
The Epstein-Barr Virus Hacks Immune Checkpoints: Evidence and Consequences for Lymphoproliferative Disorders and Cancers


Abstract
{kind=link}
1. Introduction
2. Overview of the EBV-Associated Lymphomas
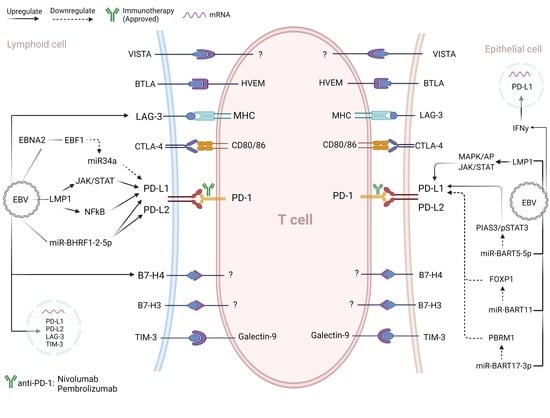
3. Interplay between Immune Checkpoints and the EBV
EBV Regulation of Immune Checkpoints: Insights Based on PD-1/PD-Ls Upregulation
4. Clinical Impact of Immune Checkpoints in Human Cancers
5. Immune Checkpoint Blockade in the EBV-Associated Cancers
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- IARC. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans A Review of Human Carcinogens—Part B: Biological Agents. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2011; Volume 100, ISBN 978-92-832-0134-2. [Google Scholar]
- IARC. IARC. IARC Working Group on the Evaluation of Carcinogenic Risk to Humans Biological Agents—Epstein-Barr Virus. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2012; Volume 100B, pp. 45–88. ISBN 9789283213192. [Google Scholar]
- Young, L.; Yap, L.-F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef]
- Hahn, A.M.; Huye, L.E.; Ning, S.; Webster-Cyriaque, J.; Pagano, J.S. Interferon Regulatory Factor 7 Is Negatively Regulated by the Epstein-Barr Virus Immediate-Early Gene, BZLF-1. J. Virol. 2005, 79, 10040–10052. [Google Scholar] [CrossRef]
- Van Gent, M.; Griffin, B.D.; Berkhoff, E.G.; Van Leeuwen, D.; Boer, I.G.J.; Buisson, M.; Hartgers, F.C.; Burmeister, W.P.; Wiertz, E.J.; Ressing, M.E. EBV Lytic-Phase Protein BGLF5 Contributes to TLR9 Downregulation during Productive Infection. J. Immunol. 2010, 186, 1694–1702. [Google Scholar] [CrossRef]
- Van Gent, M.; Gram, A.M.; Boer, I.G.J.; Geerdink, R.J.; Lindenbergh, M.F.S.; Lebbink, R.J.; Wiertz, E.J.; Ressing, M.E. Silencing the shutoff protein of Epstein-Barr virus in productively infected B cells points to (innate) targets for immune evasion. J. Gen. Virol. 2015, 96, 858–865. [Google Scholar] [CrossRef]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV Immunoevasins vIL-10 and BNLF2a Protect Newly Infected B Cells from Immune Recognition and Elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef]
- Zuo, J.; Thomas, W.A.; Haigh, T.A.; Fitzsimmons, L.; Long, H.M.; Hislop, A.D.; Taylor, G.S.; Rowe, M. Epstein-Barr Virus Evades CD4+ T Cell Responses in Lytic Cycle through BZLF1-mediated Downregulation of CD74 and the Cooperation of vBcl-2. PLoS Pathog. 2011, 7, e1002455. [Google Scholar] [CrossRef]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.H.J.; Ressing, M.E.; Rowe, M. The DNase of Gammaherpesviruses Impairs Recognition by Virus-Specific CD8 + T Cells through an Additional Host Shutoff Function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein-Barr Virus-Associated Malignancies: Roles of Viral Oncoproteins in Carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef]
- Caetano, B.F.R.; Jorge, B.A.S.; Müller-Coan, B.G.; de Oliveira, D.E. Epstein-Barr virus microRNAs in the pathogenesis of human cancers. Cancer Lett. 2020, 499, 14–23. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Gallagher, N.J.; Blake, S.M.S.; Dawson, C.W.; Young, L.S. Activation of the p38 Mitogen-activated Protein Kinase Pathway by Epstein-Barr Virus-encoded Latent Membrane Protein 1 Coregulates Interleukin-6 and Interleukin-8 Production. J. Biol. Chem. 1999, 274, 16085–16096. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Graässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRN-As. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV MicroRNAs in Primary Lymphomas and Targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef]
- Münz, C. Co-Stimulatory Molecules during Immune Control of Epstein Barr Virus Infection. Biomolecules 2021, 12, 38. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, ISBN 9789283244943. [Google Scholar]
- Stebegg, M.; Kumar, S.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The Expression Pattern of Epstein-Barr Virus Latent Genes In Vivo Is Dependent upon the Differentiation Stage of the Infected B Cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr Virus and the Origins of Associated Lymphomas. New Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A. Epstein Barr Virus-Associated Hodgkin Lymphoma. Cancers 2018, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Medeiros, L.J.; Xu-Monette, Z.Y.; Zhang, S.; O’Malley, D.P.; Orazi, A.; Zuo, Z.; Bueso-Ramos, C.E.; Yin, C.C.; Liu, Z.; et al. Epstein-Barr virus–positive nodular lymphocyte predominant Hodgkin lymphoma. Ann. Diagn. Pathol. 2014, 18, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Huppmann, A.R.; Nicolae, A.; Slack, G.W.; Pittaluga, S.; Davies-Hill, T.; Ferry, J.A.; Harris, N.L.; Jaffe, E.S.; Hasserjian, R.P. EBV May Be Expressed in the LP Cells of Nodular Lymphocyte–predominant Hodgkin Lymphoma (NLPHL) in Both Children and Adults. Am. J. Surg. Pathol. 2014, 38, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Braeuninger, A.; Küppers, R.; Strickler, J.G.; Wacker, H.-H.; Rajewsky, K.; Hansmann, M.-L. Hodgkin and Reed-Sternberg cells in lymphocyte predominant Hodgkin disease represent clonal populations of germinal center-derived tumor B cells. Proc. Natl. Acad. Sci. USA 1997, 94, 9337–9342. [Google Scholar] [CrossRef] [PubMed]
- Marafioti, T.; Hummel, M.; Foss, H.-D.; Laumen, H.; Korbjuhn, P.; Anagnostopoulos, I.; Lammert, H.; Demel, G.; Theil, J.; Wirth, T.; et al. Hodgkin and Reed-Sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000, 95, 1443–1450. [Google Scholar] [CrossRef]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef]
- Castillo, J.J.; Beltran, B.E.; Miranda, R.N.; Young, K.H.; Chavez, J.C.; Sotomayor, E.M. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2018 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 953–962. [Google Scholar] [CrossRef]
- Linke-Serinsöz, E.; Fend, F.; Quintanilla-Martinez, L. Human immunodeficiency virus (HIV) and Epstein-Barr virus (EBV) related lymphomas, pathology view point. Semin. Diagn. Pathol. 2017, 34, 352–363. [Google Scholar] [CrossRef]
- Ramos da Silva, S.; Elgui de Oliveira, D. HIV, EBV and KSHV: Viral Cooperation in the Pathogenesis of Human Malignancies. Cancer Lett. 2011, 305, 175–185. [Google Scholar] [CrossRef]
- George, L.C.; Rowe, M.; Fox, C.P. Epstein-Barr Virus and the Pathogenesis of T and NK Lymphoma: A Mystery Unsolved. Curr. Hematol. Malign- Rep. 2012, 7, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Keegan, T.H.; Glaser, S.L.; Clarke, C.A.; Gulley, M.L.; Craig, F.E.; DiGiuseppe, J.A.; Dorfman, R.F.; Mann, R.B.; Ambinder, R.F. Epstein-Barr Virus as a Marker of Survival after Hodgkin’s Lymphoma: A Population-Based Study. J. Clin. Oncol. 2005, 23, 7604–7613. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.W.; Yoon, D.H.; Suh, C.; Huh, J. Impact of the Epstein-Barr virus positivity on Hodgkin’s lymphoma in a large cohort from a single institute in Korea. Ann. Hematol. 2012, 91, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.-X.; Liang, J.-H.; Miao, Y.; Fan, L.; Wang, L.; Qu, X.-Y.; Cao, L.; Gong, Q.-X.; Wang, Z.; Zhang, Z.-H.; et al. Epstein-Barr virus positive diffuse large B-cell lymphoma predict poor outcome, regardless of the age. Sci. Rep. 2015, 5, 12168. [Google Scholar] [CrossRef]
- Keane, C.; Tobin, J.; Gunawardana, J.; Francis, S.; Gifford, G.; Gabrielli, S.; Gill, A.; Stevenson, W.; Talaulikar, D.; Gould, C.; et al. The tumour microenvironment is immuno-tolerogenic and a principal determinant of patient outcome in EBV-positive diffuse large B-cell lymphoma. Eur. J. Haematol. 2019, 103, 200–207. [Google Scholar] [CrossRef]
- June, C.H.; Ledbetter, J.A.; Gillespie, M.M.; Lindsten, T.; Thompson, C.B. T-Cell Proliferation Involving the CD28 Pathway Is Associated with Cyclosporine-Resistant Interleukin 2 Gene Expression. Mol. Cell Biol. 1987, 7, 4472–4481. [Google Scholar]
- Mueller, D.; Jenkins, M.; Schwartz, R.H. Clonal Expansion Versus Functional Clonal Inactivation: A Costimulatory Signalling Pathway Determines the Outcome of T Cell Antigen Receptor Occupancy. Annu. Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef]
- Lucca, L.E.; Dominguez-Villar, M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat. Rev. Immunol. 2020, 20, 680–693. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2017, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Kearney, E.R.; Walunas, T.L.; Karr, R.W.; Morton, P.A.; Loh, D.Y.; Bluestone, J.A.; Jenkins, M. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J. Immunol. 1995, 155, 1032–1036. [Google Scholar] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Brunner-Weinzierl, M.C.; Rudd, C.E. CTLA-4 and PD-1 Control of T-Cell Motility and Migration: Implications for Tumor Immunotherapy. Front Immunol. 2018, 9, 2737. [Google Scholar] [CrossRef]
- Green, M.; Monti, S.; Rodig, S.J.; Juszczyński, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 Activity and EBV Infection Induce PD-L1 in Hodgkin Lymphomas and Posttransplant Lymphoproliferative Disorders: Implications for Targeted Therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef]
- Ma, S.-D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef]
- Kataoka, K.; Miyoshi, H.; Sakata, S.; Dobashi, A.; Couronné, L.; Kogure, Y.; Sato, Y.; Nishida, K.; Gion, Y.; Shiraishi, Y.; et al. Frequent Structural Variations Involving Programmed Death Ligands in Epstein-Barr Virus-Associated Lymphomas. Leukemia 2019, 33, 1687–1699. [Google Scholar] [CrossRef]
- Bi, X.-W.; Wang, H.; Zhang, W.-W.; Xia, Z.; Zhang, Y.-J.; Wang, L. PD-L1 Is up-Regulated By EBV-Driven LMP1 through NF-κb Pathway and Correlates with Poor Prognosis in Natural Killer/T-Cell Lymphoma. J. Hematol. Oncol. 2016, 128, 4134. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef]
- Chan, S.Y.-Y.; Choy, K.-W.; Tsao, S.-W.; Tao, Q.; Tang, T.; Chung, G.T.-Y.; Lo, K.-W. Authentication of nasopharyngeal carcinoma tumor lines. Int. J. Cancer 2008, 122, 2169–2171. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, C.; Qin, J.; Liu, J.; Zheng, C. Genetic profiling reveals an alarming rate of cross-contamination among human cell lines used in China. FASEB J. 2015, 29, 4268–4272. [Google Scholar] [CrossRef]
- Elgui de Oliveira, D.; Marques, C.S.; Losi, V.C. “Cell Identity” Crisis: Another Call for Immediate Action. Cancer Lett. 2016, 381, 122–123. [Google Scholar] [CrossRef][Green Version]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein−Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2018, 33, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Cristino, A.S.; Nourse, J.; West, R.A.; Sabdia, M.B.; Law, S.C.; Gunawardana, J.; Vari, F.; Mujaj, S.; Thillaiyampalam, G.; Snell, C.; et al. EBV MicroRNA-BHRF1-2-5p Targets the 3’UTR of Immune Checkpoint Ligands PD-L1 and PD-L2. Blood 2019, 134, 2261–2270. [Google Scholar] [CrossRef]
- Wang, J.; Ge, J.; Wang, Y.; Xiong, F.; Guo, J.; Jiang, X.; Zhang, L.; Deng, X.; Gong, Z.; Zhang, S.; et al. EBV miRNAs BART11 and BART17-3p promote immune escape through the enhancer-mediated transcription of PD-L1. Nat. Commun. 2022, 13, 866. [Google Scholar] [CrossRef]
- Yoon, C.J.; Chang, M.S.; Kim, D.H.; Kim, W.; Koo, B.K.; Yun, S.-C.; Kim, S.H.; Kim, Y.S.; Woo, J.H. Epstein-Barr Virus-Encoded MiR-BART5-5p Upregulates PD-L1 through PIAS3/PSTAT3 Modulation, Worsening Clinical Outcomes of PD-L1-Positive Gastric Carcinomas. Gastric Cancer 2020, 23, 780–795. [Google Scholar] [CrossRef]
- Chakravorty, S.; Yan, B.; Wang, C.; Wang, L.; Quaid, J.T.; Lin, C.F.; Briggs, S.D.; Majumder, J.; Canaria, D.A.; Chauss, D.; et al. Integrated Pan-Cancer Map of EBV-Associated Neoplasms Reveals Functional Host–Virus Interactions. Cancer Res. 2019, 79, 6010–6023. [Google Scholar] [CrossRef]
- Jiang, Y.; Cai, G.; Lin, J.; Zhang, J.; Bo, Z.; Li, Y.; Wang, C.; Tong, Y. B7-H4 Is Highly Expressed in Aggressive Epstein-Barr Virus Positive Diffuse Large B-Cell Lymphoma and Inhibits Apoptosis through Upregulating Erk1/2 and Akt Signalling Pathways. Infect. Agent Cancer 2019, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Böger, C.; Behrens, H.-M.; Krüger, S.; Röcken, C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? OncoImmunology 2017, 6, e1293215. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hiếu, T.; Malarkannan, S.; Wang, L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell. Mol. Immunol. 2018, 15, 438–446. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef]
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201. [Google Scholar] [CrossRef]
- Rossille, D.; Azzaoui, I.; Feldman, A.; Maurer, M.J.; Labouré, G.; Parrens, M.; Pangault, C.; Habermann, T.M.; Ansell, S.M.; Link, B.; et al. Soluble programmed death-ligand 1 as a prognostic biomarker for overall survival in patients with diffuse large B-cell lymphoma: A replication study and combined analysis of 508 patients. Leukemia 2017, 31, 988–991. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, M.; Zhang, Y.; Meng, H.; Wang, Y.; Liu, Y.; Jing, J.; Huang, L.; Sun, M.; Zhang, Y.; et al. The Prognostic Value of Programmed Cell Death Ligand 1 Expression in Non-Hodgkin Lymphoma: A Meta-Analysis. Cancer Biol. Med. 2018, 15, 290–298. [Google Scholar] [CrossRef]
- Keane, C.; Law, S.C.; Gould, C.; Birch, S.; Sabdia, M.B.; De Long, L.M.; Thillaiyampalam, G.; Abro, E.; Tobin, J.W.; Tan, X.; et al. LAG3: A novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 1367–1377. [Google Scholar] [CrossRef]
- Choi, E.; Chang, M.S.; Byeon, S.-J.; Jin, H.; Jung, K.C.; Kim, H.; Lee, K.L.; Kim, W.; Park, J.H.; Kim, K.H.; et al. Prognostic perspectives of PD-L1 combined with tumor-infiltrating lymphocytes, Epstein-Barr virus, and microsatellite instability in gastric carcinomas. Diagn. Pathol. 2020, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Abe, H.; Kunita, A.; Saito, R.; Kanda, T.; Yamashita, H.; Seto, Y.; Ishikawa, S.; Fukayama, M. Viral loads correlate with upregulation of PD-L1 and worse patient prognosis in Epstein-Barr Virus-associated gastric carcinoma. PLoS ONE 2019, 14, e0211358. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for Classical Hodgkin’s Lymphoma after Failure of Both Autologous Stem-Cell Transplantation and Brentuximab Vedotin: A Multicentre, Multicohort, Single-Arm Phase 2 Trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Kumar, V.; Chaudhary, N.; Garg, M.; Floudas, C.S.; Soni, P.; Chandra, A.B. Current Diagnosis and Management of Immune Related Adverse Events (IrAEs) Induced by Immune Checkpoint Inhibitor Therapy. Front. Pharmacol. 2017, 8, 49. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Patel, S.S.; Weirather, J.L.; Lipschitz, M.; Lako, A.; Chen, P.-H.; Griffin, G.K.; Armand, P.; Shipp, M.A.; Rodig, S.J. The microenvironmental niche in classic Hodgkin lymphoma is enriched for CTLA-4-positive T cells that are PD-1-negative. Blood 2019, 134, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 Expression Is Characteristic of a Subset of Aggressive B-cell Lymphomas and Virus-Associated Malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, P.K.; Charu, V.; DeLisser, M.; Molina-Kirsch, H.; Natkunam, Y.; Zhao, S. Programmed death-1 ligands PD-L1 and PD-L2 show distinctive and restricted patterns of expression in lymphoma subtypes. Hum. Pathol. 2018, 71, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Gascoyne, R.D. The tumour microenvironment in B cell lymphomas. Nat. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Yoshida, M.; Yamamoto, N.; Nino, N.; Nakatani, N.; Ichikawa, T.; Nakamura, S.; Saito, A.; Kozaki, A.; Kishimoto, K.; et al. Programmed Death-1 and Programmed Death-Ligand 1 Expression Patterns in Pediatric Lymphoma. Blood 2018, 132, 5316. [Google Scholar] [CrossRef]
- Dalton, T.; Doubrovina, E.; Pankov, D.; Reynolds, I.R.C.; Scholze, H.; Selvakumar, A.; Vizconde, T.; Savalia, B.; Dyomin, V.; Weigel, C.; et al. Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 2020, 135, 1870–1881. [Google Scholar] [CrossRef]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-Cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Bin Park, G.; Song, H.; Kim, Y.-S.; Sung, M.; Ryu, J.W.; Lee, H.-K.; Cho, D.-H.; Kim, D.; Lee, W.J.; Hur, D.Y. Cell cycle arrest induced by engagement of B7-H4 on Epstein-Barr virus-positive B-cell lymphoma cell lines. Immunology 2009, 128, 360–368. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of Efficacy of Pembrolizumab between Epstein-Barr Virus‒Positive and ‒Negative Relapsed or Refractory Non-Hodgkin Lymphomas. Cancer Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef]
- Qiu, Z.-X.; Zhou, P.; Wang, K. Primary Pulmonary Lymphoepithelioma-Like Carcinoma Response Favorably to Nivolumab: A Case Report. OncoTargets Ther. 2019, 12, 8595–8600. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Knollmann, F.D.; Walby, J.A.S.; Lim, S.; Gandara, D.R.; Riess, J.W. EBV-positive Primary Pulmonary Lymphoepithelioma-like Carcinoma Response to PD-L1 Blockade. Clin. Lung Cancer 2019, 20, e238–e241. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biggi, A.F.B.; Elgui de Oliveira, D. The Epstein-Barr Virus Hacks Immune Checkpoints: Evidence and Consequences for Lymphoproliferative Disorders and Cancers. Biomolecules 2022, 12, 397. https://doi.org/10.3390/biom12030397
Biggi AFB, Elgui de Oliveira D. The Epstein-Barr Virus Hacks Immune Checkpoints: Evidence and Consequences for Lymphoproliferative Disorders and Cancers. Biomolecules. 2022; 12(3):397. https://doi.org/10.3390/biom12030397
Chicago/Turabian StyleBiggi, Alison Felipe Bordini, and Deilson Elgui de Oliveira. 2022. "The Epstein-Barr Virus Hacks Immune Checkpoints: Evidence and Consequences for Lymphoproliferative Disorders and Cancers" Biomolecules 12, no. 3: 397. https://doi.org/10.3390/biom12030397
APA StyleBiggi, A. F. B., & Elgui de Oliveira, D. (2022). The Epstein-Barr Virus Hacks Immune Checkpoints: Evidence and Consequences for Lymphoproliferative Disorders and Cancers. Biomolecules, 12(3), 397. https://doi.org/10.3390/biom12030397