
Therapeutic Approaches Targeting Proteins in Tumor-Associated Macrophages and Their Applications in Cancers


Abstract
:1. Introduction
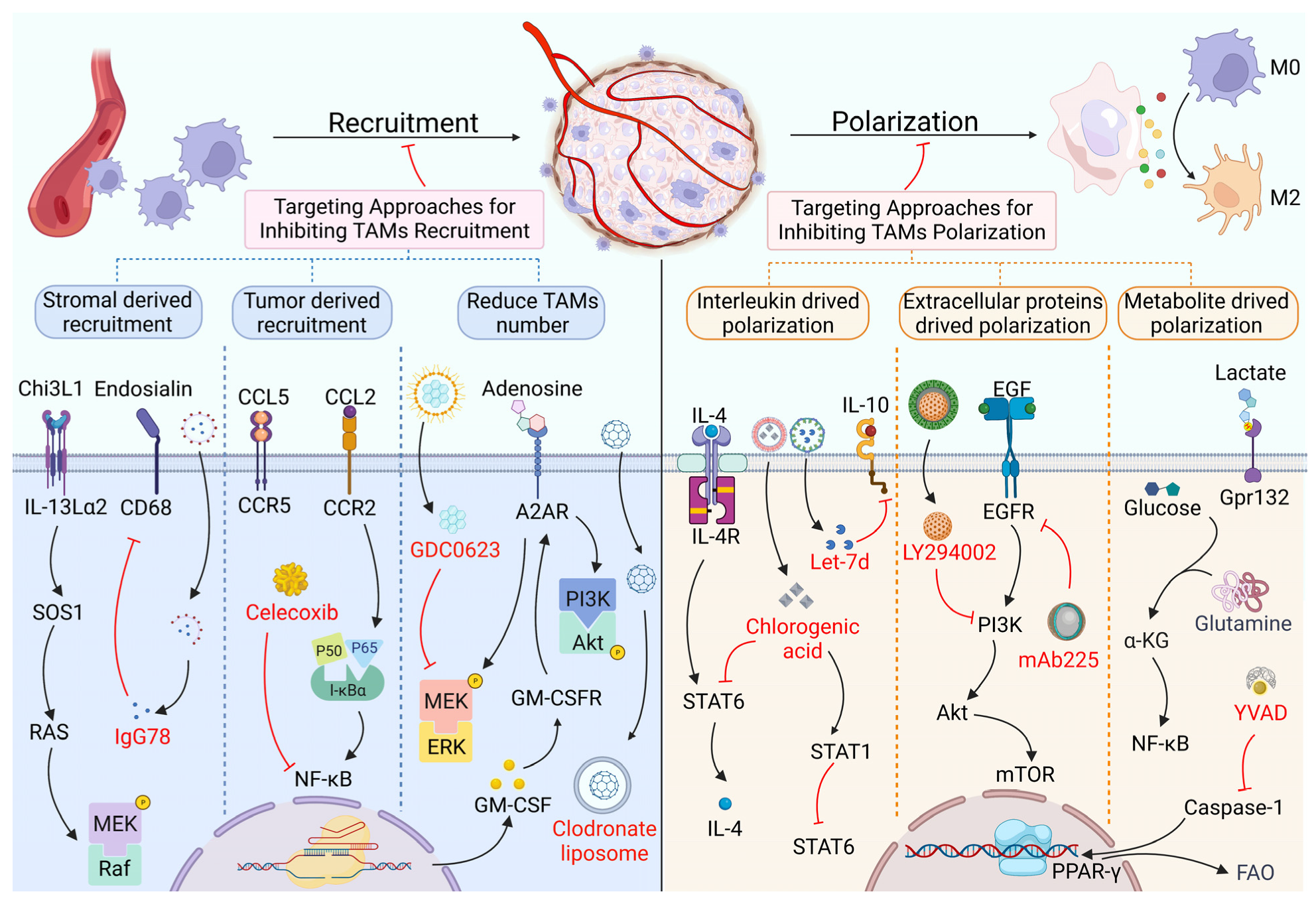
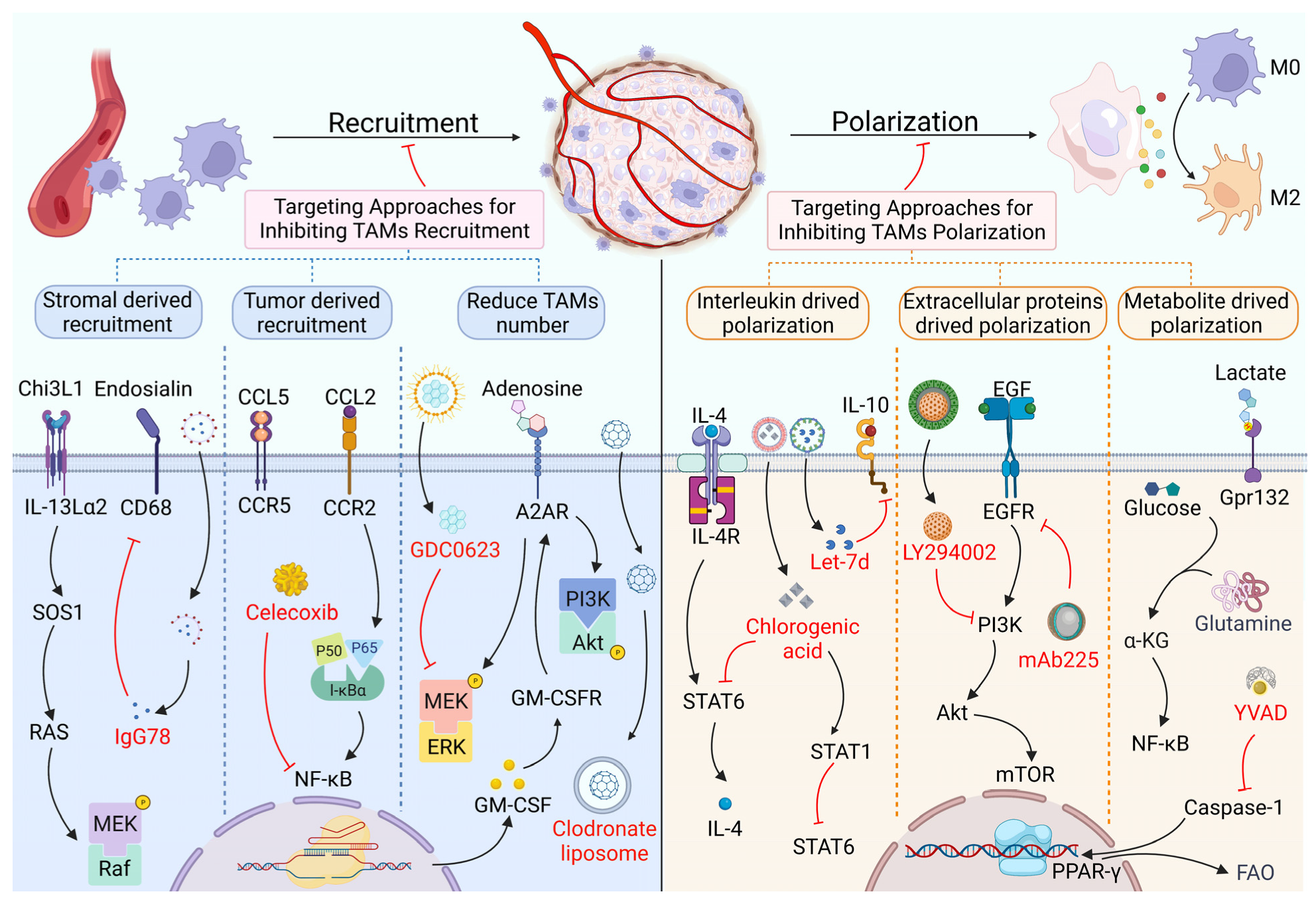
2. Targeting Proteins in the Recruitment and Polarization of TAMs
2.1. TAM Recruitment and Its Targeted Therapy Based on Proteins
2.2. TAM Polarization and Its Targeted Therapy Based on Proteins
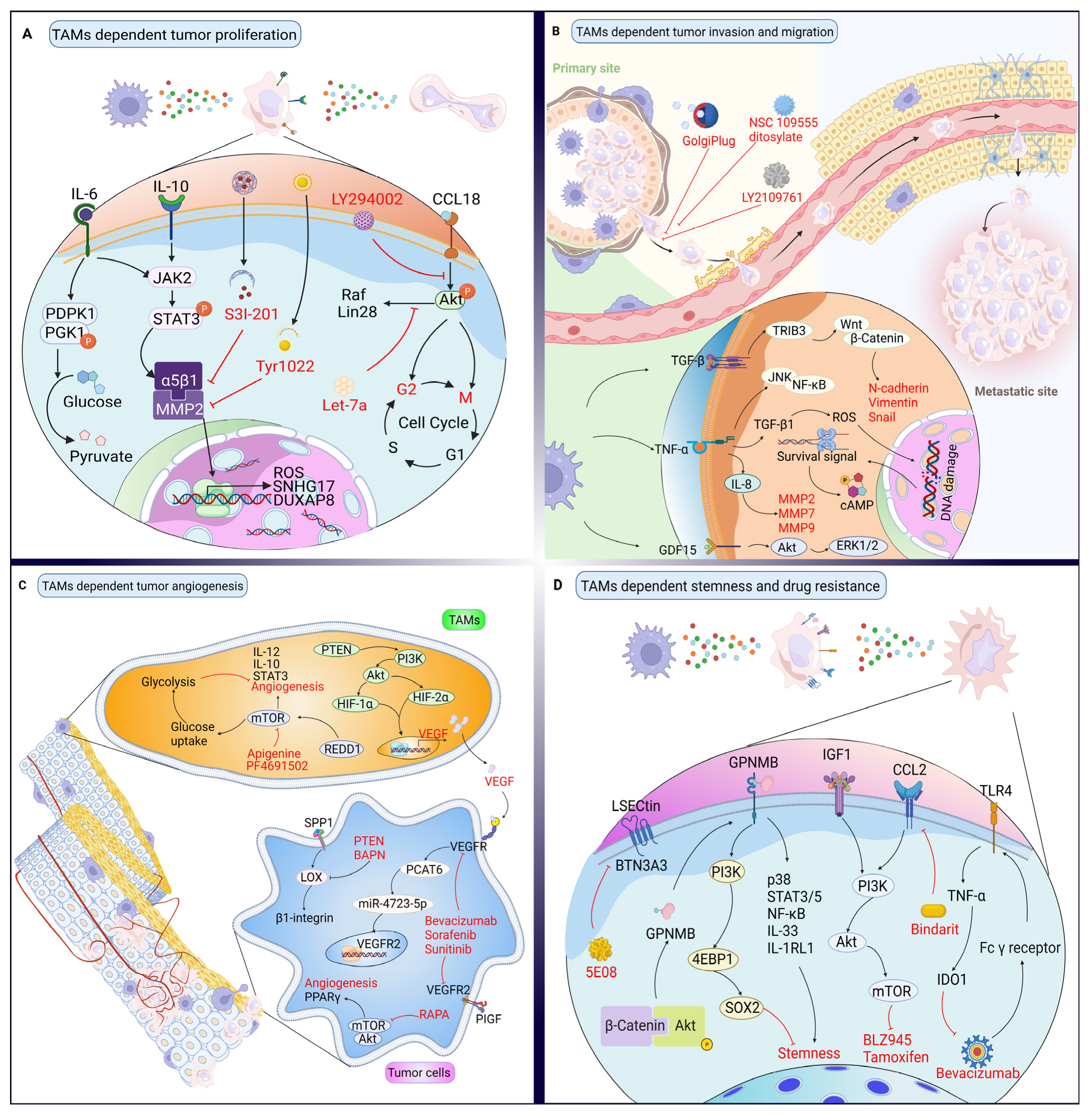
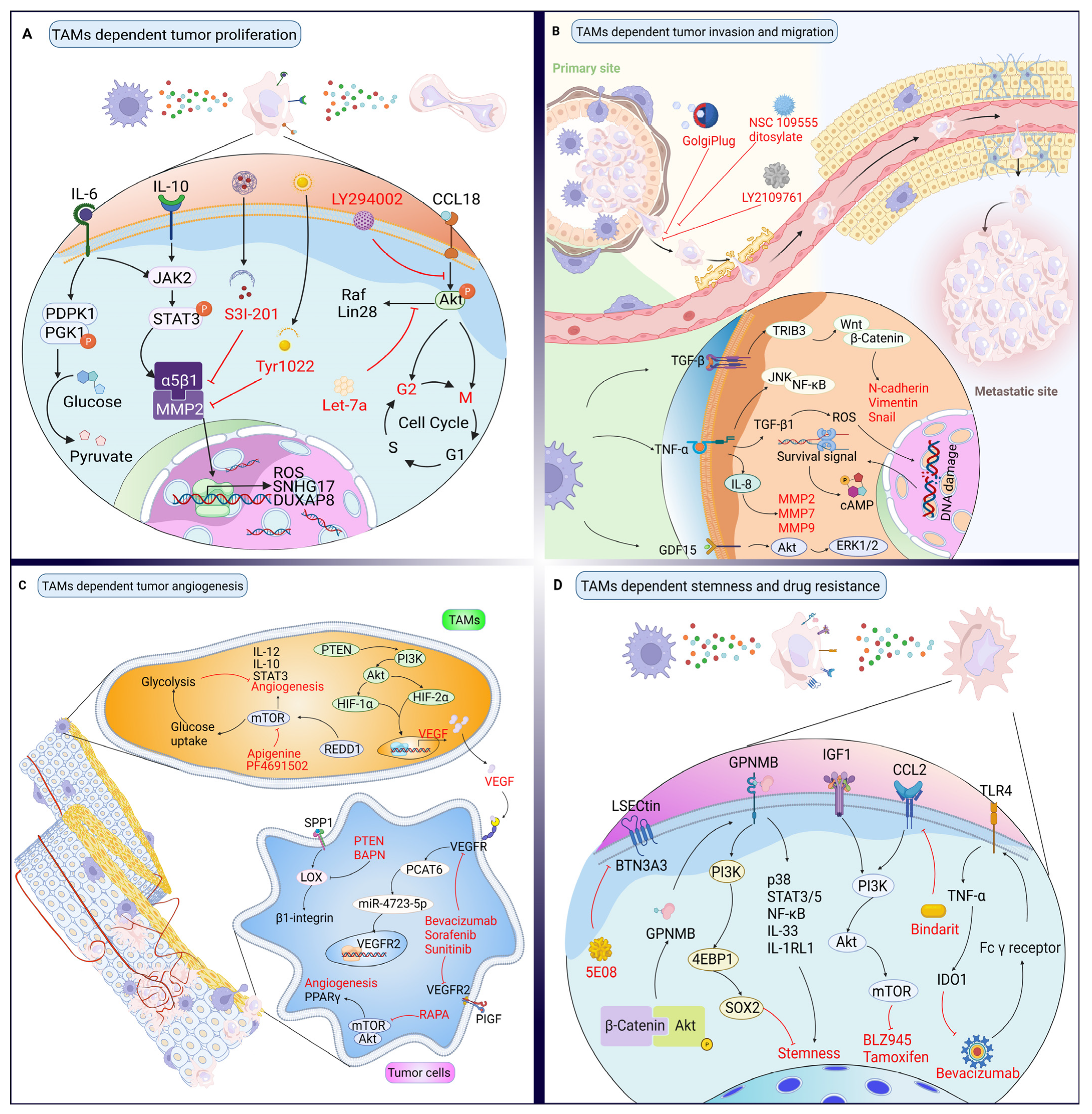
3. Targeting Proteins in the Crosstalk between TAMs and Cancer Cells
3.1. Effects of TAMs on Cancer Cell Proliferation and Their Targeted Therapy Based on Proteins
3.2. Effects of TAMs on Cancer Cell Invasion and Their Targeted Therapy Based on Proteins

{kind=link}
{kind=link}
{kind=link}
| Ligand | Effector | Tumor | Inhibitor | Anti-Tumor Mechanism | Ref. |
|---|---|---|---|---|---|
| Inhibit the proliferation of cancer cells | |||||
| IL-10 | PD-L1 | NSCLC | BFD | Decrease IL-10 induced PD-L1 expression | [88] |
| IL-10 | STAT3 | RCC | N/A | Inhibit BMP-6 induced M2 polarization | [89] |
| MCAD | Lipid | BC | Sc-98926 | Reduce LD accumulation in TAMs | [67] |
| MIF | IL-2 | CRC | NIHIII.D.9 | Decrease Treg generation and IL-2 production | [90] |
| EGFR | ILT4 | NSCLC | Human ILT4 antibody | Inhibit TAM recruitment and M2 polarization | [91] |
| MK2 | IL-1, IL-6, TNF-α | CRC | PF364402 | Inhibit IL-1β, IL-6, and TNF-α, expression | [92] |
| Inhibit the invasion of tumor | |||||
| Lactate | Gpr132 | BC | N/A | Inhibit lactate uptake and M2 macrophages activity | [61] |
| IGFBP2 | FcγRIIB | GBM | Bs-1108R | Increase CD8+ T and p-CD19+ B cells and decreases M2 macrophages | [93] |
| S100A8/A9 | MMP2, MMP9 | LCC | N/A | Decrease MMP2 and MMP9 | [94] |
| GS | Glutamine | N/A | MSO | Suppress M2 macrophages, induce T-cell recruitment | [95] |
| ATM | ATR | BC | Clone 10H11.E12 | Decrease pCREB expression | [86] |
| Inhibit the angiogenesis of tumor | |||||
| IL-10/IL-13 | N/A | RCC | Let-7d | Inhibit intratumoral macrophage M2 polarization | [47] |
| S100A7 | JAB1 | ESCC | N/A | Inhibit S1007A induced phosphorylation of ERK and FAK | [96] |
| N/A | PI3K/Akt/mTOR | HCC | Apigenin | Inhibit PI3K/Akt/mTOR pathway | [97] |
| S1PR1 | NLRP3 | BC | N/A | Inhibit S1PR1 dependent IL-1β expression | [98] |
| LOX | β1 integrin/PYK2 | GBM | BAPN | Decrease TAM-derived SPP1 | [99] |
| Inhibit the stemness of tumor | |||||
| α-KG | Jmjd-3 | N/A | BPTES | Suppressed IL-4-induced STAT6 phosphorylation | [62] |
| LSECtin | BTN3A3 | BC | 5E08 | N/A | [100] |
| CCL8 | Erk1/2 | GBM | SCH772984 | Attenuate pseudopodia formation | [101] |
| IL-8 | STAT3 | OC | IL-8 Ab | Inhibit STAT3 and increase IL-12, NO | [102] |
| CBX8 | H3K4me3 | CRC | N/A | Increased the chemosensitivity of CRC cells | [103] |
3.3. Effects of TAMs on Angiogenesis and Their Targeted Therapy Based on Proteins
3.4. Effects of TAMs on Cancer Stemness and Drug Resistance, and Their Targeted Therapy Based on Proteins
4. Targeting Proteins of TAMs in the Regulation of Tumor Immune Responses
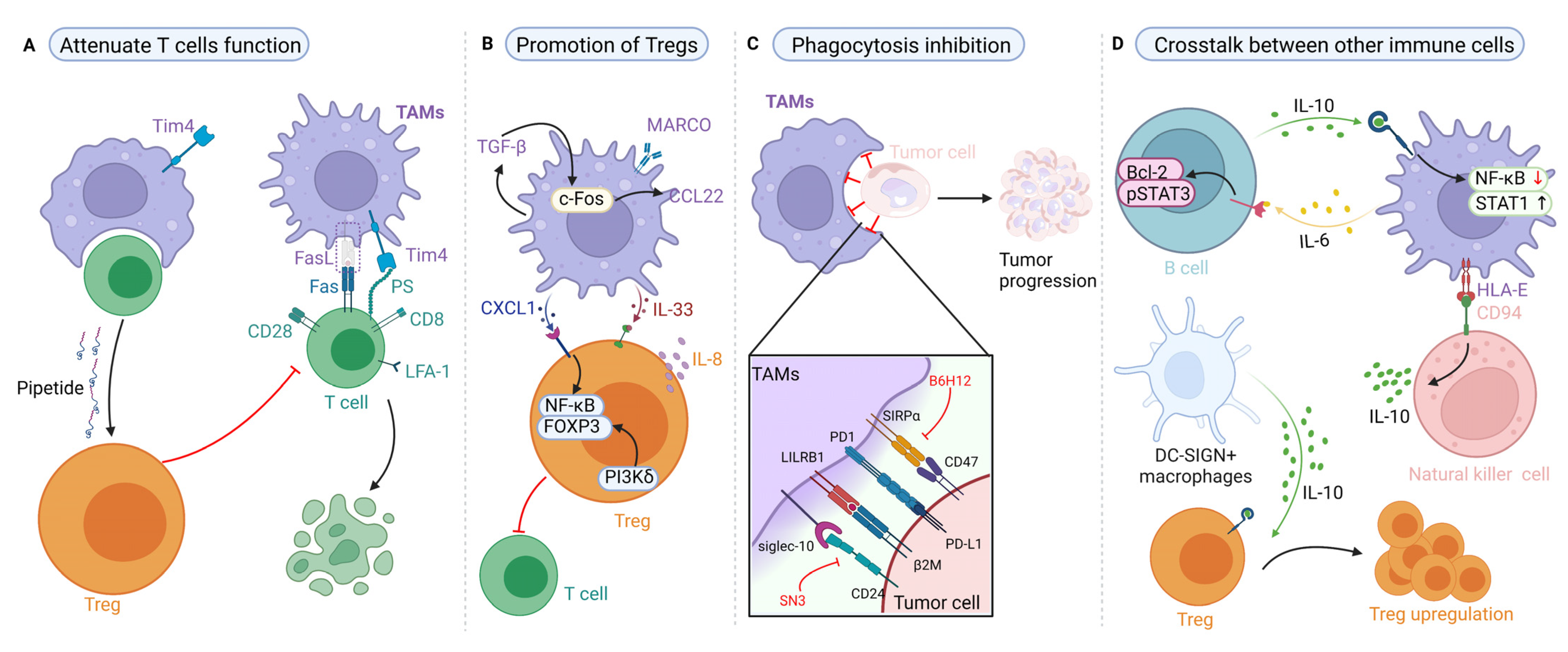
4.1. Effects of TAMs on T-Cell Immunity and Their Targeted Therapy Based on Proteins
4.2. Effects of TAMs on Regulatory T Cells (Tregs) and Their Targeted Therapy Based on Proteins
4.3. Effects of TAM-Mediated Phagocytosis and Their Targeted Therapy Based on Proteins
4.4. Effects of TAMs on Other Immune Cells and Their Targeted Therapy Based on Proteins
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, S.-Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schröder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Shono, K.; Yamaguchi, I.; Mizobuchi, Y.; Kagusa, H.; Sumi, A.; Fujihara, T.; Nakajima, K.; Kitazato, K.T.; Matsuzaki, K.; Saya, H.; et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 axes contributes to antitumor effects in a mouse model of malignant glioma. Sci. Rep. 2020, 10, 15286. [Google Scholar] [CrossRef]
- Chow, A.; Schad, S.; Green, M.D.; Hellmann, M.D.; Allaj, V.; Ceglia, N.; Zago, G.; Shah, N.S.; Sharma, S.K.; Mattar, M.; et al. Tim-4 cavity-resident macrophages impair anti-tumor CD8 T cell immunity. Cancer Cell 2021, 39, 973–988. [Google Scholar] [CrossRef]
- Ge, Z.; Ding, S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10, 590941. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Yeung, O.W.H.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.P.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.P.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef] [Green Version]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, J.; Sun, P.; Ma, Z.; Sun, P. BRD4 promotes tumor progression and NF-κB/CCL2-dependent tumor-associated macrophage recruitment in GIST. Cell Death Dis. 2019, 10, 935. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-L.; Hung, J.-Y.; Tsai, Y.-M.; Tsai, E.-M.; Huang, M.-S.; Hou, M.-F.; Kuo, P.-L. 6-shogaol, an active constituent of dietary ginger, impairs cancer development and lung metastasis by inhibiting the secretion of CC-chemokine ligand 2 (CCL2) in tumor-associated dendritic cells. J. Agric. Food Chem. 2015, 63, 1730–1738. [Google Scholar] [CrossRef]
- Regan, D.P.; Coy, J.W.; Chahal, K.K.; Chow, L.; Kurihara, J.N.; Guth, A.M.; Kufareva, I.; Dow, S.W. The Angiotensin Receptor Blocker Losartan Suppresses Growth of Pulmonary Metastases via AT1R-Independent Inhibition of CCR2 Signaling and Monocyte Recruitment. J. Immunol. 2019, 202, 3087–3102. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Wang, N.; Zheng, Y.; Yang, B.; Wang, X.; Zhang, J.; Pan, B.; Wang, Z. Aiduqing formula inhibits breast cancer metastasis by suppressing TAM/CXCL1-induced Treg differentiation and infiltration. Cell Commun. Signal. 2021, 19, 89. [Google Scholar] [CrossRef]
- Frankenberger, C.; Rabe, D.; Bainer, R.; Sankarasharma, D.; Chada, K.; Krausz, T.; Gilad, Y.; Becker, L.; Rosner, M.R. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res. 2015, 75, 4063–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauß, T.F.; Winslow, S.; Lampe, S.; Scholz, A.; Weigert, A.; Dehne, N.; von Stedingk, K.; Schmid, T.; Brüne, B. The RNA-binding protein HuR inhibits expression of CCL5 and limits recruitment of macrophages into tumors. Mol. Carcinog. 2017, 56, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Pevida, M.; Lastra, A.; Meana, Á.; Hidalgo, A.; Baamonde, A.; Menéndez, L. The chemokine CCL5 induces CCR1-mediated hyperalgesia in mice inoculated with NCTC 2472 tumoral cells. Neuroscience 2014, 259, 113–125. [Google Scholar] [CrossRef]
- Wani, N.; Nasser, M.W.; Ahirwar, D.K.; Zhao, H.; Miao, Z.; Shilo, K.; Ganju, R.K. C-X-C motif chemokine 12/C-X-C chemokine receptor type 7 signaling regulates breast cancer growth and metastasis by modulating the tumor microenvironment. Breast Cancer Res. 2014, 16, R54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Huang, L.; Ding, G.; Huang, H.; Cao, G.; Sun, X.; Lou, N.; Wei, Q.; Shen, T.; Xu, X.; et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J. Immunother. Cancer 2020, 8, e000308. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.-C.; Yip, T.-C.; Ngan, K.-C.; Cheng, W.-W.; Law, C.-K.; Chan, P.-S.; Chan, K.-C.; Wong, C.K.-C.; Wong, R.N.-S.; Lo, K.-W.; et al. Role of MIF/CXCL8/CXCR2 signaling in the growth of nasopharyngeal carcinoma tumor spheres. Cancer Lett. 2013, 335, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.; Billet, S.; Liu, C.; Haldar, S.; Choudhury, D.; Tripathi, M.; Hav, M.; Merchant, A.; Hu, T.; Huang, H.; et al. Periodontal inflammation recruits distant metastatic breast cancer cells by increasing myeloid-derived suppressor cells. Oncogene 2020, 39, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Zhang, I.Y.; Zhang, L.; Song, Y.; Liu, S.; Ren, H.; Liu, H.; Zhou, H.; Su, Y.; Yang, Y.; et al. S100B suppression alters polarization of infiltrating myeloid-derived cells in gliomas and inhibits tumor growth. Cancer Lett. 2018, 439, 91–100. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.-M.; Zhang, Z.; Hsu, J.L.; Li, C.-W.; Lim, S.-O.; Sheng, Y.-Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Chu, Y.; Li, Z.; Yu, X.; Huang, Z.; Xu, J.; Zheng, L. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol. 2021, 74, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Baumann, D.; Drebant, J.; Hägele, T.; Burger, L.; Serger, C.; Lauenstein, C.; Dudys, P.; Erdmann, G.; Offringa, R. p38 MAPK signaling in M1 macrophages results in selective elimination of M2 macrophages by MEK inhibition. J. Immunother. Cancer 2021, 9, e002319. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Zhang, J.-Q.; Liang, G.-K.; He, Q.-J.; Ding, L.; Yang, B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharm. Sin. 2017, 38, 1501–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-J.; Yang, C.-K.; Wei, P.-L.; Huynh, T.-T.; Whang-Peng, J.; Meng, T.-C.; Hsiao, M.; Tzeng, Y.-M.; Wu, A.T.; Yen, Y. Ovatodiolide suppresses colon tumorigenesis and prevents polarization of M2 tumor-associated macrophages through YAP oncogenic pathways. J. Hematol. Oncol. 2017, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Fang, Y.; Lai, Q.; Wang, S.; He, C.; Li, A.; Liu, S.; Yan, Q. CPEB3 inhibits epithelial-mesenchymal transition by disrupting the crosstalk between colorectal cancer cells and tumor-associated macrophages via IL-6R/STAT3 signaling. J. Exp. Clin. Cancer Res. 2020, 39, 132. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, J.; Gao, Y.; Wu, C.; Yi, L.; Li, G.; Qi, Y. The dual effects of a novel peptibody on angiogenesis inhibition and M2 macrophage polarization on sarcoma. Cancer Lett. 2018, 416, 1–10. [Google Scholar] [CrossRef]
- Shu, Y.; Qin, M.; Song, Y.; Tang, Q.; Huang, Y.; Shen, P.; Lu, Y. M2 polarization of tumor-associated macrophages is dependent on integrin β3 via peroxisome proliferator-activated receptor-γ up-regulation in breast cancer. Immunology 2020, 160, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Yu, W.; He, J.; Liu, W.; Yang, J.; Lin, X.; Zhang, Y.; Wang, X.; Jiang, W.; Luo, J.; et al. Reprogramming immunosuppressive myeloid cells facilitates immunotherapy for colorectal cancer. EMBO Mol. Med. 2021, 13, e12798. [Google Scholar] [CrossRef]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12, eaax6337. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Wei, Y.; Han, D.; Li, Y.; Shi, S.; Jiao, D.; Wu, J.; Zhang, Q.; Shi, C.; Yang, L.; et al. Interaction with CD68 and Regulation of GAS6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 3892–3905. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawgood, S.; Akiyama, J.; Brown, C.; Allen, L.; Li, G.; Poulain, F.R. GM-CSF mediates alveolar macrophage proliferation and type II cell hypertrophy in SP-D gene-targeted mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1148–L1156. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.L.; Nolan, K.; Strait, A.A.; Bian, L.; Nguyen, K.A.; Wang, J.H.; Jimeno, A.; Zhou, H.M.; Young, C.D.; Wang, X.J. Macrophages Promote Growth of Squamous Cancer Independent of T cells. J. Dent. Res. 2019, 98, 896–903. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Sheldrake, T.A.; Dhaliwal, K.; Kipari, T.M.J.; Marson, L.P.; Kluth, D.C.; Hughes, J. Macrophage/monocyte depletion by clodronate, but not diphtheria toxin, improves renal ischemia/reperfusion injury in mice. Kidney Int. 2012, 82, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Han, H.; Gong, Y.; Li, X.; Ji, C.; Yao, J.; Yang, J.; Hu, W.; Zhao, W.; Li, J.; et al. Let-7d inhibits intratumoral macrophage M2 polarization and subsequent tumor angiogenesis by targeting IL-13 and IL-10. Cancer Immunol. Immunother. 2021, 70, 1619–1634. [Google Scholar] [CrossRef]
- Rahal, O.M.; Wolfe, A.R.; Mandal, P.K.; Larson, R.; Tin, S.; Jimenez, C.; Zhang, D.; Horton, J.; Reuben, J.M.; McMurray, J.S.; et al. Blocking Interleukin (IL)4- and IL13-Mediated Phosphorylation of STAT6 (Tyr641) Decreases M2 Polarization of Macrophages and Protects Against Macrophage-Mediated Radioresistance of Inflammatory Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Jiang, L.; Hao, S.; Liu, Z.; Ding, S.; Zhang, W.; Yang, X.; Li, S. Activation of the IL-4/STAT6 Signaling Pathway Promotes Lung Cancer Progression by Increasing M2 Myeloid Cells. Front. Immunol. 2019, 10, 2638. [Google Scholar] [CrossRef]
- Xue, N.; Zhou, Q.; Ji, M.; Jin, J.; Lai, F.; Chen, J.; Zhang, M.; Jia, J.; Yang, H.; Zhang, J.; et al. Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci. Rep. 2017, 7, 39011. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, F.; Zhou, P.; Wang, Q.; Xu, C.; Li, Y.; Bian, L.; Liu, Y.; Zhou, J.; Wang, F.; et al. The MTOR signaling pathway regulates macrophage differentiation from mouse myeloid progenitors by inhibiting autophagy. Autophagy 2019, 15, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Chen, S.; Ouyang, M.; Li, F.; Chen, L.; Yang, J. Colon Cancer Cell Secretes EGF to Promote M2 Polarization of TAM Through EGFR/PI3K/AKT/mTOR Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819849068. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M. Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Viale, G.; Curigliano, G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients With Cancer: A Review. JAMA Oncol. 2019, 5, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, Y.; Kong, X.; Wu, R.; Peng, Q.; Zhang, Y.; Zhou, L.; Duan, L. Facilitates M2 Macrophage Polarization and Colorectal Carcinoma Progression by Activating TLR4/NF-B/S100A9 Cascade. Front. Immunol. 2021, 12, 658681. [Google Scholar] [CrossRef]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef]
- Schelbergen, R.F.; Geven, E.J.; van den Bosch, M.H.J.; Eriksson, H.; Leanderson, T.; Vogl, T.; Roth, J.; van de Loo, F.A.J.; Koenders, M.I.; van der Kraan, P.M.; et al. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann. Rheum. Dis. 2015, 74, 2254–2258. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; He, Y.; Hwang, S.; Bertola, A.; Mackowiak, B.; Ahmed, Y.A.; Seo, W.; Ma, J.; Wang, X.; Park, S.H.; et al. E-Selectin-Dependent Inflammation and Lipolysis in Adipose Tissue Exacerbate Steatosis-to-NASH Progression via S100A8/9. Cell Mol. Gastroenterol. Hepatol. 2021, 13, 151–171. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, Y.; Aishima, S.; Fujita, N.; Tanaka, Y.; Kubo, Y.; Motomura, T.; Taketomi, A.; Shirabe, K.; Maehara, Y.; Oda, Y. Tumor-associated macrophage promotes tumor progression via STAT3 signaling in hepatocellular carcinoma. Pathobiology 2013, 80, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with α5β1 integrin in glioma. Oncogene 2013, 32, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Lin, Z.; Liu, Y.; Jiang, Y.; Liu, K.; Tu, M.; Yao, N.; Qu, C.; Hong, J. Intrahepatic cholangiocarcinoma induced M2-polarized tumor-associated macrophages facilitate tumor growth and invasiveness. Cancer Cell Int. 2020, 20, 586. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lyu, Z.; Qin, Y.; Wang, X.; Sun, L.; Zhang, Y.; Gong, L.; Wu, S.; Han, S.; Tang, Y.; et al. FOXO1 promotes tumor progression by increased M2 macrophage infiltration in esophageal squamous cell carcinoma. Theranostics 2020, 10, 11535–11548. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.-X.; Zhang, D.-Z.; Fang, X.-J.; Sun, P.-S.; Xue, H.-C. Let-7a mimic attenuates CCL18 induced breast cancer cell metastasis through Lin 28 pathway. Biomed. Pharm. 2016, 78, 301–307. [Google Scholar] [CrossRef]
- Günther, C.; Zimmermann, N.; Berndt, N.; Grosser, M.; Stein, A.; Koch, A.; Meurer, M. Up-regulation of the chemokine CCL18 by macrophages is a potential immunomodulatory pathway in cutaneous T-cell lymphoma. Am. J. Pathol. 2011, 179, 1434–1442. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, W.; Wang, J.; Si, T.; Xing, W. Tumor-associated macrophage-derived transforming growth factor-β promotes colorectal cancer progression through HIF1-TRIB3 signaling. Cancer Sci. 2021, 112, 4198–4207. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Koma, Y.-I.; Kodama, T.; Nishio, M.; Shigeoka, M.; Yokozaki, H. Growth Differentiation Factor 15 Promotes Progression of Esophageal Squamous Cell Carcinoma via TGF-β Type II Receptor Activation. Pathobiology 2020, 87, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, U.; Kim, B.; Kim, S.; Han, Y.; Song, Y.S. Pro-inflammatory M1 macrophage enhances metastatic potential of ovarian cancer cells through NF-κB activation. Mol. Carcinog. 2018, 57, 235–242. [Google Scholar] [CrossRef]
- Singh, R.; Shankar, B.S.; Sainis, K.B. TGF-β1-ROS-ATM-CREB signaling axis in macrophage mediated migration of human breast cancer MCF7 cells. Cell Signal. 2014, 26, 1604–1615. [Google Scholar] [CrossRef]
- Watanabe, H.; Iwase, M.; Ohashi, M.; Nagumo, M. Role of interleukin-8 secreted from human oral squamous cell carcinoma cell lines. Oral Oncol. 2002, 38, 670–679. [Google Scholar] [CrossRef]
- Pang, L.; Han, S.; Jiao, Y.; Jiang, S.; He, X.; Li, P. Bu Fei Decoction attenuates the tumor associated macrophage stimulated proliferation, migration, invasion and immunosuppression of non-small cell lung cancer, partially via IL-10 and PD-L1 regulation. Int. J. Oncol. 2017, 51, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Lee, G.T.; Woo, S.H.; Ha, Y.-S.; Kwon, S.J.; Kim, W.-J.; Kim, I.Y. BMP-6 in renal cell carcinoma promotes tumor proliferation through IL-10-dependent M2 polarization of tumor-associated macrophages. Cancer Res. 2013, 73, 3604–3614. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Kim, H.-R.; Leng, L.; Kang, I.; Jorgensen, W.L.; Cho, C.-S.; Bucala, R.; Kim, W.-U. Role of macrophage migration inhibitory factor in the regulatory T cell response of tumor-bearing mice. J. Immunol. 2012, 189, 3905–3913. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gao, A.; Zhang, F.; Yang, Z.; Wang, S.; Fang, Y.; Li, J.; Wang, J.; Shi, W.; Wang, L.; et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics 2021, 11, 3392–3416. [Google Scholar] [CrossRef] [PubMed]
- Phinney, B.B.; Ray, A.L.; Peretti, A.S.; Jerman, S.J.; Grim, C.; Pinchuk, I.V.; Beswick, E.J. MK2 Regulates Macrophage Chemokine Activity and Recruitment to Promote Colon Tumor Growth. Front. Immunol. 2018, 9, 1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Song, C.; Shen, F.; Zhang, J.; Song, S.W. IGFBP2 promotes immunosuppression associated with its mesenchymal induction and FcγRIIB phosphorylation in glioblastoma. PLoS ONE 2019, 14, e0222999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene 2016, 35, 5735–5745. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zheng, S.; Liu, C.; Wang, X.; Zhang, G.; Wang, F.; Wang, S.; Huang, J.; Mao, S.; Lei, Y.; et al. S100A7 as a potential diagnostic and prognostic biomarker of esophageal squamous cell carcinoma promotes M2 macrophage infiltration and angiogenesis. Clin. Transl. Med. 2021, 11, e459. [Google Scholar] [CrossRef]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharm. 2018, 103, 699–707. [Google Scholar] [CrossRef]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.-C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Li, J.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884. [Google Scholar] [CrossRef]
- Liu, D.; Lu, Q.; Wang, X.; Wang, J.; Lu, N.; Jiang, Z.; Hao, X.; Li, J.; Liu, J.; Cao, P.; et al. LSECtin on tumor-associated macrophages enhances breast cancer stemness via interaction with its receptor BTN3A3. Cell Res. 2019, 29, 365–378. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Dang, W.-Q.; Cao, M.-F.; Xiao, J.-F.; Lv, S.-Q.; Jiang, W.-J.; Yao, X.-H.; Lu, H.-M.; Miao, J.-Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2020, 100, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Cui, Y.; Li, X.; Cao, X.; Chen, A.; Xu, C.; Cao, J.; Luo, X. Co-culture of ovarian cancer stem-like cells with macrophages induced SKOV3 cells stemness via IL-8/STAT3 signaling. Biomed. Pharm. 2018, 103, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kang, M.; Zhang, B.; Meng, F.; Song, J.; Kaneko, H.; Shimamoto, F.; Tang, B. mA modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol. Cancer 2019, 18, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol. Cancer Res. 2014, 12, 1520–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, F.; Ruan, S.; Wang, J.; Xia, Y.; Le, K.; Xiao, X.; Hu, T.; Wang, Q. M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2. Cell Death Dis. 2020, 11, 728. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Narang, A.S.; Varia, S. Role of tumor vascular architecture in drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 640–658. [Google Scholar] [CrossRef] [PubMed]
- Konerding, M.A.; Malkusch, W.; Klapthor, B.; van Ackeern, C.; Fait, E.; Hill, S.A.; Parkins, C.; Chaplin, D.J.; Presta, M.; Denekamp, J. Evidence for characteristic vascular patterns in solid tumours: Quantitative studies using corrosion casts. Br. J. Cancer 1999, 80, 724–732. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Nagy, J.A.; Dvorak, J.T.; Dvorak, A.M. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am. J. Pathol. 1988, 133, 95. [Google Scholar]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar] [PubMed]
- Paku, S.; Paweletz, N. First steps of tumor-related angiogenesis. Lab. Investig. 1991, 65, 334–346. [Google Scholar] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Ma, T.; Shen, X.-N.; Xia, X.-F.; Xu, G.-D.; Bai, X.-L.; Liang, T.-B. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Tao, T.; Liu, S.; Yang, X.; Chen, X.; Liang, J.; Hong, R.; Wang, W.; Yang, Y.; Li, X.; et al. An RFC4/Notch1 signaling feedback loop promotes NSCLC metastasis and stemness. Nat. Commun. 2021, 12, 2693. [Google Scholar] [CrossRef]
- Allavena, P.; Anfray, C.; Ummarino, A.; Andón, F.T. Therapeutic Manipulation of Tumor-associated Macrophages: Facts and Hopes from a Clinical and Translational Perspective. Clin. Cancer Res. 2021, 27, 3291–3297. [Google Scholar] [CrossRef]
- Liguori, M.; Digifico, E.; Vacchini, A.; Avigni, R.; Colombo, F.S.; Borroni, E.M.; Farina, F.M.; Milanesi, S.; Castagna, A.; Mannarino, L.; et al. The soluble glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness and metastasis via CD44 and IL-33. Cell Mol. Immunol. 2021, 18, 711–722. [Google Scholar] [CrossRef]
- Maric, G.; Annis, M.G.; MacDonald, P.A.; Russo, C.; Perkins, D.; Siwak, D.R.; Mills, G.B.; Siegel, P.M. GPNMB augments Wnt-1 mediated breast tumor initiation and growth by enhancing PI3K/AKT/mTOR pathway signaling and β-catenin activity. Oncogene 2019, 38, 5294–5307. [Google Scholar] [CrossRef]
- Gomez, K.E.; Wu, F.; Keysar, S.B.; Morton, J.J.; Miller, B.; Chimed, T.-S.; Le, P.N.; Nieto, C.; Chowdhury, F.N.; Tyagi, A.; et al. Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells. Cancer Res. 2020, 80, 4185–4198. [Google Scholar] [CrossRef]
- Sohn, S.-H.; Kim, B.; Sul, H.J.; Choi, B.Y.; Kim, H.S.; Zang, D.Y. Foretinib Inhibits Cancer Stemness and Gastric Cancer Cell Proliferation by Decreasing CD44 and c-MET Signaling. Onco Targets 2020, 13, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020, 111, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ji, X.; Kang, N.; Zhou, J.; Liang, X.; Li, J.; Han, T.; Zhao, C.; Yang, T. Tumor necrosis factor α inhibition overcomes immunosuppressive M2b macrophage-induced bevacizumab resistance in triple-negative breast cancer. Cell Death Dis. 2020, 11, 993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macro.ophage-medi.iated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J.; Wu, X.; Liang, Z. PMA treated THP-1-derived-IL-6 promotes EMT of SW48 through STAT3/ERK-dependent activation of Wnt/β-catenin signaling pathway. Biomed. Pharm. 2018, 108, 618–624. [Google Scholar] [CrossRef]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Möller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Wang, G.-Z.; Wang, Y.; Huang, H.-Z.; Li, W.-T.; Qu, X.-D. Hypoxia-induced HMGB1 expression of HCC promotes tumor invasiveness and metastasis via regulating macrophage-derived IL-6. Exp. Cell Res. 2018, 367, 81–88. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Xu, J.-Y.; Shi, X.-Y.; Huang, W.; Ruan, T.-Y.; Xie, P.; Ding, J.-L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic.c cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Xu, L.; Wang, Y.; Jiang, Q.; Liu, Z.; Zhang, J.; Zhou, Q.; Zeng, H.; Tong, S.; Wang, T.; et al. Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur. Urol. 2019, 75, 752–763. [Google Scholar] [CrossRef]
- Nie, W.; Yu, T.; Sang, Y.; Gao, X. Tumor-promoting effect of IL-23 in mammary cancer mediated by infiltration of M2 macrophages and neutrophils in tumor microenvironment. Biochem. Biophys. Res. Commun. 2017, 482, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Franzè, E.; Laudisi, F.; Di Grazia, A.; Marônek, M.; Bellato, V.; Sica, G.; Monteleone, G. Macrophages produce and functionally respond to interleukin-34 in colon cancer. Cell Death Discov. 2020, 6, 117. [Google Scholar] [CrossRef]
- Liu, C.; Yao, Z.; Wang, J.; Zhang, W.; Yang, Y.; Zhang, Y.; Qu, X.; Zhu, Y.; Zou, J.; Peng, S.; et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2020, 27, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Lopez, L.; Kong, Y.W.; Sriram, G.; Patterson, J.C.; Rosenberg, S.; Morandell, S.; Haigis, K.M.; Yaffe, M.B. MAPKAP Kinase-2 Drives Expression of Angiogenic Factors by Tumor-Associated Macrophages in a Model of Inflammation-Induced Colon Cancer. Front. Immunol. 2020, 11, 607891. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zheng, S.; Yan, Z.; Deng, Z.; Wang, R.; Zhang, B. CCL18 promotes the invasion and metastasis of breast cancer through Annexin A2. Oncol. Rep. 2020, 43, 571–580. [Google Scholar] [CrossRef]
- Pham, T.-H.; Bak, Y.; Kwon, T.; Kwon, S.-B.; Oh, J.-W.; Park, J.-H.; Choi, Y.-K.; Hong, J.T.; Yoon, D.-Y. Interleukin-32θ inhibits tumor-promoting effects of macrophage-secreted CCL18 in breast cancer. Cell Commun. Signal. 2019, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, N.; Li, Q.; Zhang, W.; Ke, F.; Leng, Q.; Wang, H.; Chen, J.; Wang, H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 2011, 6, e19495. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, B.; Zhang, Z.; Zhang, W.; Liu, Y. Response gene to complement 32 expression in macrophages augments paracrine stimulation-mediated colon cancer progression. Cell Death Dis. 2019, 10, 776. [Google Scholar] [CrossRef]
- Wang, D.; Wang, R.; Huang, A.; Fang, Z.; Wang, K.; He, M.; Xia, J.-T.; Li, W. Upregulation of macrophage migration inhibitory factor promotes tumor metastasis and correlates with poor prognosis of pancreatic ductal adenocarcinoma. Oncol. Rep. 2018, 40, 2628–2636. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, Z.; Ma, R.; Zhang, Y.; Zhao, L.; Yan, Y.; Lv, X.; Zhang, L.; Su, P.; Bi, J.; et al. lncRNA-Xist/miR-101-3p/KLF6/C/EBPα axis promotes TAM polarization to regulate cancer cell proliferation and migration. Mol. Nucleic Acids 2021, 23, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Luput, L.; Licarete, E.; Sesarman, A.; Patras, L.; Alupei, M.C.; Banciu, M. Tumor-associated macrophages favor C26 murine colon carcinoma cell proliferation in a.an oxidative stress-dependent manner. Oncol. Rep. 2017, 37, 2472–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.J.; Lee, G.T.; Lee, J.-H.; Iwakura, Y.; Kim, W.-J.; Kim, I.Y. Mechanism of pro-tumorigenic effect of BMP-6: Neovascularization involving tumor-associated macrophages and IL-1a. Prostate 2014, 74, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Pagano, E.; Elias, J.E.; Schneditz, G.; Saveljeva, S.; Holland, L.M.; Borrelli, F.; Karlsen, T.H.; Kaser, A.; Kaneider, N.C. Activation of the GPR35 pathway drives angiogenesis in the tumour microenvironment. Gut 2021, 71, 509–520. [Google Scholar] [CrossRef]
- Schuette, V.; Embgenbroich, M.; Ulas, T.; Welz, M.; Schulte-Schrepping, J.; Draffehn, A.M.; Quast, T.; Koch, K.; Nehring, M.; König, J.; et al. Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proc. Natl. Acad. Sci. USA 2016, 113, 10649–10654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-S.; Shiau, A.-L.; Su, B.-H.; Hsu, T.-S.; Wang, C.-T.; Su, Y.-C.; Tsai, M.-S.; Feng, Y.-H.; Tseng, Y.-L.; Yen, Y.-T.; et al. Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J. Hematol. Oncol. 2020, 13, 62. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Dan, H.; Liu, S.; Liu, J.; Liu, D.; Yin, F.; Wei, Z.; Wang, J.; Zhou, Y.; Jiang, L.; Ji, N.; et al. RACK1 promotes cancer progression by increasing the M2/M1 macrophage ratio via the NF-κB pathway in oral squamous cell carcinoma. Mol. Oncol. 2020, 14, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Zhu, W.-W.; Yu, G.-Y.; Wang, X.; Gao, C.; Zhou, X.; Lin, Z.-F.; Shao, W.-Q.; Wang, S.-H.; Lu, M.; et al. S100 calcium-binding protein A9 from tumor-associated macrophage enhances cancer stem cell-like properties of hepatocellular carcinoma. Int. J. Cancer 2021, 148, 1233–1244. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Restifo, N.P. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zhou, H.; Krueger, J.; Kaplan, C.; Lee, S.-H.; Dolman, C.; Markowitz, D.; Wu, W.; Liu, C.; Reisfeld, R.A.; et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J. Clin. Investig. 2006, 116, 2132–2141. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.R.; Mandell, J.B.; Beatty, K.M.; Harvey, S.A.K.; Rizzo, M.J.; Previte, D.M.; Thorne, S.H.; McKenna, K.C. Splenectomy promotes indirect elimination of intraocular tumors by CD8+ T cells that is associated with IFNγ- and Fas/FasL-dependent activation of intratumoral macrophages. Cancer Immunol. Res. 2014, 2, 1175–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Green, M.D.; Li, S.; Sun, Y.; Journey, S.N.; Choi, J.E.; Rizvi, S.M.; Qin, A.; Waninger, J.J.; Lang, X.; et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 2021, 27, 152–164. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ruffell, B. Cavity macrophages stop anti-tumor T cells. Cancer Cell 2021, 39, 900–902. [Google Scholar] [CrossRef]
- Xia, H.; Li, S.; Li, X.; Wang, W.; Bian, Y.; Wei, S.; Grove, S.; Wang, W.; Vatan, L.; Liu, J.R.; et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI Insight 2020, 5, e141115. [Google Scholar] [CrossRef]
- McGrath, M.M. Diverse roles of TIM4 in immune activation: Implications for alloimmunity. Curr. Opin. Organ Transpl. 2018, 23, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Fleur, L.; Botling, J.; He, F.; Pelicano, C.; Zhou, C.; He, C.; Palano, G.; Mezheyeuski, A.; Micke, P.; Ravetch, J.V.; et al. Targeting MARCO and IL37R on Immunosuppressive Macrophages in Lung Cancer Blocks Regulatory T Cells and Supports Cytotoxic Lymphocyte Function. Cancer Res. 2021, 81, 956–967. [Google Scholar] [CrossRef]
- Gyori, D.; Lim, E.L.; Grant, F.M.; Spensberger, D.; Roychoudhuri, R.; Shuttleworth, S.J.; Okkenhaug, K.; Stephens, L.R.; Hawkins, P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 2018, 3, e120631. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8 T Cell-Derived Interferon-γ. Immunity 2019, 51, 381–397. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- offe, A.M.; Bakalar, M.H.; Fletcher, D.A. Macrophage phagocytosis assay with reconstituted target particles. Nat. Protoc. 2020, 15, 2230–2246. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody.....y target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Ye, T.; Liu, J.; Zhao, W.; Gao, S.; Wang, S.; Wu, F.; Zhou, H. The hypothesis of tumor-associated macrophages mediating semi-phagocytosis of cancer cells in distant metastasis. Future Oncol. 2021, 17, 1125–1129. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef]
- Zhao, J.; Zhong, S.; Niu, X.; Jiang, J.; Zhang, R.; Li, Q. The MHC class I-LILRB1 signalling axis as a promising target in cancer therapy. Scand. J. Immunol. 2019, 90, e12804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Wong, S.-C.; Puaux, A.-L.; Chittezhath, M.; Shalova, I.; Kajiji, T.S.; Wang, X.; Abastado, J.-P.; Lam, K.-P.; Biswas, S.K. Macrophage polarization to a unique phenotype driven by B cells. Eur. J. Immunol. 2010, 40, 2296–2307. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, M.; Andersson, E.; Villabona, L.; Seliger, B.; Lundqvist, A.; Kiessling, R.; Masucci, G.V. HLA-dependent tumour development: A role for tumour associate macrophages? J. Transl. Med. 2013, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cao, Y.; Li, R.; Gu, Y.; Chen, Y.; Qi, Y.; Lv, K.; Wang, J.; Yu, K.; Lin, C.; et al. Poor clinical outcomes of intratumoral dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin-positive macrophages associated with immune evasion in gastric cancer. Eur. J. Cancer 2020, 128, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Park, H.-J.; Chi, G.-Y.; Choi, Y.-H.; Park, S.-H. The root bark of Morus alba L. regulates tumor-associated macrophages by blocking recruitment and M2 polarization of macrophages. Phytother. Res. 2020, 34, 3333–3344. [Google Scholar] [CrossRef]
- Bhaskaran, N.; Ghosh, S.K.; Yu, X.; Qin, S.; Weinberg, A.; Pandiyan, P.; Ye, F. Kaposi’s sarcoma-associated herpesvirus infection promotes differentiation and polarization of monocytes into tumor-associated macrophages. Cell Cycle 2017, 16, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Liao, Q.; Zeng, Z.; Guo, X.; Li, X.; Wei, F.; Zhang, W.; Li, X.; Chen, P.; Liang, F.; Xiang, B.; et al. LPLUNC1 suppresses IL-6-induced nasopharyngeal carcinoma cell proliferation via inhibiting the Stat3 activation. Oncogene 2014, 33, 2098–2109. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhong, Y.-B.; Shao, J.; Zhang, C.; Shi, C. Tumor-associated macrophages promote human hepatoma Huh-7 cell migration and invasion through the Gli2/IGF-II/ERK1/2 axis by secreting TGF-β1. Cancer Biol. 2020, 21, 1041–1050. [Google Scholar] [CrossRef]
- Liu, D.; Song, L.; Wei, J.; Courtney, A.N.; Gao, X.; Marinova, E.; Guo, L.; Heczey, A.; Asgharzadeh, S.; Kim, E.; et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J. Clin. Investig. 2012, 122, 2221–2233. [Google Scholar] [CrossRef] [Green Version]
| Target | Inhibitor | Tumor | Study Design | Anti-Tumor Mechanism | Ref. |
|---|---|---|---|---|---|
| Inhibit recruitment of TAMs | |||||
| CCL2 | Celecoxib | GBM | C57BL/6 J mice + Eagle medium F-12 | Decrease pNF-κB expression | [9] |
| 6-Shogaol | BC | MDA-MB-231/A549/4T1 cell line + Leibovitz’s L-15, F-12K, etc. medium | Decrease CCL2 by inhibiting STAT3 activation | [18] | |
| CCR2 | Losartan | BC | 4T1-Luc, etc. cell line + ICR, etc. mice | Inhibit CCL2-induced p-ERK1/2 | [19] |
| CXCL1 | Aiduqing | BC | 4T1/293 T cell line + BALB/c mice + DMEM/RPMI-1640 | Decrease Tregs differentiation and infiltration | [20] |
| CCR5 | Maraviroc | BC | MDA-MB-436/4T1.2 cell line + DMEM | Inhibit TAM recruitment | [21] |
| CCL5 | HuR | BC | MCF-7/MDA-MB-231 cell line + DMEM | Inhibit CCL5 expression | [22] |
| CCR1 | J113863 | FA | NCTC 2472 cell line + NCTC 135 medium + C57BL/6, C3H/He mice | Inhibit thermal hyperalgesia | [23] |
| CXCR7 | CCX771 | BC | 4T1 cell line + DMEM + BALB/c mice | Reduce p-STAT3 activation | [24] |
| CXCL8 | IFN-γ | PC | BxPC-3, etc. cell line + C57BL/6 mice | Inhibit macrophages traffic | [25] |
| ACPP Antibody | NPC | C666-1 cell line + RPMI 1640 medium | Inhibit PI3K/AKT pathway | [26] | |
| IL-1β | Anakinra | BC | 4T1 cell line + α-MEM + BALB/c mice | Inhibit CCL5, CXCX12 expression | [27] |
| IL-6 | Siltuximab | OC | Tissue from ovarian cancer patients + endotoxin-free RPMI/DMEM medium | Reduce cytokine and chemokine, inhibit IL-6 signaling | [28] |
| S100B | Duloxetine | GLA | GL261-Luc/KR158B cell line + DMEM + CX3CR1GFP mice | Decrease CCL2 expression | [29] |
| CSF-1R | PLX3397 | HCC | Hep3B/HepG2/THP-1, etc. cell line + OPN knockout C57BL/6 mice | Inhibit PPARγ activity to reduce TAM numbers | [30] |
| A2A | SCH58261 | HCC | Tissue from HCC patients | Reduce Akt and ERK phosphorylation to reduce TAM numbers | [31] |
| MEK | GDC-0623 | PC | PDA30364 cell line + pan monocyte isolation kit | Exterminate M2 macrophages | [32] |
| Inhibit the polarization of TAMs | |||||
| STAT6 | Gefitinib | LLC | Cells from Chinese Academy of Sciences + DMEM + C57BL/6 mice | Inhibit IL-13/STAT6 pathway | [33] |
| CSF-1R | BLZ945 | GLA | U-87 MG, etc. cell line + RCAS-hPDGF-B/Nestin-Tv-a; Ink4a/Arf−/− mice | Inhibit heterotypic signaling | [34] |
| YAP | Ovatodiolide | CRC | HT-29, etc. cell line + Serum-Free Medium + NOD, SCID, BALB/c mice | Suppress IL-6 induced pathway | [35] |
| IL-6R | CPEB3 | CRC | SW480/HCT116/LoVo, etc. cell line + BALB/c mice | Inhibit epithelial-mesenchymal transition | [36] |
| Ang-2 | AS16 | SA | Plasmid pPIC3.5K + BMMY + SD rat | Inhibit M2 polarization | [37] |
| Integrin β3 | Sc-7312 | BC | 4T1/HEK293T cell line + RPMI-1640 and DMEM + BALB/c mice | Inhibit integrin β3 induced PPARγ activity | [38] |
| EP4 | TP-16 | CRC | CT26/4T1/HCT116 cell line + DMEM and F12 medium + C57BL/6, etc. mice | Reprogram IMCs, enhance tumor elimination | [39] |
| CD206 | RP-182 | PC | CD206high M2-like macrophages + KPC, KP16 mice | Reduce M2-like TAMs, improve antitumor immune responses | [40] |
| PlGF | HRG | BT | T241/Panc02 cell line + C57BL/6, BALB/c mice | Promote vessel normalization, improve tumor perfusion | [41] |
| Factor | Cancer | Recipient | Influence on Tumor | Biochemical Mechanism | Ref. |
|---|---|---|---|---|---|
| Cytokines | |||||
| IL-1β | HCC | Tumor | Promote tumor migration | NLRP3 dependent FAO/ROS/IL-1β axis | [123] |
| IL-6 | CRC | Tumor | Promote tumor invasion and migration | Regulate JAK2/STAT3/miR-506-3p/FoxQ1 axis | [124] |
| CRC | Tumor | Promote tumor invasion and migration | Activate the Wnt/β-catenin pathway | [125] | |
| BC | TAMs | Promote tumor development | Activate the gp130/STAT3 pathway | [126] | |
| HCC | Tumor | Promote tumor invasion and metastasis | Activate IL-6/ERK and STAT3 pathway | [127] | |
| IL-8 | OC | Tumor | Promote tumor stemness | Activate the IL-8/STAT3 pathway | [102] |
| IL-10 | PC | Tumor | Promote tumor migration | Activate TLR4/IL-10 to express MMP2 and MMP9 | [128] |
| NSCLC | Tumor | Promote tumor invasion | Induce PD-L1 expression | [88] | |
| BC | DC | Attenuate CD8+ T-cell cytotoxicity | Decrease IL-12 expression | [129] | |
| IL-23 | KC | Treg | Promote tumor immune evasion | Increase IL-10, TGF-β expression, and Treg activity | [130] |
| BC | TAMs | Promote tumor angiogenesis | Increase IL-10, TGF-β, VEGF expression | [131] | |
| IL-34 | CRC | TAMs | Promote tumor growth | Increase IL-6 expression | [132] |
| Chemokines | |||||
| CCL2 | BC | Tumor | Promote drug resistance | Activate the PI3K/Akt/mTOR pathway | [120] |
| CCL5 | CRC | Tumor | Promote tumor immune escape | Activate the p65/STAT3-CSN5-PD-L1 pathway | [133] |
| CCL8 | GBM | Tumor | Promote tumor invasion and stemness | Activate the ERK1/2 pathway | [101] |
| CXCL12 | CRC | Tumor | Promote tumor angiogenesis | Activate the MK2 pathway | [134] |
| CCL18 | BC | Tumor | Promote tumor invasion and metastasis | Activate the AnxA2/PI3K/Akt/GSK3β/Snail pathway | [135] |
| BC | Tumor | Promote tumor metastasis | Activate the PKCδ/STAT3, NF-κB pathway | [136] | |
| CCL20 | CRC | Treg | Promote Treg recruitment | CCL20/CCR6 couple | [137] |
| CCL22 | NSCLC | Treg | Promote Treg recruitment | Increase IL-8 expression | [138] |
| Others | |||||
| TNF-α | BC | Tumor | Promote tumor EMT and migration | Increase cAMP and CREB expression | [86] |
| TGF-β | CRC | TAMs | Promote tumor proliferation | Increase RGC-32, COX2 expression | [139] |
| LSECtin | BC | Tumor | Promote tumor stemness | N/A | [100] |
| MIF | CRC | N/A | Promote tumor growth | Increase Tregs generation | [90] |
| PDAC | N/A | Promote tumor metastasis | Activate AKT, ERK, and express cyclin-D1, MMP2 | [140] | |
| Xist | BC | TAMs | Promote tumor proliferation | lncRNA-Xist/miR-101-3p/KLF6/C/EBPα axis | [141] |
| ROS | CRC | N/A | Promote tumor proliferation | Activate NF-κB, AP-1, EGR-1 | [142] |
| MCP-1 | CRC | Tumor | Promote tumor growth, invasion | Activate the MK2 pathway | [92] |
| BMP-6 | PC | TAMs | Promote tumor angiogenesis and growth | Increase IL-1a expression through Smad1, NF-κB | [143] |
| GPR35 | CRC | Tumor | Promote tumor angiogenesis and growth | Na/K-ATPase-dependent ion pumping | [144] |
| CD206 | CRC | N/A | Attenuate CD8+ T-cell cytotoxicity | Inhibit CD45 phosphatase activity | [145] |
| Oct4 | LC | TAMs | Promote tumor growth | Increase M-CSF expression | [146] |
| Chi3L1 | BC | Tumor | Promote tumor metastasis | Activate the CHI3L1/IL-13Rα2/ERK/JNK axis | [147] |
| RACK1 | OSCC | TAMs | Promote tumor development | Regulate NF-κB pathway | [148] |
| GPNMB | BC | Tumor | Promote tumor stemness | Increase IL-33, CD44 expression | [116] |
| S100A9 | HCC | Tumor | Promote tumor stemness | Activate AGER/NF-κB axis | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, D.; Liu, X.; Mu, J.; Yang, J.; Wu, F.; Zhou, H. Therapeutic Approaches Targeting Proteins in Tumor-Associated Macrophages and Their Applications in Cancers. Biomolecules 2022, 12, 392. https://doi.org/10.3390/biom12030392
Wu D, Liu X, Mu J, Yang J, Wu F, Zhou H. Therapeutic Approaches Targeting Proteins in Tumor-Associated Macrophages and Their Applications in Cancers. Biomolecules. 2022; 12(3):392. https://doi.org/10.3390/biom12030392
Chicago/Turabian StyleWu, Deyang, Xiaowei Liu, Jingtian Mu, Jin Yang, Fanglong Wu, and Hongmei Zhou. 2022. "Therapeutic Approaches Targeting Proteins in Tumor-Associated Macrophages and Their Applications in Cancers" Biomolecules 12, no. 3: 392. https://doi.org/10.3390/biom12030392
APA StyleWu, D., Liu, X., Mu, J., Yang, J., Wu, F., & Zhou, H. (2022). Therapeutic Approaches Targeting Proteins in Tumor-Associated Macrophages and Their Applications in Cancers. Biomolecules, 12(3), 392. https://doi.org/10.3390/biom12030392