1. Introduction
Congenital Disorders of Glycosylation (CDG) are multisystemic metabolic disorders caused by defects in N-linked, or O-linked oligosaccharides, shared substrates, glycophosphatidylinositol (GPI) anchors, or dolichols pathways [
1]. There are two types of CDGs: type I CDGs are diagnosed when there are defects in the assembly or transfer of the dolichol-linked glycan in either the cytosol or the endoplasmic reticulum (ER). Type II CDGs are diagnosed when there are processing defects of the glycan in the ER or the Golgi apparatus.
ALG13 gene (Asparagine-linked glycosylation homolog 13) dysfunction is related to type I CDGs. The product of the
ALG13 gene, the Alg13 protein, is involved in the N-glycosylation process. Several mutations of the
ALG13 gene have been reported, related mostly to epilepsy [
2,
3,
4,
5,
6,
7], but also to kidney failure [
8]. Considering ALG13-CDG, the c.320A>G (p.Asn107Ser) variant is by far the most frequent in affected female heterozygotes [
7]. To date, it has been described in 46 females and only three males [
2,
4,
5,
6,
7,
9,
10]. Yet, the details of the effect of this mutation on glycosylation process remains unknown.
The Alg13 protein is a highly conserved X-linked uridine diphosphate (UDP)-N-acetylglucosaminyltransferase required for the transfer of N-acetylglucosamine (GlcNAc) onto the extending lipid-linked oligosaccharide (LLO) structure, dolichol-P-P GlcNAc [
11]. This early step in the LLO synthesis pathway is crucial for the N-glycosylation process, as was shown by studies on yeasts. The yeast Alg13 protein is localized mostly in the cytoplasm. However, it can be also found in the ER as a part of a heterodimer, while it binds with an ER transmembrane protein Alg14. The ER-bound complex of Alg13-Alg14 is crucial for both cell viability and proper N-glycosylation, since lack of this interaction causes profound defects in both [
11]. Other studies showed that the yeast Alg13 and Alg14 proteins are distantly related to the bacterial MurG UDP-GlcNAc glycosyltransferase which is involved in peptidoglycan and are implicated to playing a role in N-glycosylation [
12,
13].
In early 2005, Chantret et al. [
12] suggested that for the second step of N-glycosylation in eukaryotes, two proteins homologous to the N-terminal and C-terminal domains of the bacterial MurG are required. Later that year, Gao et al. confirmed their results, pointing out Alg13 and Alg14 as the proteins involved, and showed that Alg14 functions as a membrane anchor that recruits Alg13 to the cytosolic face of the ER, where catalysis of GlcNAc2-PP-dol occurs [
13]. Wang et al. speculated that since the sugar donor binding domain and the sugar acceptor binding domain exist separately in Alg13 and Alg14 in some organisms, their bipartitioning can offer many advantages, such as flexibility in the protein activity regulation and the ability to recruit substrates in both the cytosol and the membrane [
14]. They proposed Alg13 as a general UDP-GlcNAc carrier, regulating the delivery of UDP-GlcNAc to a variety of membrane-associated proteins involved in complex carbohydrate synthesis. The modification of the binding affinity could provide vital consequences. Moreover, Wang et al. analyzed the behavior of yeast Alg13 in the presence of the UDP-GlcNAc substrate. They showed that the residues located on the two helices are the most disturbed by the presence of the substrate [
14].
Protein N-glycosylation is a two-step process: it starts with the transfer of an oligosaccharide from a lipid-linked oligosaccharide onto proteins and is initiated by UDP-GlcNAc:dolichol phosphate
N-acetylglucosamine-1-phosphate transferase (GPT) encoded by
DPAGT1. It was reported that deficiencies of
DPAGT1 are also related to a group of CDG disorders (formerly Ij type) [
13]. The second step differs between prokaryotes and eukaryotes. It has been previously shown that prokaryotes require a monomeric enzyme corresponding to MurG, while in the case of eukaryotes the role of bacterial MurG is carried out by two enzymes, such as Alg13 and Alg14.
There is a high sequence similarity between the prokaryotic and eukaryotic enzymes involved in the first step of N-glycosylation [
15]. The product of the
DPAGT1 gene, the GPT enzyme, is mostly membrane-bound in the ER. For example, the hamster GPT enzyme, which was examined by Dan et al. [
16], consists of 10 membrane units, so-called spans, connected by loops, two of which have a lumenal orientation (loop 6/7 and the C-terminal region), while two others (loops 1/2 and 9/10) are cytosolic. Moreover, loop 9/10 has highly conserved elements that are essential for enzyme function; the C-terminal of the 10 span plays a role in the stabilization of the GPT [
17], while spans 2 and 7 contain potential dolichol recognition sequences. Their more recent results suggest that GPT may occur in an oligomeric state [
18]. It should be noted here that the GPT protein, a product of the
DPAGT1 gene is called Alg7 in
S. cerevisiae [
16], and its interaction with the Alg13 and Alg14 has already been analyzed by Chantret et al. [
12], as well as Noffz et al. [
19], who were first to show the Alg7-Alg13-Alg14 interaction in eukaryotes. Moreover, their results suggest that the Alg7-Alg13-Alg14 complex might be present in a multimeric form. Interestingly, they also showed that the Alg7 and Alg13 are not directly in contact, but rather they associate by close contact with Alg14 [
19].
Taken together all the above mentioned information, a clear mechanism appears; however, it still requires experimental confirmation. N-glycosylation is a process which takes place on the endoplasmic reticulum, and in the case of eukaryotes requires the presence of three enzymes, Alg7, Alg13, and Alg14, which can create a complex together. They may share a common binding site cavity, or the cavity is separated by some flexible regions, which act as molecular gates [
20]. Alg7 is mostly bound in the membrane of the ER, and it facilitates the first step of the process. The Alg14 protein is in close contact with the Alg7. The only soluble protein in this complex, the Alg13, acts as a transporter, delivering the substrate directly into the Alg7-Alg14 complex. Since the investigated mutation is localized on the human Alg13 loop which participates in both Alg13-Alg14 dimer formation and the substrate binding, we hypothesized that it can have multi-level consequences: (i) influence the substrate carrier properties, (ii) modify the activity/selectivity of the Alg13-Alg14 dimer, and finally (iii) disrupt the dimer formation.
Contrary to other known variants related to
ALG13 dysfunctions, where transferrin isoelectric focusing (IEF) revealed notably increased asialo- and disialotransferrin fractions, in the patients harboring the p.Asn107Ser mutation, a normal glycosylation pattern was observed (in four individuals), similarly to mass spectrometry (MS) results, previously described in one individual [
2]. There is, however, one report showing MS evidence of the slightly reduced glycosylation with the absence of one glycan [
5]. Since the laboratory data are ambiguous, and the number of reported c.320A>G ALG13-CDGs is still very limited, we would like to present different IEF patterns observed in the transferrin IEF results among the female patients described in our previous publication [
10], in order to expand the reported phenotypic variability resulting from the c.320A>G
ALG13 variant. We complemented the report by an exhaustive homology modeling study of the human Alg13 protein and the complex formation with Alg14. We ran in total 6 µs of molecular dynamics (MD) simulations to examine the behavior of the Alg13 protein, its complex with the UDP-GlcNAc substrate and the Alg13-Alg14 heterodimer in the presence of the substrate. We identified a conformational change which is crucial for the substrate transport and heterodimer formation that was not observed in the Asn107Ser variant of the human Alg13 protein. We hope that our results shed light on the role of the human Alg13 protein.
3. Results
This section is divided into experimental and computational parts. The first part presents the transferrin isoform patterns of our three patients obtained by IEF. We observed differences in the transferrin isoform patterns between our patients, as well as between samples taken from the same patient at different times. These observations have led us to search for differences in the mechanism of action between the native and Asn107Ser variants of the Alg13 protein. Therefore, the experimental part is complemented by the computational part. The latter describes the homology modeling of human Alg13 protein, the molecular dynamics simulations of the apo (“empty”) structure of Alg13, and its complex with the UDP-GlcNAc substrate. We also created an Alg13-Alg14 complex homology model based on bacterial MurG proteins with a bound substrate, and finally we ran MD simulations of the Alg13-Alg14 heterodimer in the presence of the substrate.
3.1. Clinical Data
For detailed descriptions of the patients, please refer to our previous article by Paprocka et al. [
10]. Here we present the anthropometric results and the transferrin IEF data, as well as a clinical update.
During the latest assessment, Patient 1 was 3 years and 8 months of age. She attended a kindergarten for children with special needs, to which she adapted very well. She needed foot orthoses, but was able to walk independently with some coordination issues. She has a broad-based gait with uplifted forearms. Her speech was still severely delayed, she could say only “mama”. Seizures were not observed and antiepileptic drugs had been reduced.
Patient 2, now 6 years old, started to walk independently at the age of four, and it is no longer a wide-based gait but more natural. She is continuously rehabilitated and undergoes special education with a psychologist, neurologist, SI therapist, hand therapy, and group social therapy. There are still problems with eating—she often chokes, does not eat on her own (cannot use a spoon/fork), and cannot drink from a cup despite constant training at home and in therapy classes—she drinks from a bottle. The patient still does not speak, and her imitation ability is very limited. For two years, she has had alternative communication introduced. When asked, she can often show the correct answer (but only those she is regularly shown and told); she distinguishes between symbols yes/no and often shows adequately to the situation. Unfortunately, the proband understands very little, can only react to her name, and addresses regularly repeated requests such as “hands up”. She does not report basic physiological needs and is still diapered. As for the patient’s medications, she is still on Sabril and Lamitrin S (for which the dosage is increasing, trends toward monotherapy). Epileptic seizures occur only during sleep, but they are quite frequent—every second/third night. They appear as follows: the patient wakes up during the night, and for a few minutes, she periodically blinks her eyes and tenses her body, after which she falls asleep. Seizures very rarely require Relsed infusion. The patient has been attending a private kindergarten for the second year now. Although the progress is slow, she is very eager to participate in the classes and even demands continuing when they are over. The patient has a very gentle and cheerful disposition, smiles often and loves to cuddle. She is very friendly; she demonstrates many behaviors on the autism spectrum, but the diagnosis has not been made.
Patient 3, aged 7 years and 9 months, is the most severely delayed. She is wheelchaired, cannot speak and suffers from everyday epileptic attacks, especially during bedtime. As the patient cannot swallow solid pieces, she only eats liquid food. Her diet is high in carbohydrates, mainly potatoes, and therefore a ketogenic diet is not possible.
3.2. Anthropometric Measurements
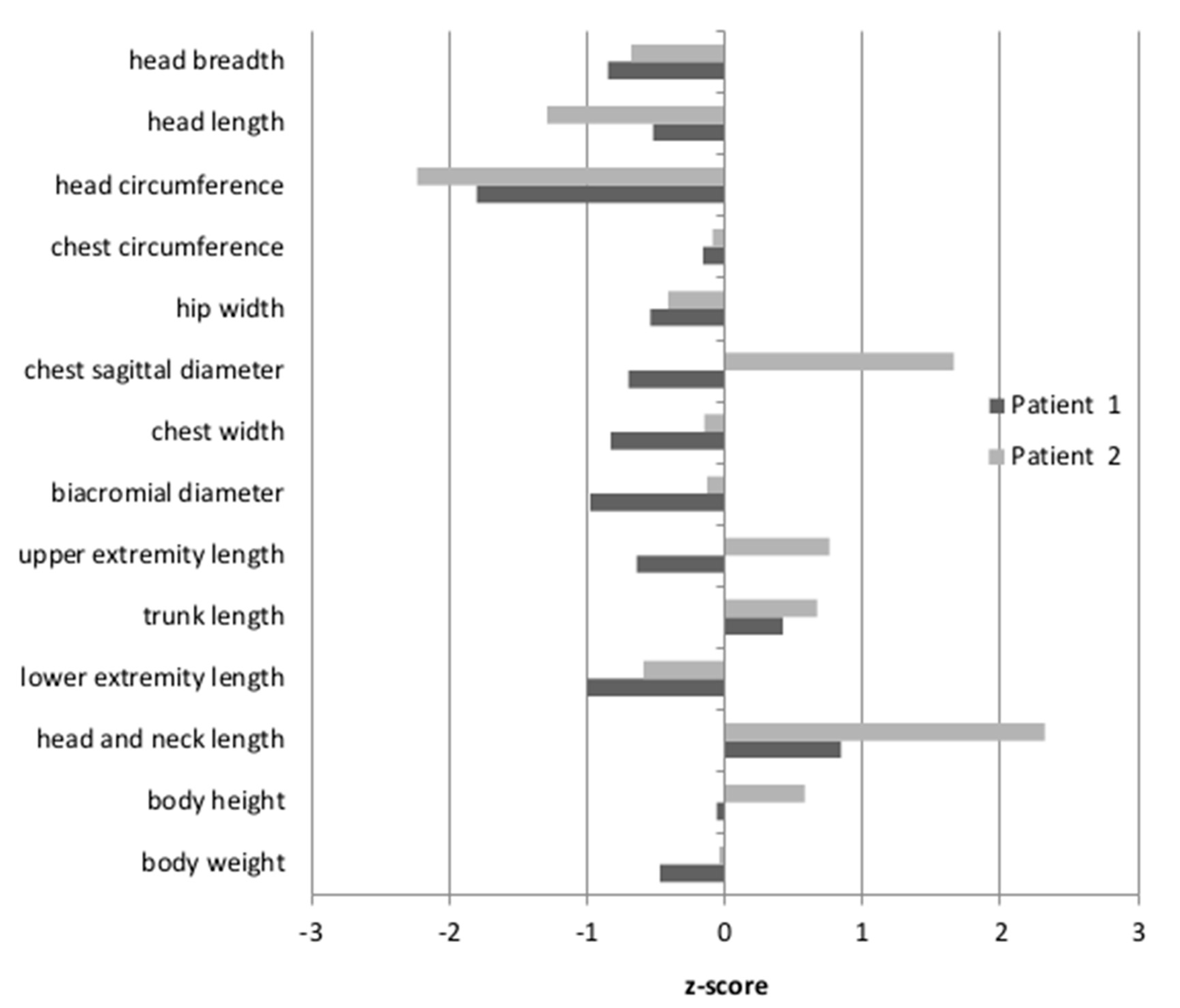
Anthropometric measures were undertaken for Patients 1 and 2, while Patient 3 was unavailable. The results were then standardized, based on the mean and the standard deviation of the examined feature in a given age group of healthy Polish children. The resulting z-scores for Patient 1 and Patient 2 are shown in
Figure 1. The body height of the patients was between the 50th and 75th percentile, therefore within the normal range (z-score −0.06–0.58). For Patient 2, the length of the head and neck were above the 97th percentile (z-score 2.32). For Patient 1, only the tendency for an elongated head and neck was observed, although the measured values fell within the normal range (z-score 0.84). Moreover, only Patient 2 had a convex chest (the z-score for the sagittal chest diameter was 1.66). The head circumferences in both patients were below the 3rd percentile (Patient 1: z-score −1.8; Patient 2: z-score −2.23). Other body proportion parameters of both patients did not differ from their healthy peers.
3.3. Transferrin Isoforms Pattern Detection by IEF
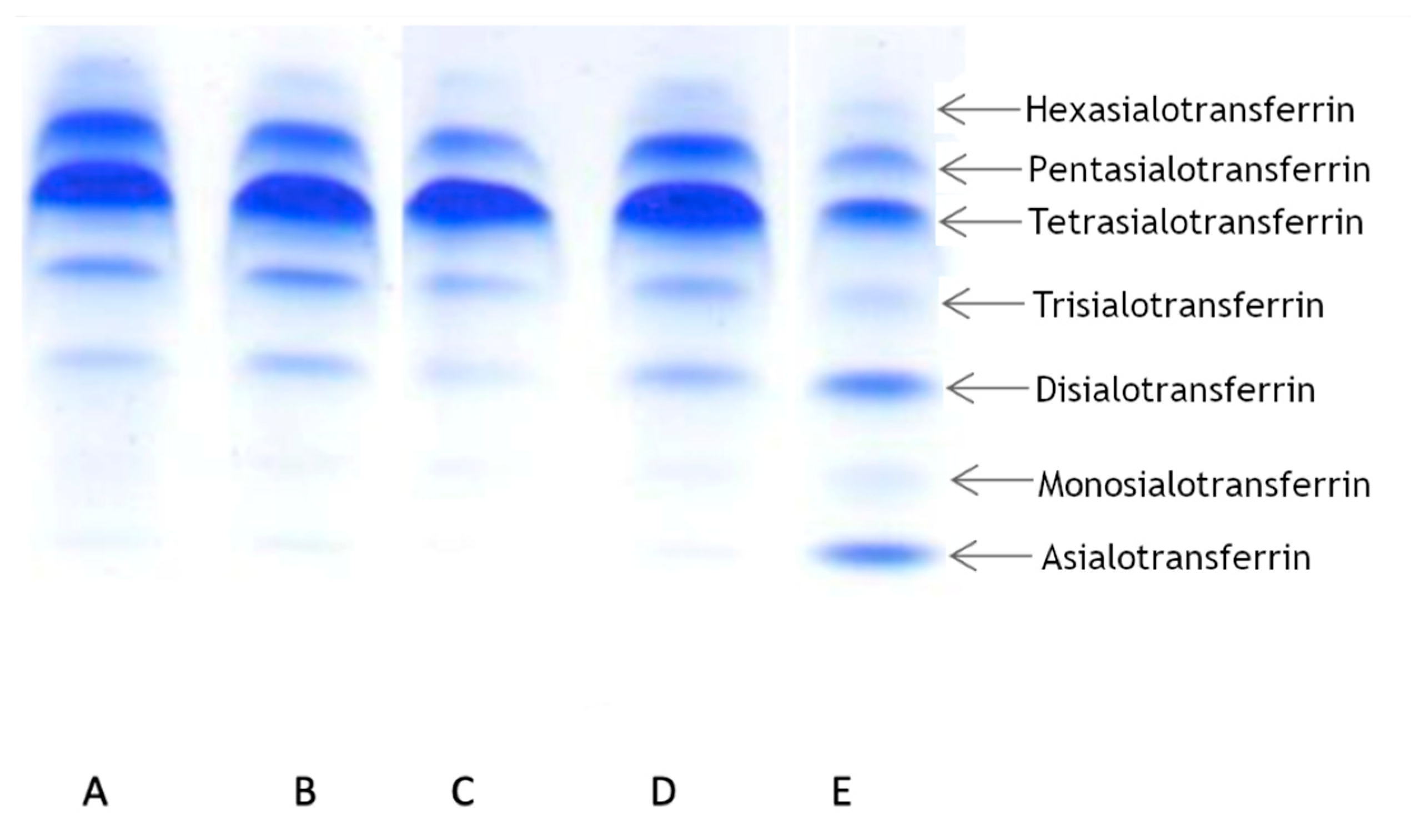
To determine the transferrin pattern, we used IEF. For each patient we ran two analyses at different times. Patient 1 showed a normal transferrin pattern in both trials. Patients 2 and 3 displayed abnormalities in at least one trial. Patient 2 displayed a slight elevation of asialo-, monosialo-, and disialo fraction in the first trial, while in the second trial the pattern was normal. Patient 3 displayed a normal pattern in the first trial, but in the second trial we observed a slight elevation in only the disialo- fraction. The results are summarized in
Table 1 and presented in
Figure 2.
In light of the available publications on the subject, these results seemed surprising. Therefore, we also analyzed samples using another method (CE), which confirmed the results (
Table 2). Patient 1 displayed normal value of transferrin level, while two other patients displayed increased levels of transferrin. These findings encouraged us to examine the mechanism of Alg13 protein dysfunction, so we used an in silico approach to determine the structure of human Alg13 and the Alg13-Alg14 heterodimer.
3.4. Alg13 Protein Modeling
Since the crystal structure of human Alg13 protein remains unknown, we created a homology model using the I-TASSER webserver, which we will refer to as hAlg13_hm. The model had a structural similarity with the yeast Alg13 structures described by Wang et al. [
14] and Raman et al. [
43], despite relatively low sequence similarity (28%; human Alg13 protein UniProt ID: Q9NP73). The model was evaluated by ProSA and PROCHECK to assess its overall quality. Both tools identified that the model has good quality (ProSA Z-score: −7.62, PROCHECK overall score: −0.58). It is worth noting that the sequence of human Alg13 protein was also shorter (165 aa) relative to the yeast Alg13 (224 aa in Wang’s model and 201 aa in Raman’s model, respectively, UniProt ID: P53178), as we modeled the short
ALG13-is2 isoform of human Alg13 protein. These two factors can substantially impact the quality of the hAlg13_hm model. Both
Saccharomyces cerevisiae Alg13 models were obtained as a set of ensembles, so for our analysis we used only the structure of the first ensemble (
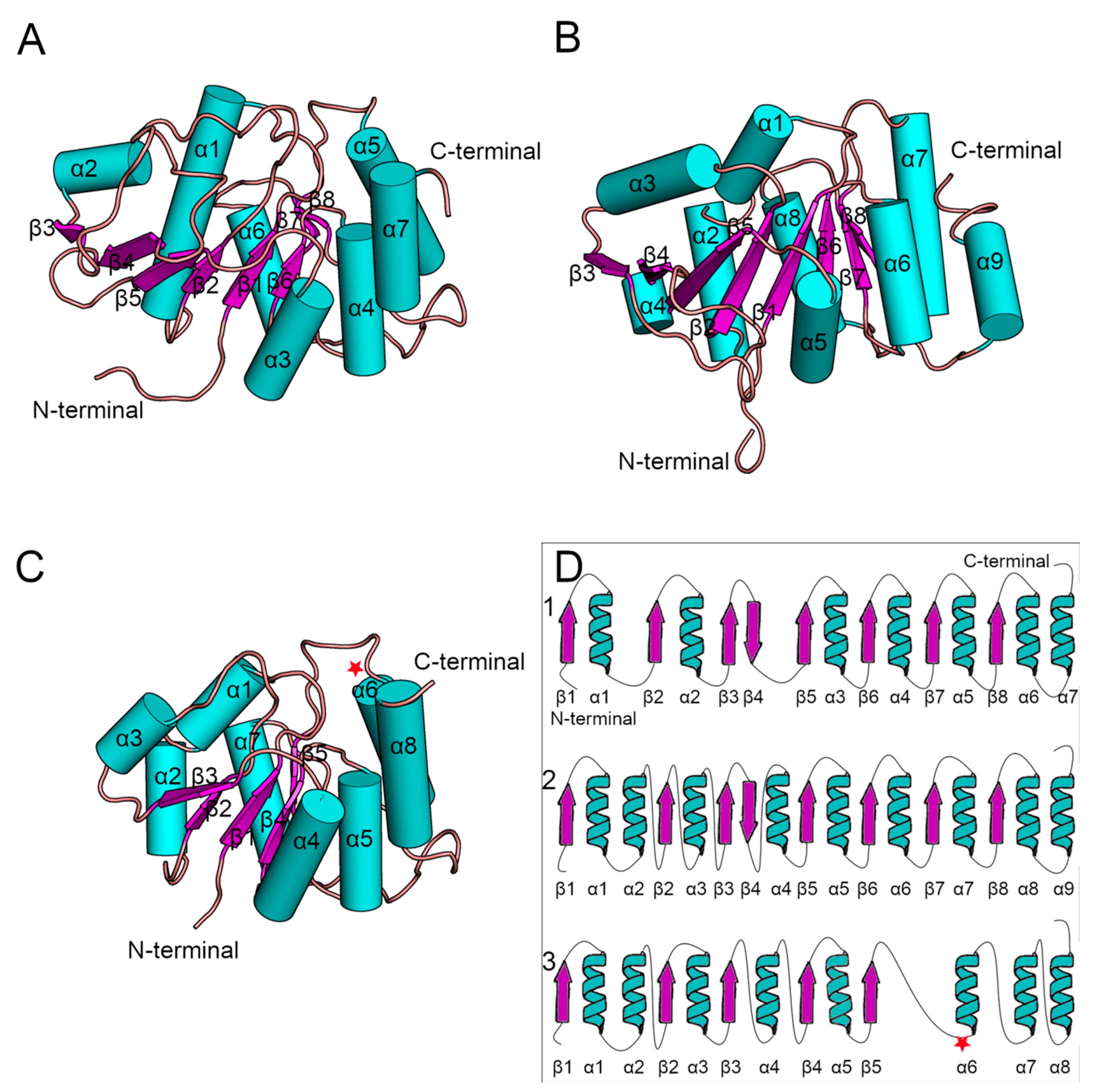
Figure 3). Additionally, their sequences were identical when compared using BLAST [
44]. The yeast Alg13 structures displayed a high overall similarity with two major differences: (i) the α1 helix of Wang’s model was divided into two helices, namely α1 and α2, in Raman’s model, and (ii) the α4 helix in Raman’s model was inserted between two β-strands, namely β4 and β5, while in Wang’s model the β4 and β5 strands were separated only by a short loop. While the yeast Alg13 had a common β-strand pattern (eight mostly parallel β-strands with an antiparallel β4 strand) surrounded by up to nine helices, the hAlg13_hm was relatively smaller. It comprised five parallel β-strands and eight α-helices. It was most similar to Raman’s model with the same pattern of the two consecutive α1 and α2 helices. The 107 residue was located on the loop between the β5 strand and α6 helix.
3.5. Analysis of the Mutability of the Asn107 Position c.320A>G p.107N>S
A single-point mutation changing asparagine into serine in position 107 of the human Alg13 is caused by a substitution of the second adenine with guanine in the AAT codon (c.320A>G). According to the results of bioinformatic analyses using the PredictSNP webserver to assess the effect of the Asn107Ser mutation, the mutation is deleterious, although the protein is produced (
Supplementary Table S2). However, we could speculate that the introduction of the substitution in the protein structure should display a neutral or stabilizing character. To assess the effect of the mutation on the protein structure and folding, we calculated the differences in the Gibbs free energy of protein folding between the native protein and the mutant variants using the FoldX suite. Interestingly, the introduction of serine was found to have a potentially destabilizing effect for protein folding (1.4 kcal/mol, measured as ΔΔG between the native protein and the mutant). Since the destabilizing effect is small, the protein can still be produced; however, it seems to have an effect on Alg13 function. Given the ambiguous results of transferrin IEF in our patients, we decided to run several experiments and analyze the dynamics of the created model of the human Alg13 structure with and without the UDP-GlcNAc substrate, as well as analyzing the dynamics of the Alg13-Alg14 complex.
3.6. Comparison of the Dynamics of Native Alg13 and the Asn107Ser Variant
To investigate the behavior and effect of the introduced mutation on the human Alg13 protein, we ran multiple repetitions of molecular dynamics simulations of the previously built model and the model with the introduced Asn107Ser mutation. To describe the simulations’ results, two commonly used metrics were used, RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation), to measure the quality of each simulation and the model itself. RMSD measures the overall protein stability; it is used to detect conformational changes. RMSF provides information on the average flexibility of a single residue during the simulation time. RMSD and RMSF for the native protein and mutant variant are shown in
Supplementary Figure S1. Moreover, since the residue at position 107 is located on a loop, we determined the flexibility of this region by measuring the distance between the Cα atom of the 107 residue and the most stable part of the protein, which was the β4 strand (based on the RMSF values). Higher values suggested a higher flexibility of the region hosting the Asn107Ser mutation, which may be related to problems during substrate transport or complex formation.
Human Alg13 protein is relatively stable. The RMSD plots of the native Alg13 structure fluctuated around 4–5 Å and suggested that the protein does not undergo rapid conformational changes. On the contrary, the RMSD plots of the Asn107Ser variant substantial and rapid conformational changes. In three out of five simulations the RMSD values exceeded 8 Å, while for the two other simulations they were fluctuating near 6 Å (
Supplementary Figure S1). However, in one simulation the RMSD values at first reached 10 Å, but after the first 20 ns they decreased near 4 Å and remained on the same level till the end of the simulation. These differences in protein behavior are visible also on the RMSF plots (
Supplementary Figure S1). While the RMSF values of the native Alg13 protein share the same pattern in all MD simulations, the RMSF plot of the Asn107Ser variant shows differences between each run. For the native Alg13 protein the most flexible region is its C-terminal with α7 and α8 helices. The same tendency is visible on the RMSF plot of the Asn107Ser variant; however, the RMSF values are higher relative to the RMSF values of the native structure. We also observed that the final positions of residue 107 in both the native and the mutant variant were different from the starting positions.
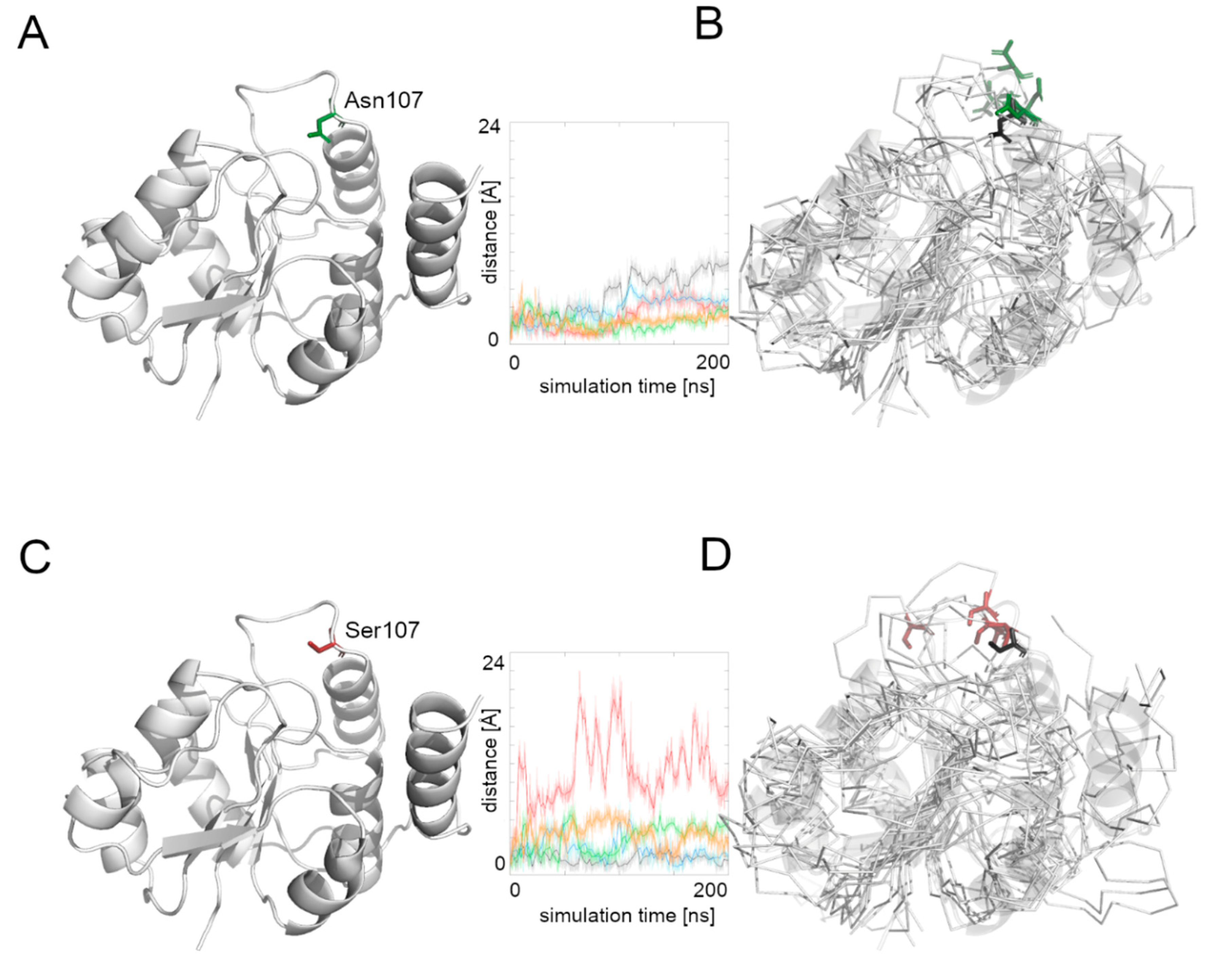
We then measured the distance between the starting position of residue’s 107 Cα atom and its position during the whole simulation time (
Figure 4). The Asn107 residue’s Cα atom was stable for the first half of the most MD simulations, and then it moved away from the starting point position, but the maximal distance did not exceed 10 Å. In the case of the Asn107Ser variant, the Cα atom of the 107 residue was more flexible during the whole MD simulation and in one case moved far away (more than 20 Å) from its starting position. This observation suggests flexibility of the loop on which the residue 107 is located, and also that the Asn107Ser variant shows greater flexibility for this region relative to the native protein. It also suggests that the substrate binding and its further transport may be less effective in the case of the Asn107Ser variant. Therefore, in the next step we focused on substrate transport analysis.
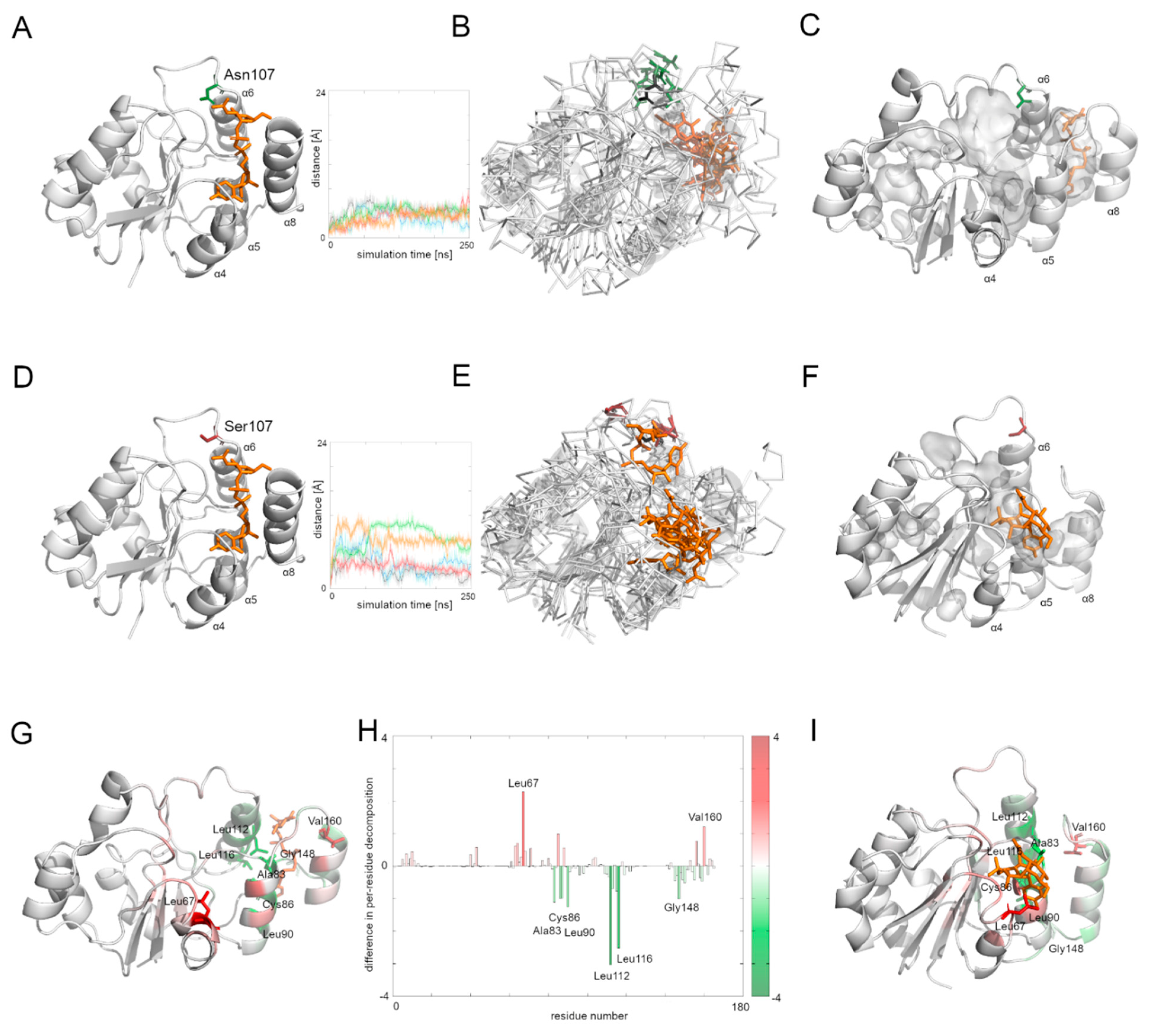
3.7. Comparison of the Dynamics of the Native Alg13 and the Asn107Ser Variant in a Complex with UDP-GlcNAc
To investigate the influence of the UDP-GlcNAc substrate on the Alg13 protein behavior, we ran five repetitions of the native Alg13and the Asn107Ser variant complexed with UDP-GlcNAc. The position of the substrate was modeled according to the similar crystal structure of the P. aeruginosa MurG protein (PDB ID: 3s2u).
The human Alg13 protein’s complex with the substrate was relatively stable in both the native protein and the Asn107Ser variant. Interestingly, the RSMD and RMSF plots of the native Alg13 suggest that in the presence of the substrate the protein gained flexibility in the C-terminal and the region comprising the α3 helix and β3 strand (
Supplementary Figure S2). On the other hand, the RMSD and RMSF plots of the Asn107Ser variant suggest that its structure became more rigid. The RMSD values for the Asn107Ser variant are lower than those of the native structure, and the same tendency is observed in the case of the RMSF plots. However, in the case of the RMSF plot of the Asn107Ser variant, a high peak is observed in the loop region hosting the mutation that is not present on the RMSF plot of the native protein (
Supplementary Figure S2). We measured the distance between the starting position of the Cα atom of the 107 residue during the whole simulation time in both protein-substrate complexes (
Figure 5). In the case of the native Alg13 protein in the presence of the UDP-GlcNAc substrate, we observed that the Cα atom moved away in each simulation and the distance fluctuated around 6 Å suggesting that its position was stable. This was not observed in the case of the Asn107Ser variant, in which the distances were greater than in the native protein; in two simulations they exceeded 10 Å. Additionally, when we compared the ending positions of the 107 residue in both Alg13 proteins, we saw that in the case of the native Alg13 protein the position of the 107 residue remained similar in each simulation, while in the case of the Asn107Ser variant the ending positions of the 107 residue were different. Moreover, we observed differences in the UDP-GlcNAc substrate movements. In the case of the native Alg13 the substrate moved into a cavity which was created by a conformational change of α8 helix shifting toward α5 and α6 helices. The UDP-GlcNAc substrate remained in that cavity for most of the simulation time. This behavior of the α5, α6, and α8 helices was not observed in the case of the Asn107Ser variant (
Figure 5E,F). It is also worth noting that the α8 helix is thought to be related to the interaction with Alg14 protein [
13]. Therefore, our observation might suggest the potential effect of the Asn107Ser variant on Alg13-Alg14 complex formation in the presence of the substrate.
Using MMPBSA and MMGBSA approaches we analyzed the free energy of the substrate binding in both the native and the Asn107Ser variant of the human Alg13 protein (
Supplementary Table S3 and Figure S3). The results suggest that in both systems, the binding was stable. We also analyzed the per-residue contribution of the human Alg13 protein with the substrate. While in the case of the native human Alg13 protein only the C-terminal region interacted with the UDP-GlcNAc substrate, in the case of the Asn107Ser variant both the N-terminal and C-terminal, as well as the middle regions of the protein, had contact with the substrate. We also observed that in the complex with the native Alg13 the substrate was located in a cavity created by hydrophobic and negatively charged residues, such as: Leu112 (−3.1 kcal/mol), Leu116 (−2.5 kcal/mol), Leu87 (−1.5 kcal/mol), Leu90 (−1.2 kcal/mol), Ala83 (−1.2 kcal/mol), Cys86 (−1.1 kcal/mol), and Pro147 (−1.0 kcal/mol). We also observed several residues with positive values of per-residue contribution which were repulsing the substrate; however, none of them reached the level of +1 kcal/mol. In the case of the Asn107Ser variant the substrate was positioned differently, and the contributing residues were mostly hydrophobic, polar and positively charged, such as: Leu67 (−2.3 kcal/mol), Val160 (−1.3 kcal/mol), Leu163 (−1.2 kcal/mol), Phe156 (−1.1 kcal/mol), and Ser85 (−1.0 kcal/mol). Similarly, as in the case of the native Alg13 protein, we observed several repulsing residues with a contribution of lower than +1 kcal/mol. We also measured the differences between per-residue contribution between the native protein and the Asn107Ser variant of the human Alg13 protein by subtracting the contribution values of the mutant variant from the native protein values (
Figure 5H). The regions on the plot for which the contribution values are positive (shown as red bars) represent the region in which the substrate interacted with the Asn107Ser variant, which shows that in the case of the Asn107Ser variant the substrate binds in different region than in the native protein. Those results suggest that for the Asn107Ser variant the substrate transport is disturbed. In the next step we analyzed the Alg13-Alg14 dimer with the UDP-GlcNAc substrate to gain a whole picture of the substrate transport.
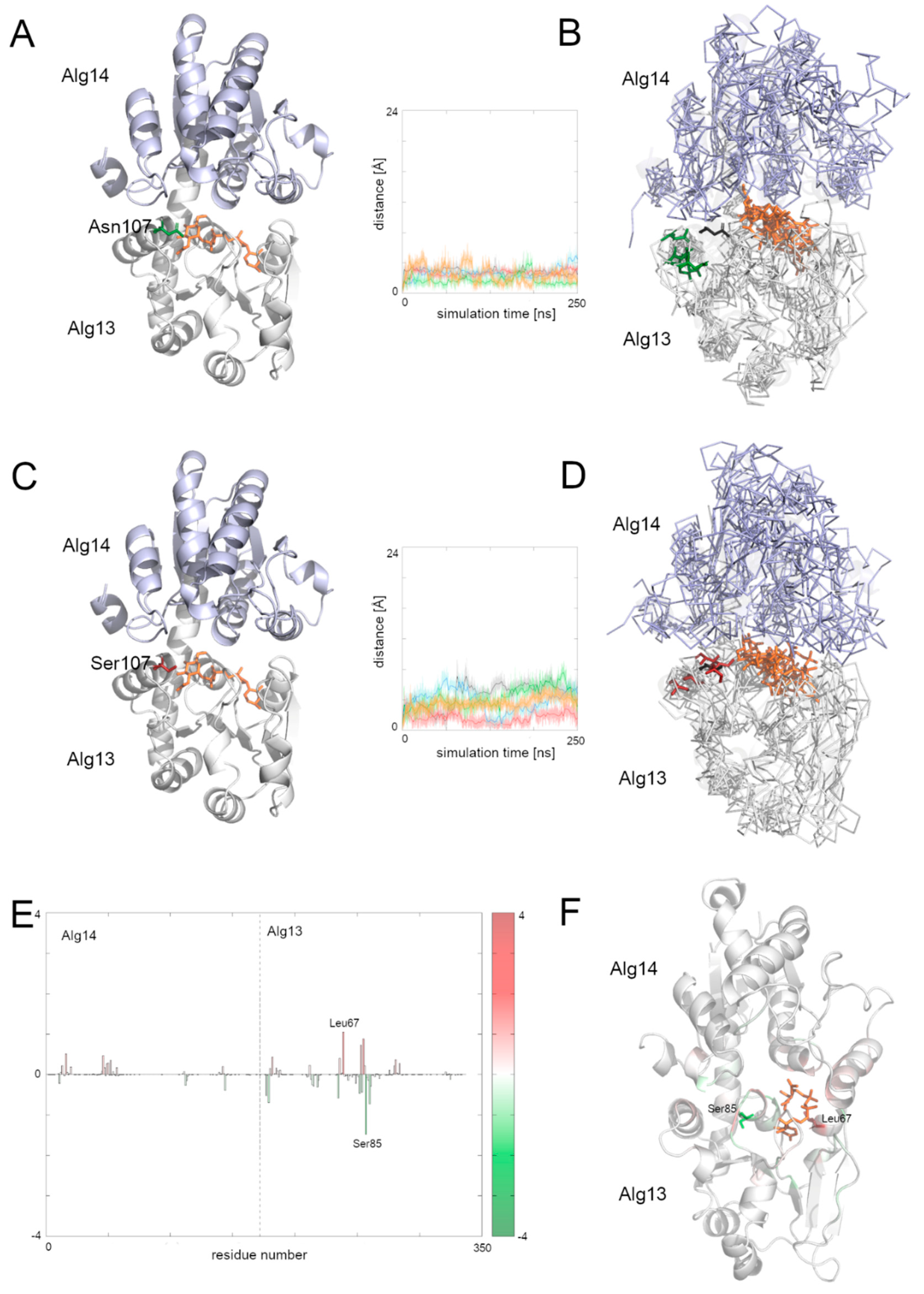
3.8. Comparison of the Dynamic of the Native Alg13 and Asn107Ser Variant in the Alg13-Alg14 Complex with UDP-GlcNAc
To examine the behavior of the Alg13-Alg14 dimer, we needed to create another homology model, this time of the dimer. We employed the Modeller 9v19 to create a homology model based on two templates of the MurG protein complexed with the substrate—E. coli MurG and P. aeruginosa MurG. The position of the substrate was modeled according to the P. aeruginosa MurG which had better resolution. The human Alg14 protein structure was downloaded from the SWISS-MODEL repository (ID: Q96F25). This model was also evaluated using ProSA and PROCHECK tools, which assessed its overall quality as good (ProSA Z-score: −5.1, PROCHECK overall score: −0.25). The resulting dimer structure was also assessed using the same tools (ProSA Z-score: −1.42, PROCHECK overall score: −0.15) indicating that the dimer model has good quality; however slightly lower than the quality of Alg13 and Alg14 models separately.
Although the bacterial MurG protein consists of two domains forming a monomer, the human Alg13-Alg14 dimer structurally resembles it. The human Alg14 overlaps with the N-terminal domain, while the Alg13 overlaps with the C-terminal domain. However, the sequence similarity between those proteins is relatively low (below 28% identity). The N-terminal domain of MurG as well as the human Alg14 model, consists of six parallel β-strands surrounded by five α-helices in the case of MurG and seven in the case of the human Alg14 structure. The C-terminal domain of MurG also consists of six parallel β-strands surrounded by seven α-helices and an additional α-helix which interacts with the β-sheet of the N-terminal domain. As previously shown [
6], in the Alg13-Alg14 complex the α8 helix of Alg13, which interacts with the Alg14 protein, adopts the same conformation as the last α-helix of the MurG structure.
To examine the behavior of the system, we analyzed the RMSD of the two domains separately as well as for the whole complex. We also divided the RMSF plot in order to show each domain separately (
Supplementary Figure S3). In general, both complexes with the native structure and with the Asn107Ser variant remained stable during the simulation time. Especially the dimer with the native Alg13 domain remained stable, with one exception: in one case the RMSD value increased up to 7 Å after 40 ns and remained at this level till the end of simulation. The RMSF plots of both complexes share mostly the same pattern of high peaks; however the RMSF values differ substantially (
Supplementary Figure S4). This suggests that even though the structures are relatively stable, they may display different network of interactions. We therefore also checked the binding free energies of the substrate to the Alg13-Alg14 heterodimer using MMPBSA and MMGBSA (
Supplementary Table S4) and the per-residue contribution to the substrate binding (
Figure 6 and
Supplementary Figure S5). Our results suggest that in both heterodimers (i.e., with the native Alg13 and the Asn107Ser variant) the substrate was stably bound.
Supplementary Figure S5 shows that the biggest differences in the contribution to the substrate binding were found in the C-terminal and the middle region of the Alg13 domain. The results show that in the case of the heterodimer with the native Alg13 residues with the highest contribution were hydrophobic and polar, such as: Ser85 of Alg13 (−1.5 kcal/mol), His14 of Alg14 (−1.4 kcal/mol), Glu88 of Alg13 (−1.3 kcal/mol), Thr7 of Alg13 (−1.1 kcal/mol), and Phe5 of Alg13 (−1.1 kcal/mol). On the other hand, the highest contribution in the case of the Alg13-Alg14 dimer with the Asn107Ser variant have the following residues: Leu67 of Alg13 (−1.5 kcal/mol), His14 of Alg14 (−1.4 kcal/mol), Thr16 of Alg14 (−1.3 kcal/mol), Gly13 of Alg14 (−1.2 kcal/mol), Ala83 of Alg13 (−1.1 kcal/mol), and Thr10 of Alg13 (−1.0 kcal/mol). Interestingly, even though the substrate remained in the same cavity in both systems, it interacted with different residues. Only His14 of the Alg14 domain was identified as high contributing in both cases. It is also worth noticing that the Alg13 residue 107 was not involved in the substrate binding. We also compared the distances between the starting position of the Cα atom of the Alg13 107 residue during the whole MD simulation time in both heterodimers (
Figure 6). We observed that the overall distance was similar between both heterodimers, which suggests that in the presence of the UDP-GlcNAc substrate and the Alg14 protein, the Alg13 107 residue displayed similar behavior. This also suggests that the Alg13-Alg14 complex may be functional, and that the most crucial step is substrate transport.
5. Conclusions
A single-point mutation c.320A>G in the ALG13 gene results in the Asn107Ser variant of the Alg13 protein. According to the bioinformatic analyses, the mutation itself is not deleterious; however, it causes a multisystemic metabolic disorder, called ALG13-CDG, in the patients with the c.320A>G mutation.
Analysis of five repetitions of MD simulations of the homology model of the human Alg13, both as an apo structure or in complex with the UDP-GlcNAc substrate suggests that the Asn107Ser variant alters flexibility of the loop hosting this mutation. Such influence was not observed for the Alg13-Alg14 dimer. The observations from the MD simulations, as well as data from a thorough literature search allowed us to speculate about a plausible mechanism for the role of the Asn107Ser variant of the Alg13 protein during N-glycosylation. While Alg14 is membrane-bound, Alg13 acts as a transporter, it binds the substrate and moves it to Alg14. In the presence of the substrate, Alg13 and Alg14 form a heterodimer in which the second step of the N-glycosylation occurs. Our results suggest that in the case of the Asn107Ser variant of the human Alg13 protein, the Alg13-Alg14 complex remains functional, but the process of the substrate binding to Alg13 and its further transport might be disturbed, thus disturbing the N-glycosylation process.
We also showed the IEF results of three female patients diagnosed with ALG13-CDG caused by c.320A>G. Since the ALG13-CDG caused by c.320A>G is very rare, we would like to report on further female patients with different spectra of clinical severity, with abnormal patterns of serum transferrin.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}