030406
Chicago/Turabian StyleGaládová, Helena, Zoltán Polozsányi, Albert Breier, and Martin Šimkovič. 2022. "Sulphoraphane Affinity-Based Chromatography for the Purification of Myrosinase from Lepidium sativum Seeds" Biomolecules 12, no. 3: 406. https://doi.org/10.3390/biom12030406
APA StyleGaládová, H., Polozsányi, Z., Breier, A., & Šimkovič, M. (2022). Sulphoraphane Affinity-Based Chromatography for the Purification of Myrosinase from Lepidium sativum Seeds. Biomolecules, 12(3), 406. https://doi.org/10.3390/biom12030406