1. Introduction
Prostate cancer is the second most common cancer in males worldwide and the most common cancer in males in the UK, with approximately 1,111,000 and 43,436 new cases per year, respectively [
1,
2]. Diagnostic techniques, however, are still reliant on the inherently flawed PSA blood test, which has limited associated risks in comparison to other invasive testing, such as prostate biopsies, which may result in a subsequent infection and urinary incontinence [
3,
4]. It is, therefore, important to identify novel biomarkers that can be used to improve the accuracy of low invasive testing. Of greater significance is the current inability to differentiate between low-risk progression and high-risk progression prostate cancer at an early curable stage [
5], whereby low-risk progression prostate cancers are those that are unlikely to grow or metastasize outside of the prostate for many years and, therefore, have a limited risk of morbidity or mortality, whilst high-risk progression prostate cancers are those that are likely to grow and progress to metastatic disease, resulting in an increased morbidity and mortality [
6]. The problem with not being able to identify high-risk progression prostate cancer is that it results in the overtreatment of low-risk progression prostate cancer and the unnecessary associated morbidity [
5]. This highlights the necessity of identifying biomarkers to categorize tumours that are likely to progress at an early stage to ensure treatment is provided.
The hepatitis A virus cellular receptor (HAVcR-1) is the cellular receptor for the hepatitis A virus and hepatotropic picornavirus, the cause of acute hepatitis A in humans [
7]. HAVcR-1 is also termed T-cell immunoglobulin and a mucin domain containing molecule 1 (TIM-1) and kidney injury molecule-1 (KIM-1). HAVcR-1 is expressed on every tested human organ, including the liver, small intestine, colon and spleen, as well as high expression in the kidney and testis, however, the natural function of HAVcR-1 has not been fully investigated [
7].
HAVcR-1 has been found to be upregulated in certain cancers, including breast, ovarian, colon and renal cancer [
8,
9,
10]. HAVcR-1 is, therefore, a molecule of interest for cancer diagnosis and a potential target for cancer therapies. HAVcR-1 is proteolytically cleaved proximal to the cell membrane to release an ectodomain [
11,
12]. This HAVcR-1 ectodomain can be secreted into urine from certain tissue types, and this release is increased in renal cell carcinoma [
13]. The HAVcR-1 ectodomain is, therefore, a potential biomarker for certain cancers. HAVcR-1 expression and ectodomain release in cancer is still poorly categorized. There is little known about its usefulness as a biomarker for prostate cancer diagnosis, progression and prognosis. Furthermore, there is a lack of study into the release of the HAVcR-1 ectodomain into the circulation and the use of this as a potential biomarker for use in blood tests for cancer diagnosis and monitoring.
HAVcR-1 has been linked to tight junctions (TJs), which have an important role in the prevention of cancer metastasis. Evidence currently suggests that HAVcR-1 overexpression seen in cancer is linked to TJ disruption and, therefore, links HAVcR-1 to cancer metastasis [
9]. A number of virus receptors have been found to be associated with junctional structures, including TJs and adherens junctions (AJs), and investigations into the association of HAVcR-1 with junctional structures found via immunoprecipitation that the 50 kDa HAVcR-1 associates with the C terminal of Zonula Occludens-1 (ZO-1) and, to a lesser extent, Zonula Occludens-2 (ZO-2), as well as the N-terminal of occludin and RhoC (Ras homolog gene family, member C, Rho GTPase) [
14]. Due to the importance of these molecules in TJs, it is possible that HAVcR-1 is also involved in the TJ complex in endothelial and epithelial cells. The overexpression and knockdown analysis of HAVcR-1 in a human umbilical cord cell line (HECV cells) suggests the importance of HAVcR-1 expression in the hepatocyte growth factor (HGF)-mediated breakdown of TJ, as shown by the decreased trans-endothelial resistance (TER) in HAVcR-1 overexpressed HECV cells in comparison to HAVcR-1 knockdown HECV endothelial cells when treated with HGF [
14]. The dual immunofluorescence of HAVcR-1 and ZO-1 showed an increased expression and concentrated disruption of ZO-1 in cell–cell junctions in knockdown HECV cells in comparison to wild-type HECV cells when treated with HGF. Therefore, it appears likely that both HGF and HAVcR-1 act on the same pathway responsible for the integrity and maintenance of TJs [
14]. The overexpression of HAVcR-1 in cell lines results in decreased TJs, and HAVcR-1 overexpression in cancer is likely to also result in decreased TJs, which may mediate metastasis.
The role of HAVcR-1 in cancer development and progression is an active area of research; however, the role of HAVcR-1 in prostate cancer has not been fully investigated. Therefore, in this study, we sought to determine whether HAVcR-1 and/or the HAVcR-1 ectodomain would provide an effective biomarker for prostate cancer diagnosis and that through the dysregulation of epithelial cell adhesion, HAVcR-1 would contribute to the development and progression of the metastatic disease of prostate cancer. Furthermore, this work focuses on evaluating whether there is a potential to use HAVcR-1 as a prostate cancer biomarker and/or a viable therapeutic option to prevent/treat prostate cancer metastasis and to establish the role of HAVcR-1 in cellular adhesion complexes, cellular behaviour and the effects of HGF in conjunction with HAVcR-1 expression on cellular adhesion complexes and cellular behaviour.
2. Materials and Methods
2.1. Cell Lines
This study used PZ-HPV-7 (an immortalized prostate epithelial cell line), CA-HPV-10 (an immortalized prostate adenocarcinoma cell line) and LNCaP, PC-3 and Du145 (metastatic prostate cancer cell lines). In addition, the HECV vascular endothelial cell line was used. All cell lines were purchased from the American Tissue Culture Collection (ATCC) (Manassas, VA, USA) at the commencement of this study. PC-3, Du145 and HECV cell lines were maintained in DMEM medium (Sigma-Aldrich, Gillingham, Dorset, UK). LNCaP clone FGC cell line was maintained in RPMI-1640 medium (Sigma-Aldrich, Gillingham, Dorset, UK). PZ-HPV-7 and CA-HPV-10 cell lines were maintained in Keratinocyte-SFM (Sigma-Aldrich, Gillingham, Dorset, UK).
2.2. Polymerase Chain Reaction (PCR) Primers
Primers used were designed using Primer-BLAST available from NCBI. Reverse primers used for quantitative polymerase chain reaction (qPCR) included the addition of a z-sequence on the 5′ end of the primer (Amplifuor™ Uniprimer™) consisting of a 3′ complimentary sequence that pairs with the z-sequence (ACTGAACCTGACCGTACA) present in qPCR reverse primers, as well as a 5′ hairpin loop labelled with a fluorophore reporter (FAM). Custom-designed primers were synthesized by Sigma-Aldrich (Gillingham, Dorset, UK), diluted to 100 µm in PCR H2O and stored at −20 °C. Forward and reverse primers and forward primers for qPCR were further diluted 1:10 before use. Reverse qPCR primers were further diluted 1:100 before and all diluted primers were temporarily stored at 4 °C. Full sequences were as follows: Target Gene Forward (F) and Reverse (R) Primer Sequences 5′-3′ Cycle Number was 30. HAVcR1 F: CAACAACAAGTGTTCCAGTG 436 bp; R: GCATTTTGCAAAGCTTTAAT. GAPDH F: GGCTGTTTTAACTCTGGTA 475 bp; R: GACTGTGGTCATGAGTCCTT. ZO-1 F: CCACATACAGATACGAGTCCTC 533 bp; R: TGGCTTATGCTGAGATGAAGG. ZO-2 F: CTGACATGGAGGAGCTGA; R: GAGACCATACTCTTCGTTCG. CLDN1 F: ATGGCCAACGCGGGGC 636 bp; R: TCACACGTAGTCTTTCC. CLDN2 F: TATAGCACCCTTCTGGGCCT 432 bp; R: CCTTGGAGAGCTCCTTGTGG. CLDN3 F: ATGCAGTGAAGGTGTACGA; R: TGGTGGCCGTGTACTTCTTC. CLDN4 F: TGGGAGGGCCTCTGGATGAA 422 bp; R: TGGTGGCCGTGTACTTCTTC. CLDN7 F: ATAACCCTTTGATCCCTACC 113 bp; R: ACTGAACCTGACCGTACAACAGG. CLDN9 F: CTTCATCGGCAACAGCATCG 339 bp; R: AAGTCCTGGATGATGGCGTG. JAM1 F: AACAAGATCACAGCTTCCTA 600 bp; R: CTTACTCGAAGTCCCTTTCT. OCLN F: ATGTCATCCAGGCCTC 579 bp; R: ATAGACAATTGTGGCA. α-catenin F: CACAGAGAAGGTTCTGGAAG 518 bp; R: CCGATGTATTTTTGAGTGGT. β-catenin F: AAAGGCTACTGTTGGATTGA 649 bp; R: TCCACCAGAGTGAAAAGAAC. Cyclin D1 F: CGGTGTCCTACTTCAAATGT 721 bp; R: ACCTCCTCCTCCTCCTCT. EPLIN F: TCAAACTAAGATTCTCCGGG 30 875; R: TCGGGGCATCTTCTACC. GSK3β F: ATGTTTCGTATATCTGTT 534 bp; R: GGTGGAGTTGGAAGCTGATG. Primer sequences used in qPCR, reverse primer z-sequences are highlighted in bold. Target Gene Forward (F) and Reverse (R) Primer Sequences 5′-3′. HAVcR-1 F: GACAATGTTTCAACGA 99 bp; R: ACTGAACCTGACCGTACATGGAGGAACAAA. GAPDH F: CTGAGTACGTCGTGGAGTC 93 bp; R: ACTGAACCTGACCGTACACAGAGATGATGACCCTTTTG.
2.3. Antibodies
Primary antibodies were purchased and used at a dilution of 1:500 (unless indicated otherwise) as follows: HAVcR-1 Pab13202, (Abnova, Heyford, Oxon, UK); TIM-1/HAVcR-1 AF1817, E-cadherin 17029 (R&D Systems, Abingdon, Oxon, UK); TIM-1 (N-13 HAVcR-1) SC47495, GAPDH SC32233, Claudin-1 SC17658, Claudin-7 SC177670, Occludin SC8145, ZO-1 SC8146, PKM2 SC65176, cyclin D1 SC753 (Insight Biotechnology LTD, Middlesex, UK); α-catenin C1620 (BD Transduction Laboratories, San Jose, CA, USA); β-catenin 8415 (Sigma-Aldrich, Gillingham, Dorset, UK); Eplin A300-103A (Bethyl Labs, Montgomery, TX, USA). Secondary antibodies were prepared as per manufacturer’s instructions and were purchased as follows: Anti-Mouse IgG A4416, anti-Rabbit IgG A6154, anti-Goat IgG A5420 (Sigma-Aldrich, Gillingham, Dorset, UK); Biotinylated anti-Mouse IgG (Vector Labs, Orton Southgate, Peterborough, UK); AlexaFluor 488 anti-Rabbit, anti-Mouse, anti-Goat and AlexaFluor 594 anti-Rabbit, anti-Goat, DAPI (Thermo Fisher Scientific, Cramlington, UK).
2.4. Human Prostate Cancer Serum Samples
Prostate cancer serum samples (n = 237) and breast cancer serum samples (n = 148) were obtained from Wales Cancer Bank (WCB). Whole blood (n = 9) was obtained from the Welsh Blood Service or obtained from male volunteers with informed consent (n = 4). Serum from volunteers was extracted via centrifugation at 15,000× g for 10 min and stored at −80 °C.
2.5. Patient Tissue Samples
Prostate cancer samples (n = 2) and normal control samples (n = 2) were collected at the University Hospital of Wales between January 2003 and 2006. Sections were collected with informed patient consent and with ethical approval from the South East Wales Research Ethics Committee (Panel C) under the project title “Hepatocyte growth factor (HGF) and its regulators on the behavior of invasive/metastatic prostate cancer”. Ethics no: 03/5048.
2.6. HAVcR-1 Knockdown/Overexpression
Knockdown of HAVcR-1 was achieved using ribozyme technology. The HAVcR-1 specific ribozyme insert was cloned into the plasmid using the pEF6/V5-His TOPO TA expression kit (Invitrogen, Paisley, Scotland, UK). Ribozymes and the expression cassette were constructed as described in work by Martin et al. (2011) [
14].
2.7. RNA Extraction and PCR
Cells were lysed and RNA extracted using EZ-RNA kit (Geneflow, Staffordshire, UK). RNA was reverse transcribed to cDNA using the GoScript™ Reverse Transcription System (Promega, Southampton, UK). PCR was carried out using GoTaq Green Master Mix (Promega, Southampton, UK) with specific primers detailed above using a thermocycler geneAmp PCR system 2700 (Thermo Fisher Scientific, Cramlington, UK). All genes were normalized to the GAPDH housekeeping gene; thus, for every cDNA sample, a PCR reaction with primers specific for GAPDH was carried out. Furthermore, for every primer set, a negative control PCR reaction was carried out, whereby the PCR mix contained no cDNA. Agarose gel electrophoresis was used to separate DNA fragments according to size. Samples were loaded onto a 2% (w/v) agarose gel (Sigma-Aldrich, Gillingham, Dorset, UK). Precision FAST 2X qPCR Master Mix with ROX (Primer Design, Southampton, UK) and Amplifuor™ Uniprimer™ Universal System (Intergen Company®, Bronx, NY, USA) were used to carry out qPCR. qPCR was carried out for each sample using primers specific to the housekeeping gene GAPDH, which was then used for normalizing. The qPCR mixes were compiled in triplicate in a microamp® Fast Optical 96-well reaction plate with barcode (Applied Biosystems, Carlsbad, CA, USA) and covered with MicroAmp® Optical Adhesive film (Thermo Fisher Scientific, Cramlington, UK). qPCR was carried out using the StepOne Plus Real-Time PCR System (Thermo Fisher Scientific, Cramlington, UK). The cycle at which the fluorescent signal reached a particular threshold, known as the CT value, was then given and this was then analysed using ∆∆ CT normalized to the GAPDH housekeeping gene.
2.8. Protein Detection and Analysis
Cell media were aspirated, and cells were washed with PBS before being lysed with lysis buffer. The amount of lysis buffer used depended on culture size. Cell lysates were then incubated on ice for 5 min and transferred to microfuge tubes. Cell lysates were then rotated for 30 min on a Labinoco LD79 Test-tube Rotator (Wolf Laboratories, York, UK) prior to centrifugation at 12,000× g for 15 min at 4 °C. Supernatant (protein lysate) was then transferred into a fresh microfuge tube and either stored at −20 °C ready for protein sample quantification or equal volumes of LaemmLi 2 X Concentrate (Sigma-Aldrich, Gillingham, Dorset, UK) added prior to boiling at 100 °C for 10 min. The Bio-Rad DC™ Protein Assay Kit (Bio-Rad, Hertfordshire, UK) was used for protein sample quantification. SDS-PAGE was undertaken on an acrylamide gel composed of a 10% (v/v) running gel and 5% (v/v) stacking gel in an OmniPAGE VS10DYS Vertical Electrophoresis System (OmniPAGE, Cleaver Scientific Ltd., Rugby, UK). Samples were then transferred from the acrylamide gel to a PVDF Transfer Membrane (Merck Millipore, Sigma-Aldrich, Gillingham, Dorset, UK) using the Mini Trans-Blot® Cell (Bio-Rad, Hertfordshire, UK) wet transfer system. Standard Western blotting was carried out. EZ-ECL Chemiluminescent Detection Kit (Geneflow, Staffordshire, UK) was used for protein visualization.
2.9. Kinexus™ Antibody Microarray Protein Preparation
In preparation for a Kinexus™ Antibody Microarray, PZ-HPV-7 pEF6 and PZ-HPV-7 HAVcR1EXP cells were cultured in 10 cm dishes. When confluent cells were washed twice in PBS, 100 μL Kinexus™ Antibody array lysis buffer was added to lyse cells, followed by quantification using fluorescamine reagent (F9015, Sigma-Aldrich, Gillingham, Dorset, UK). Protein samples were then diluted to 4 mg/mL using Kinexus™ Antibody array lysis buffer to a final volume of 300 μL. Samples were then stored at −20 °C prior to being shipped to Kinexus Bioinformatics, Vancouver, Canada, for the Kinexus™ Antibody Microarray. The Kinexus™ KAM880 Protein Array service provided by Kinexus Bioinformatics Ltd. (Vancouver, BC, Canada) was utilized for this project. The Kinexus™ KAM880 Protein Array uses microarray chips which contain two sets of 877 antibodies. The percentage change from control (%CFC) was calculated, whereby treated referred to PZHPV-7 HAVcR-1EXP and control referred to PZ-HPV-7 pEF6. Percentage error, Z-scores and Z-ratios were also calculated. Significance was based on z-values of ≤−1.65 or ≥1.65.
2.10. Visualization of Proteins Using Immunofluorescence
Cells were seeded into 8-well glass Millicell EZ slides (Merck Millipore, Sigma-Aldrich, Gillingham, Dorset, UK) at 5 X 104 cells per well in 500 μL medium. Slides were then incubated at 37 °C in a 95% (v/v) humidified atmosphere of 5% (v/v) CO2 until cells were confluent. Once confluent, culture medium was removed and cells were washed with PBS and fixed in 500 μL 100% ice-cold ethanol per well and left at −20 °C. Ethanol was then removed and cells were wash 3 times with PBS for 5 min per wash and permeabilized by adding 500 μL 0.1% Triton X-100 (Sigma-Aldrich, Gillingham, Dorset, UK) per well for 1–5 min, depending on protein of interest, at room temperature. Cells were then washed 3 times in PBS for 5 min per wash and blocked using blocking buffer, consisting of 7.5% (v/v) donkey serum (D9663, Sigma-Aldrich, Gillingham, Dorset, UK) in PBS, at 300 μL per well for 6 h at room temperature. Primary and secondary antibodies were used according to manufacturer’s instructions, slides were mounted with FluorSave™ (345789, Sigma-Aldrich, Gillingham, Dorset, UK) and a cover slip. Slides were then left to set in the dark at 4 °C overnight and visualized/imaged using the Hamamatsu Orca ER digital camera and the Olympus BXSA microscope at 100× magnification. Merged images were then created using Adobe Photoshop software.
2.11. Immunohistochemical Staining (IHC) of Patient Tissues
Cryosections were stored at −80 °C. These were allowed to thaw at room temp for approximately 15 min prior to being fixed with dried acetone (10162180, Fisher Scientific, Loughborough, UK) for 15 min, air dried for 15 min and washed 3 times with PBS for 5 min per wash. Cryosections were then incubated with blocking diluent (0.1% (v/v) BSA, 0.01% (v/v) Marvel, 10% (v/v) horse serum and 90% (v/v) PBS) for 1 h in a humidified box at room temperature. Sections were then incubated in a humidified chamber for 1 h in primary antibody diluted in blocking diluent to a final concentration of 2 µg/mL or blocking diluent for negative controls. Sections were again washed 3 times in PBS for 5 min per wash and then incubated for 30 min in ABC biotinylated secondary antibody diluted in blocking diluent in a humidified chamber for 30 min. Sections were washed 3 times in PBS for 5 min per wash, incubated in a humidified chamber for 30 min in ABC reagent provided in the Vectastain Universal Elite ABC kit (Vector, Peterborough, UK), washed 3 times in PBS and developed with diaminobenzidine substrate (DAB) (Abcam, Cambridge, UK) (90% (v/v) 10% (v/v) DAB and 6 μL hydrogen peroxide for 10 min). Sections were then washed in H2O, counterstained in Ehrlich’s haematoxylin for 5–10 min and washed in H2O. Visualization and imaging of sections was performed using the Leica DM10000LED microscope with a MC120 HD camera and Leica Application Suite (version 3.0.0) software (Leica Microsystems, UK). Localization and intensity of staining were judged blindly by two people independent of one another.
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA was performed using Human TIM-1 (HAVCR1) ELISA Kit (Thermo Fisher Scientific, Cramlington, UK). Serum samples were diluted 1:2 in Diluent B and 100 μL of each sample and provided standards were placed into appropriate wells of the provided 96-well plate. The absorbance was measured on an ELx800 Absorbance Reader (BioTek, Swindon, UK) at 450 nm. The absorbance of standards was then used to form a standard curve and this was used to calculate the protein concentration of samples.
2.13. In Vitro Cell Function Assays to Ascertain Changes in Cell Behaviour after HAVcR-1 Knockdown
Growth assays—Cells were seeded in triplicate into 24-well plates at 1 X 103 cells per well in 1 mL of cell medium and incubated at 37 °C in a 95% (v/v) humidified atmosphere of 5% (v/v) CO for 1, 3 and 5 days. After incubation, cells were washed with PBS and then fixed, stained and imaged (crystal violet, V5265, Sigma-Aldrich, Gillingham, Dorset, UK). Cell growth was presented as the fold change in cell number from the 1 day time point. Adhesion assays—Conducted using Matrigel™ basement membrane matrix (BD Biosciences, Oxford, UK) diluted to 0.05 mg/mL in cell medium and 100 μL was loaded into each well of a 96-well plate. This was then dehydrated at 56 °C. Following rehydration with serum-free media, cells were seeded at 5000 cells per well in 200 μL of cell medium and incubated for 30 min. The cell medium was then discarded, the cells were washed with PBS and stained using crystal violet before counting or die removal using 10% (v/v) acetic acid, and absorbances were measured at 540 nm using the ELx800 Absorbance Reader (BioTek, Swindon, UK). Invasion potential—Assessed using a standard invasion model, with a 0.8 µm pore ThinCert™ 24-well plate inserts (Greiner Bio-One Ltd., Gloucestershire, UK). Matrigel™ basement membrane matrix (BD Biosciences, Oxford, UK) was diluted in serum-free medium to 0.5 mg/mL and 100 μL loaded into each insert to replicate the extracellular matrix. Cell seeding was at 3 X 104 cells per insert in 500 μL of serum-free medium. Cells that had invaded the Matrigel™ layer and had migrated through to the underside of the ThinCert™ 24-well plate inserts were stained using crystal violet before counting. Changes in TJ function—Assessed using transepithelial resistance (TER) and paracellular permeability (PCP). TER used 0.4 µm pore ThinCert™ 24-well plate inserts (Greiner Bio-One Ltd., Gloucestershire, UK) in 24-well plates. Cells were seeded into inserts at 5 X 103 cells per insert in 500 μL of cell medium with 1.5 mL medium in the well outside of the insert. Cells were incubated until confluent. Resistance across the membrane was then measured in triplicate immediately afterwards using the EVOM² Epithelial Volt/Ohm Meter (World Precision Instruments, Hitchin, Hertfordshire, UK). Where treatments were applied, resistance was also measured every hour after the initial measurement for 10 h. Data were then converted to Ω·cm2 by the multiplication of measured resistance by the surface area of the ThinCert™ 24-well plate inserts. PCP used 0.4 µm pore ThinCert™ 24-well plate inserts (Greiner Bio-One Ltd., Gloucestershire, UK) in 24-well plates. Cells were seeded into inserts at 5 X 103 cells per insert. Medium was then replaced and 0.2 mg/mL of tetramethylrhodamine isothiocyanate (TRITC)–dextran conjugate with an average molecular weight of 40 kDa (42874, Sigma-Aldrich, Gillingham, Dorset, UK) and 0.2 mg/mL of fluorescein isothiocyanate (FITC)–dextran conjugate with an average molecular weight of 10 kDa (FD10S, Sigma-Aldrich, Gillingham, Dorset, UK) was added to each insert. Immediately after media change and every hour thereafter until 10 h, 20 μL of cell medium from outside of the inserts was transferred into a black 96-well cell culture microplate (Greiner Bio-One) in duplicate. Fluorescence was then measure using the GloMax® Multi Detection System (Promega, Southampton, UK) at excitation 520 and emission 580–640 for TRITC–dextran and excitation 940 and emission 510–570 for FITC–dextran. Measurements were then normalized to the 0 h time point measurement via subtraction and a statistical analysis was performed. Standard wound healing/scratch model to assess migration—Briefly, cells were seeded in quadruplicate into 24-well plates at 5 X 103 cells per well, a manual scratch was performed and images were then taken at 5× magnification to give the 0 h time point. Images were then taken every hour after this up to 10 h. Images were then analysed using ImageJ software to give wound area, which was then used to calculate the percentage change in area from the 0 h time point (presented as percentage wound closure). Electric cell–substrate impedance sensing (ECIS) to assess changes in cell behaviour—ECIS was performed using 96W1E+ plates (ECIS Cultureware™, IBIDI, Martinsried, Germany) and the ECIS® Z-theta model instrument (IBIDI, Martinsried, Germany). Wells were inoculated with 5 X 104 cells per well in 300 μL recommended cell medium. The behaviour for cell monolayers was then electrically monitored at 7 predefined frequencies (1, 2, 4, 8, 16, 32 and 64 kHz). At 25 h, an electrical wound of 60,000 Hz and 3000 µA was applied to the cell monolayers for 30 s. The resulting changes to the cell monolayers were continuously monitored, at the frequencies stated previously, during wounding and for 17 h post wounding. The data collected were analysed as fold change from time 0 h for initial attachment and spreading and from time 25 h for wound healing for resistance and capacitance at 1 kHz and 64 kHz, respectively, as well as well as for Rb and alpha. Effect of HGF—Functional assays where HGF treatment used cells were either treated with 40 ng/mL HGF or an equal amount of 0.1% BSS in PBS as the control. Treatment was conducted at time 0 in all cases with this being at seeding for growth, invasion and adhesion, immediately after scratch formation with migration, at the same time as fluorescent dextran conjugates for PCP and immediately after base line (time 0 h) readings for TER.
2.14. Statistical Analyses
Microsoft Excel was used for statistical analysis of the majority of data utilizing a two-tailed unpaired Student’s t-test. For patient serum samples, Graphpad Prism (version 6, GraphPad Software Inc., San Diego, CA, USA) was used, whereby a D’Agostino–Pearson omnibus K2 normality test was performed on columns to assess normality. If data were of a normal distribution, a two-tailed unpaired Student’s t-test or T-test with Welsh correction was performed for the comparison of two datasets or a one-way ANOVA for the comparison of three or more datasets. If data were not of a normal distribution, a Mann–Whitney U test was performed for the comparison of two datasets or a Kruskal–Wallis test was performed for the comparison of three or more datasets. Linear regression analysis using Graphpad Prism 6 software was utilized to address significance between slopes. In all cases, p values of ≤0.05 were considered significant and represented on graphs by *. Where p ≤ 0.01 or p ≤ 0.001, the representation of ** or *** was used, respectively.
4. Discussion
Prostate cancer is a significant problem throughout the world, and due to the high-incidence rates can result in a large proportion of people burdened with the disease. Current diagnostic testing fails to meet the requirements for effective screening and lack an understanding of the progression of prostate cancer to metastatic disease. There is still a requirement for novel biomarkers to improve the diagnosis, monitoring and prognostic indicators to increase our understanding of disease progression, with the hopes of developing therapeutic targets for the treatment or prevention of metastatic prostate cancer.
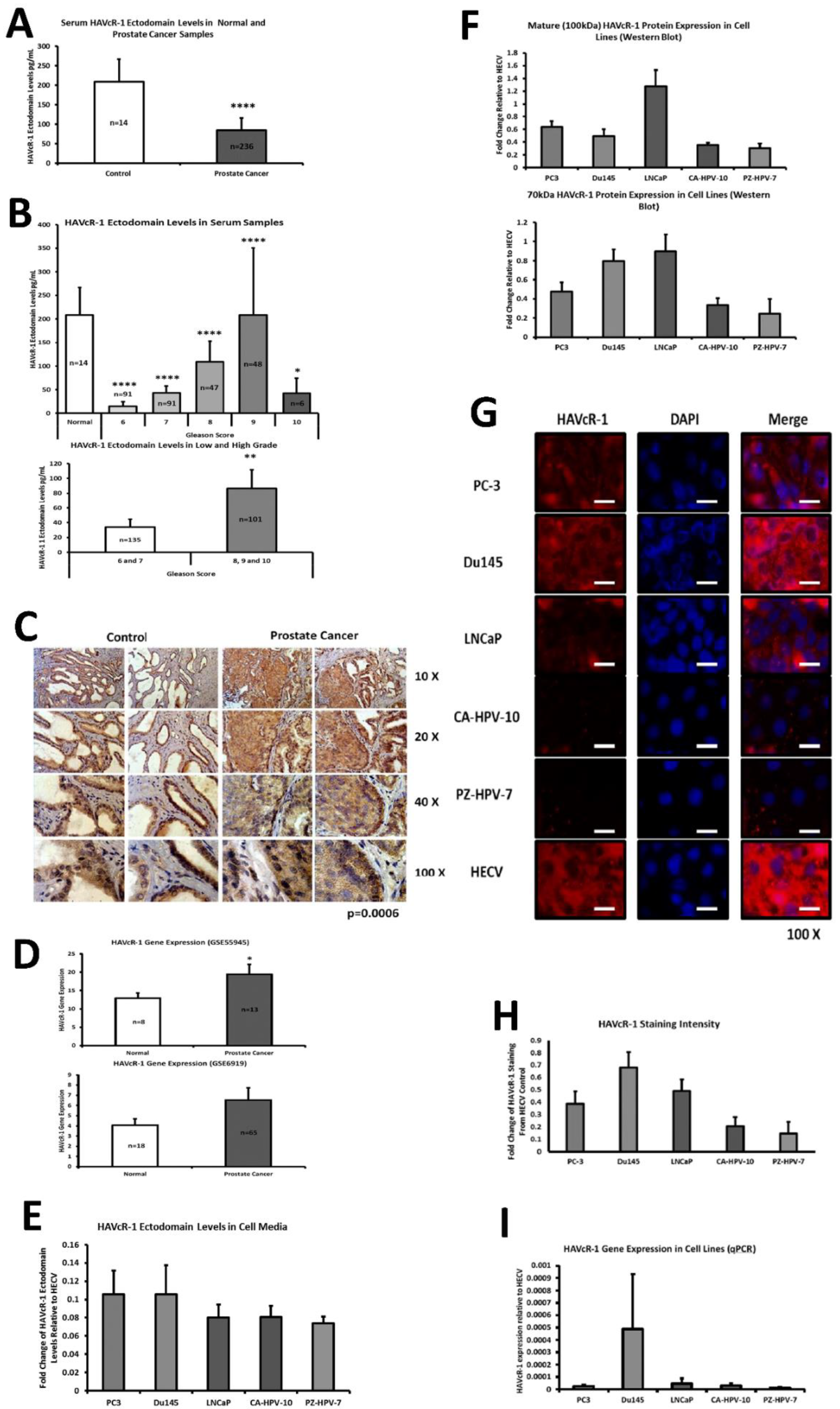
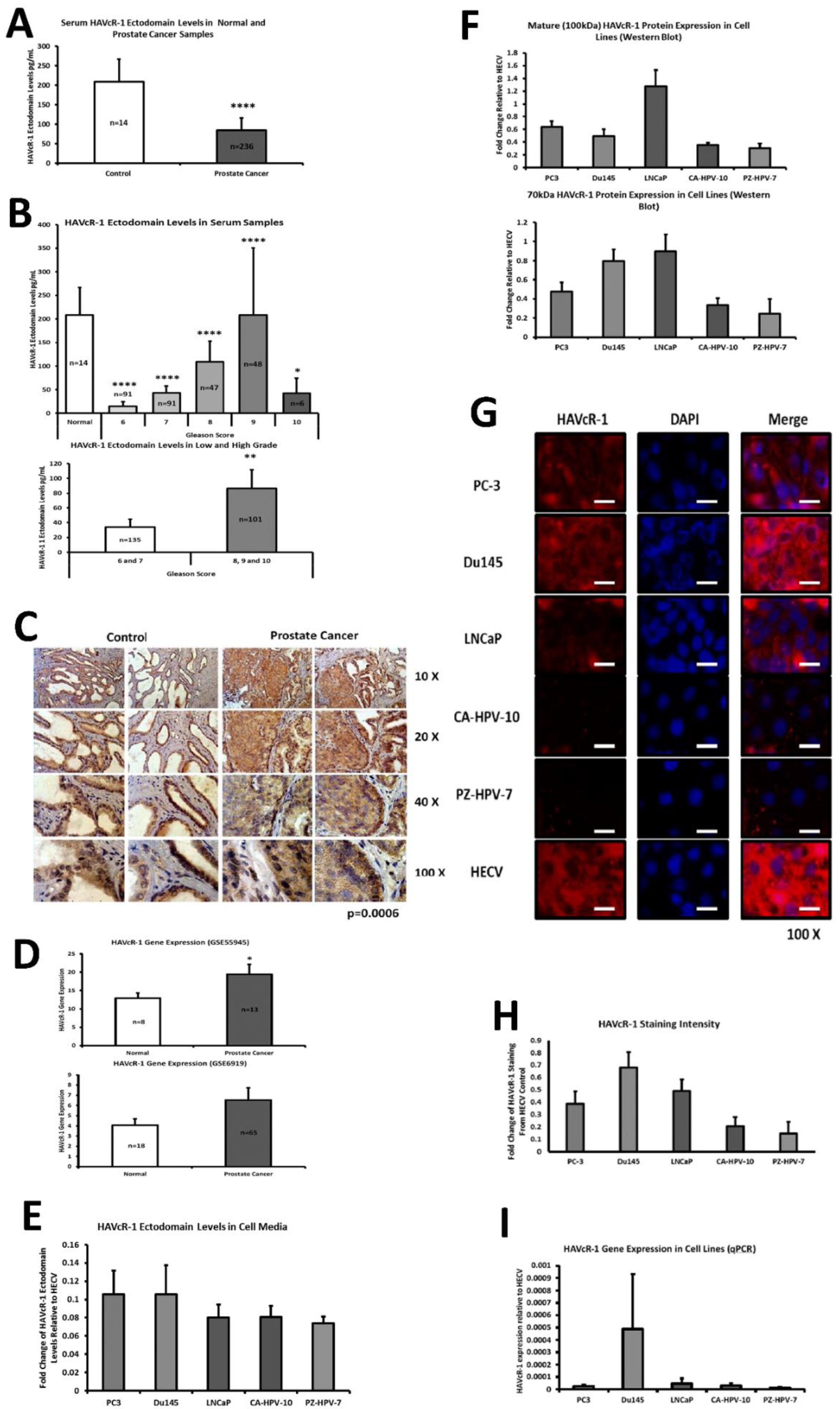
This current study initially examined serum HAVcR-1 ectodomain levels in prostate cancer, which showed decreased serum HAVcR-1 ectodomain levels in comparison to healthy control levels, and a significance decrease in serum HAVcR-1 ectodomain levels in the Gleason score of 6, 7, 8, 9 and 10 prostate cancers, in comparison to healthy control levels. This, therefore, presents HAVcR-1 as a potential diagnostic biomarker, which would be of particular interest where the current biomarker (PSA) is highly nonspecific [
3]. Whether this is unique to prostate cancer has yet to be determined, and it may be possible that serum levels decrease in a variety of cancer types and, thus, HAVcR-1 may need to be used in conjunction with other biomarkers such as PSA in the instance of prostate cancer or followed by further testing to determine the cancer type. A more recent study has shown that HAVcR-1 was significantly upregulated at the mRNA and protein level in gastric cancer tissues compared to the adjacent normal tissues, and was an independent indicator of shorter overall survival, an important indicator that HAVcR-1 could be considered as a novel prognostic indicator in this cancer type [
15]. It is also important to note that these are preliminary results and further testing is required prior to HAVcR-1 being used in this capacity, including greater sample numbers and trials to investigate whether serum HAVcR-1 levels can accurately diagnose prostate cancer as well as stage the disease. In clear cell renal cell carcinoma, there is a known link between the HAVcR-1 ectodomain shedding effect invasiveness and tumour malignancy, and it would, therefore, be interesting to investigate if a similar effect is seen in prostate cancer [
16].
This study also aimed to assess the total HAVcR-1 expression in prostate cancer, which revealed a significant increase in HAVcR-1 protein expression in prostate cancer tissue samples in comparison to normal control samples. This result was unsurprising as it has previously been documented that there is increased HAVcR-1 staining in prostate cancer tissue samples [
17]. Furthermore, using GEO datasets, HAVcR-1 overexpression in prostate cancer was also shown at the gene level. HAVcR-1 overexpression has previously been observed in breast cancer, ovarian cancer and renal cell carcinoma; therefore, providing further evidence that HAVcR-1 is not specific to a certain cancer type [
16,
18]. Interestingly, this increase in total HAVcR-1 protein expression was the opposite of the observed decreased serum HAVcR-1 ectodomain levels. Two possible explanations for this are that either the cleavage event that resulted in the release of the ectodomain was decreased in prostate cancer or there was a decreased entry of the HAVcR-1 ectodomain into the circulation in prostate cancer. A decreased cellular cleavage appears unlikely due to previously documented increased urinary HAVcR-1 levels with the occurrence of prostate cancer [
13]. In regard to decreased entry into the circulation as HAVcR-1 is expressed in prostate glandular epithelial cells, it would be expected that, similarly to PSA, the disruption of the normal prostate architecture that occurs with prostate cancer progression would cause increased entry into the circulation [
19]. A possible explanation as to why this was not the case is that the HAVcR-1 ectodomain is sequestered within the tumour. HAVcR-1 is expressed on the surfaces of CDK4+ T cells, CDK8+ T cells, natural killer (NK) cells, NKT cells, dendritic cells, B cells and mast cells [
20,
21]. HAVcR-1 is also a co-stimulatory molecule with ligand binding, resulting in the activation, proliferation and cytokine production of T cells and the activation of NKT cells [
21,
22,
23] and ligands, including TIM-4 and phosphatidylserine (PS) [
20,
22,
23]. Because HAVcR-1 can bind PS, it is possible that the released HAVcR-1 ectodomain was sequestered within the tumour, binding to TIM-4 and PS, preventing the activation of infiltrating immune cells [
20]. If this was the case, the release of the HAVcR-1 ectodomain may contribute to the nonresponsiveness of many tumour-infiltrating immune cells and would be of interest for future study. HAVcR-1 is overexpressed in prostate cancer and, therefore, the staining of prostate biopsies could be used to aid in prostate cancer diagnosis; however, it would be of interest to investigate whether there is any correlation between the total HAVcR-1 expression and disease prognosis, as this would have more clinical benefit.
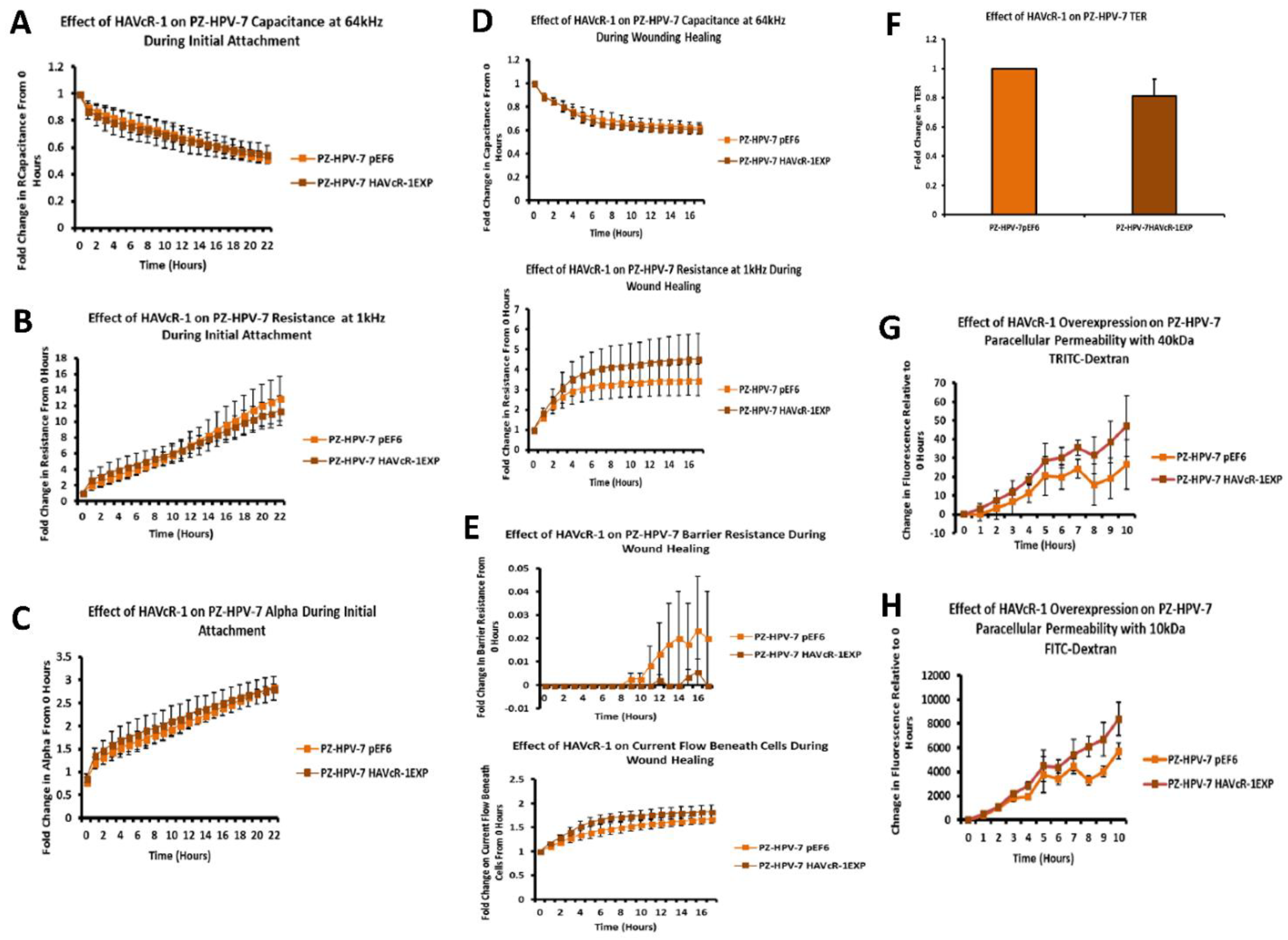
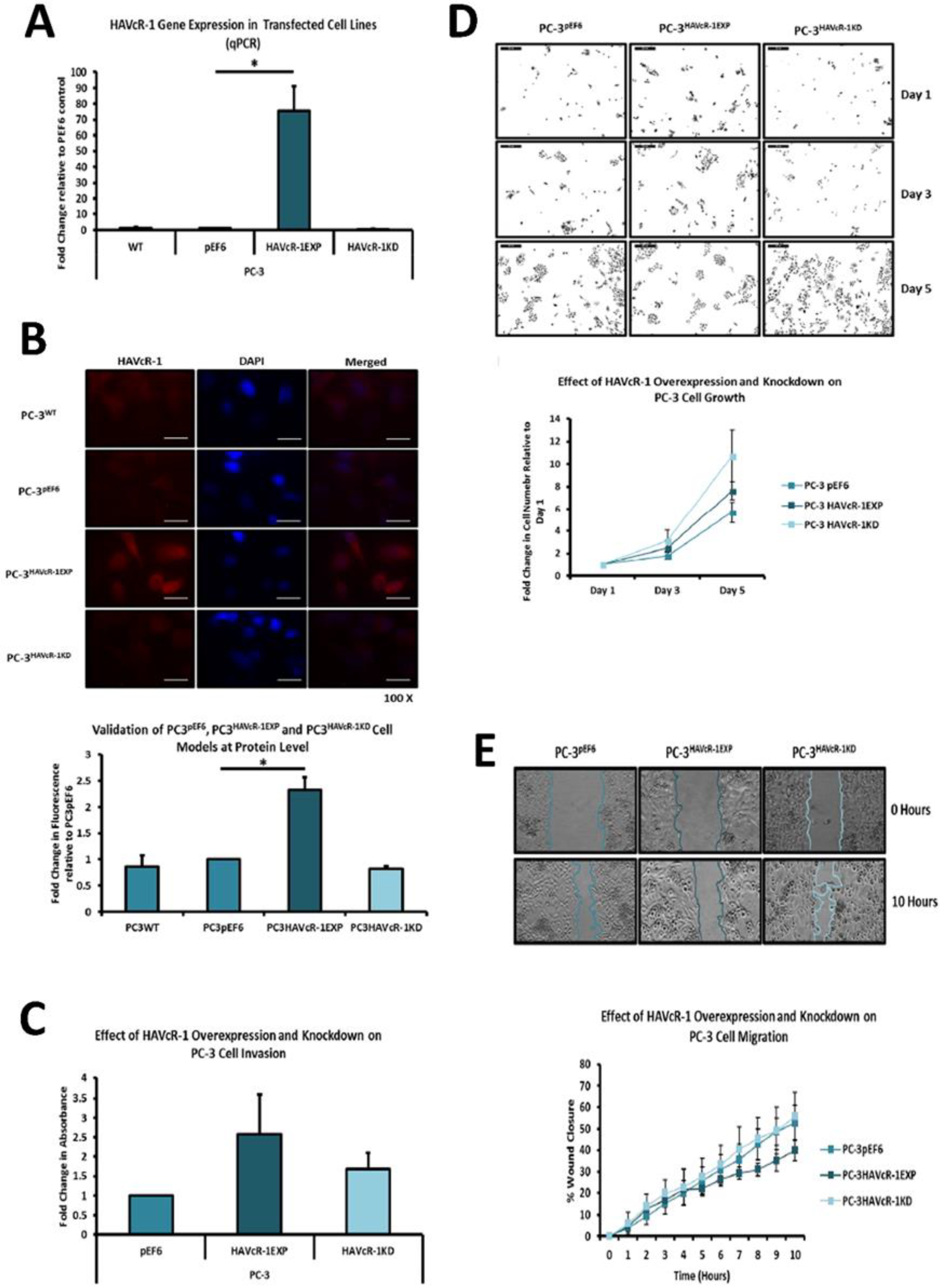
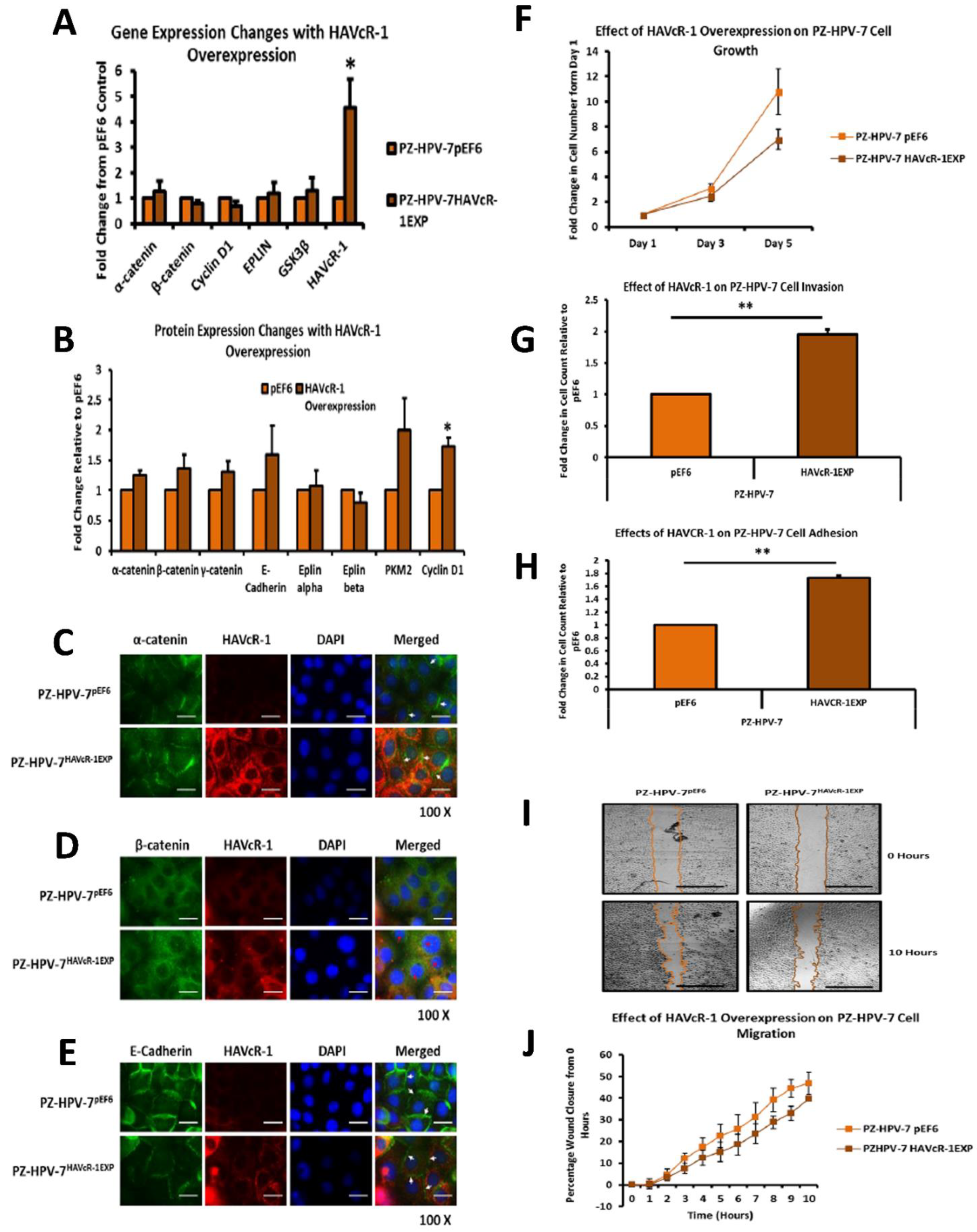
PC-3, Du145 and LNCaP were assessed to model metastatic disease, CA-HPV-10 to model localised disease, and PZ-HPV-7 to model normal prostate epithelia. HAVcR-1 gene expression increased in PC-3, Du145, LNCaP and CA-HPV-10 cell lines in comparison to PZ-HPV-7, with this change being significant in PC-3 cells in comparison to LNCaP, CA-HPV-10 and PZ-HPV-7 cells. There was also a consistent increase in the total HAVcR-1 protein expression in PC-3, Du145, LNCaP and CA-HPV10 cells in comparison to PZ-HPV-7 cells. Therefore, a similar trend was seen in cell lines as in the clinical samples, that HAVcR-1 is overexpressed at the protein and gene level in prostate cancer. Moreover, there was no change in HAVcR-1 ectodomain levels found in cell media between cell lines; thus, conferring with the clinical data theory that the variation in serum HAVcR-1 ectodomain levels with the occurrence of prostate cancer is not due to a variation in the amount of HAVcR-1 cleavage. Cell line expression, therefore, agreed with the clinical data to a suitable degree that they could be used for further study into the effect of HAVcR-1 in prostate cancer.
We also set out to evaluate the effect of HAVcR-1 on prostate cancer cell behaviours that were imperative for metastasis to occur. Consistent with previous HAVcR-1 studies in colorectal cancer, HAVcR-1 had no significant effect on cell growth [
10]. However, unlike the colorectal study, which showed that increased HAVcR-1 decreased invasion and adhesion, this study showed no significant change in either with HAVcR-1 overexpression or knockdown. HAVcR-1 overexpression in colorectal cells resulted in no change in cell migration; however, overexpression in PC-3 showed a potential decrease in wound healing and, therefore, proposes HAVcR-1 as a tumour suppressor [
10].
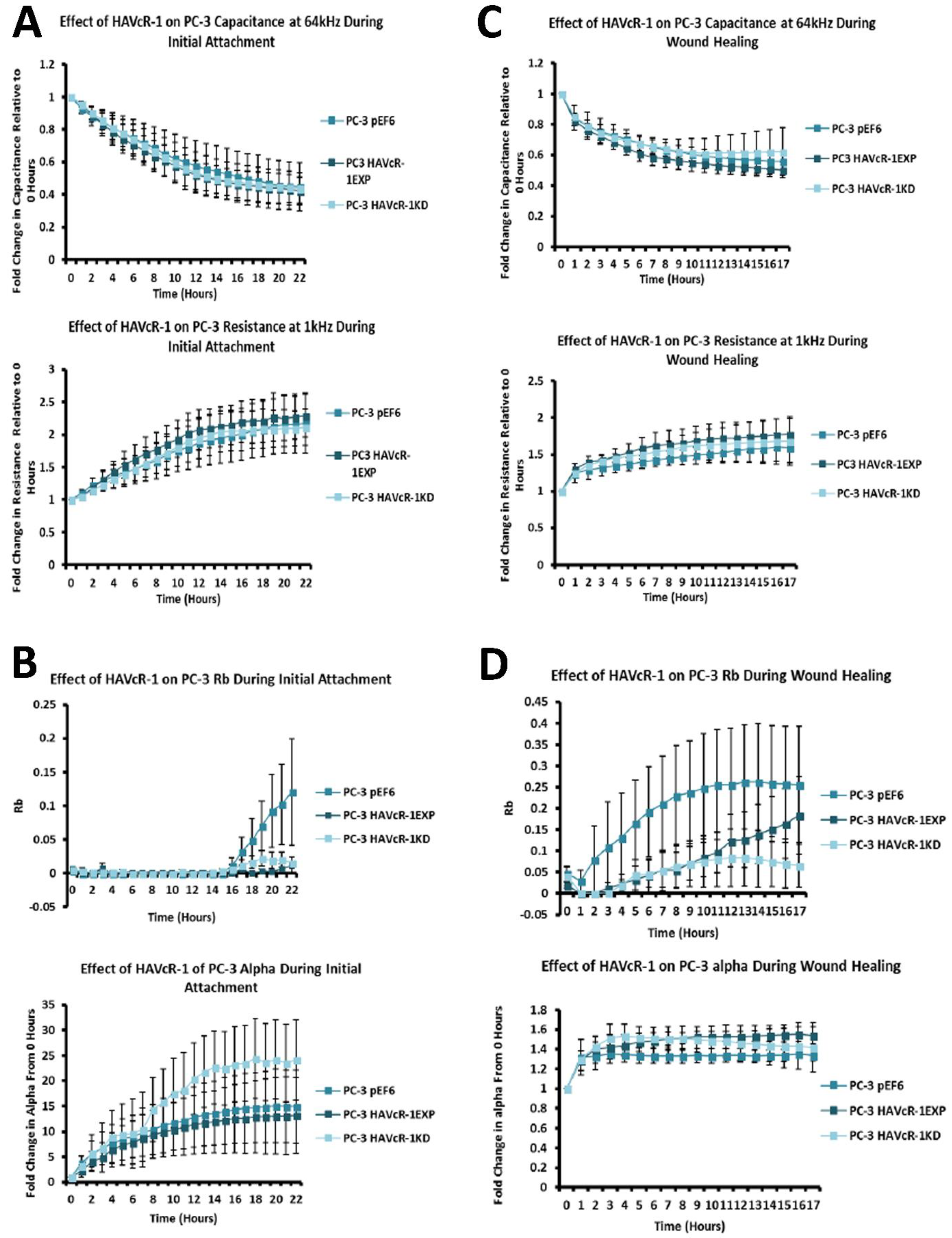
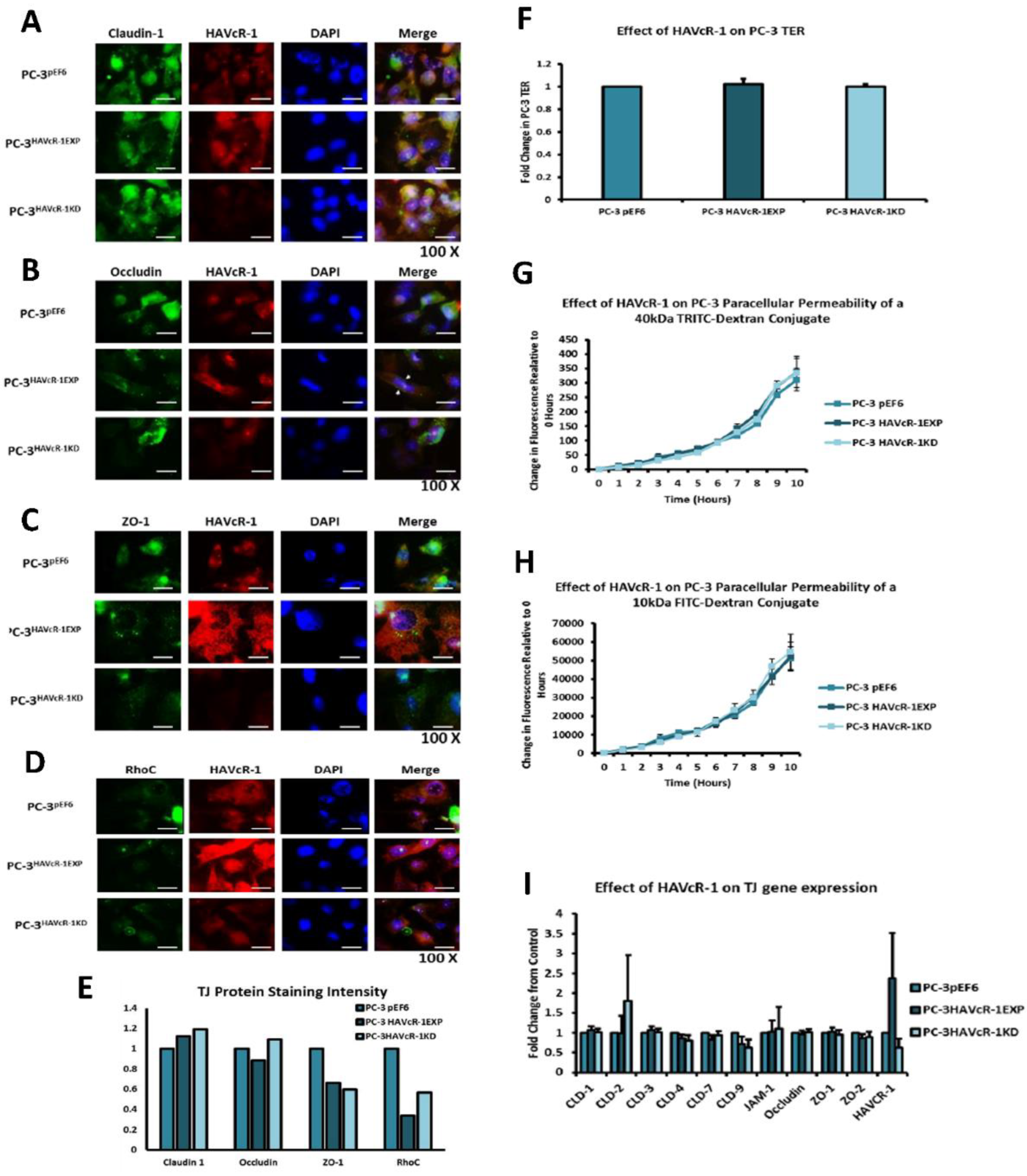
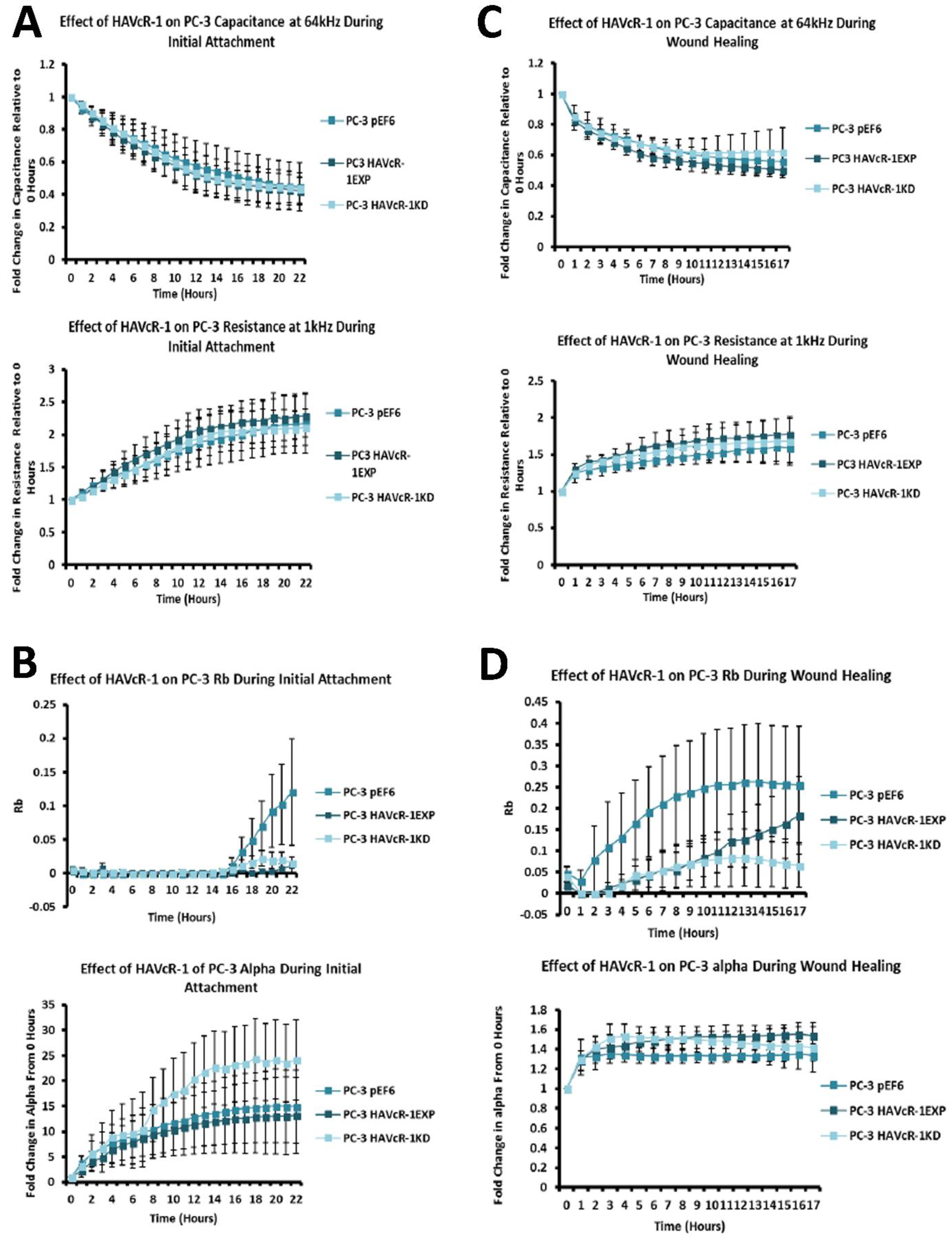
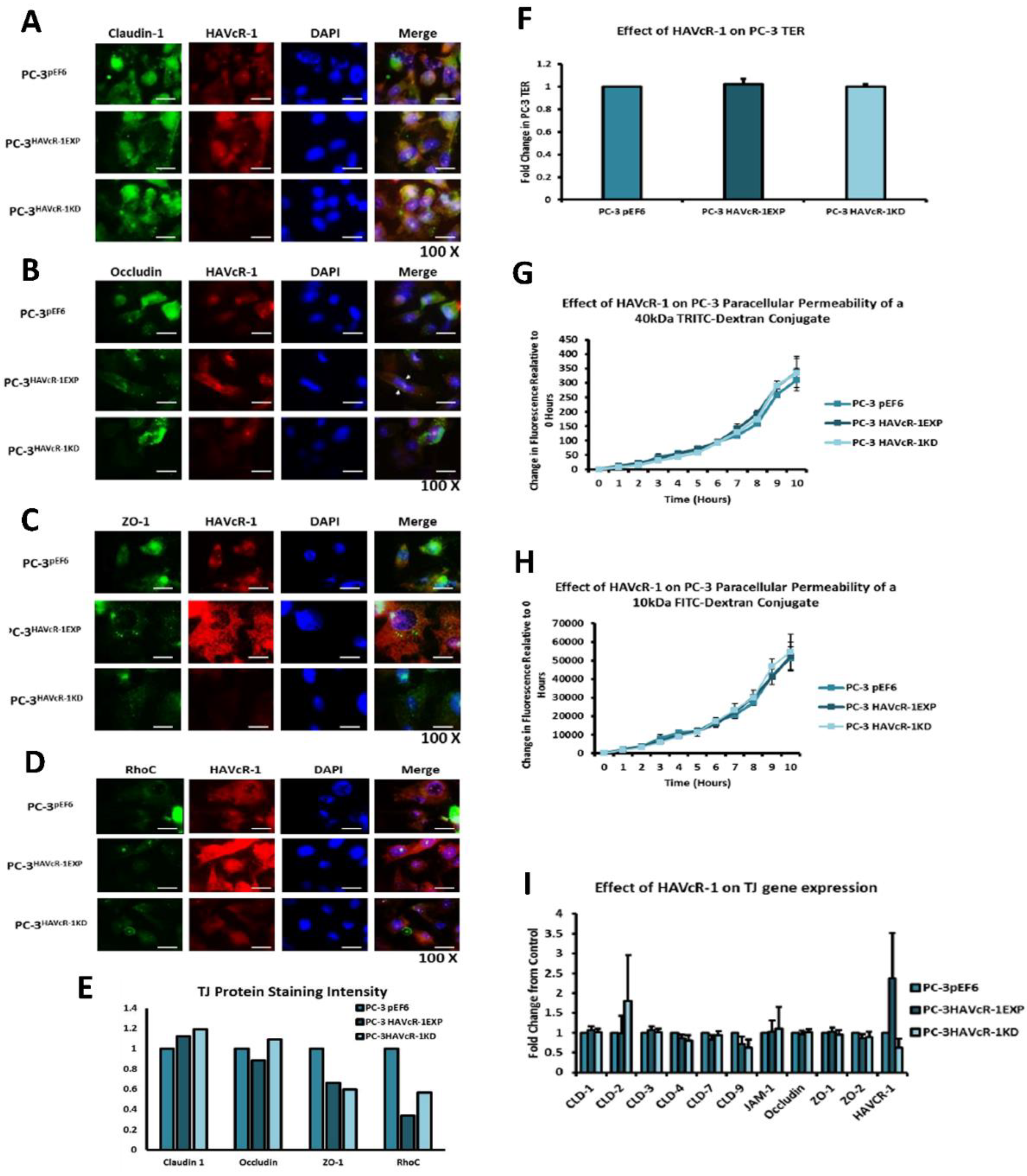
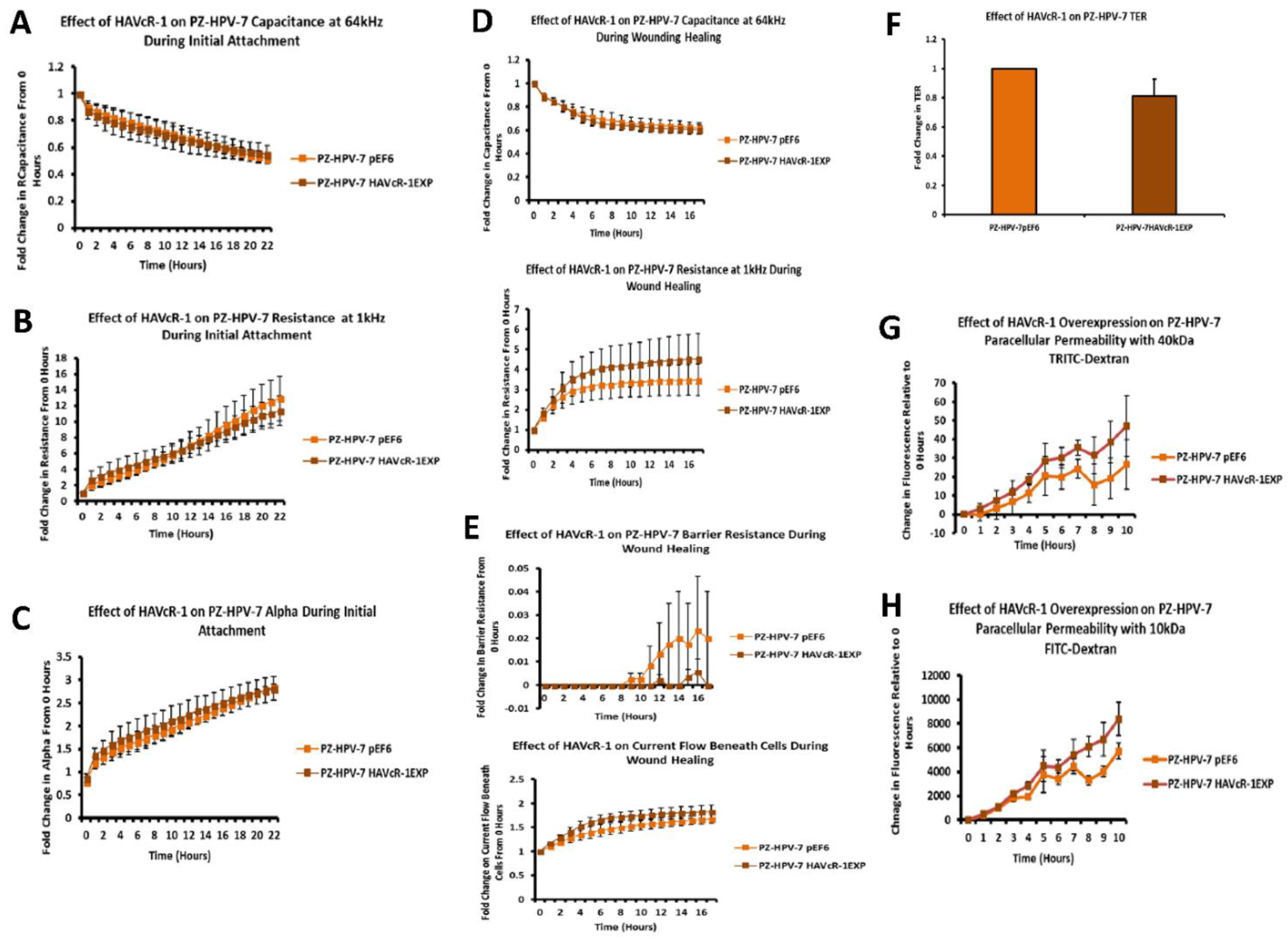
In regard to TJ functions and the potential role that HAVcR-1 might have, PC3HAVcR-1EXP and PC3HAVcR-1KD cell models were utilised in a series of assays for the measurement of TJ integrity. Overall, these assays showed no change in resistance with PC3HAVcR-1EXP or PC3HAVcR-1KD, suggesting that HAVcR-1 has no effect on TJ integrity and is inconsistent with the hypothesis that the increased HAVcR-1 expression seen in prostate cancer is important for metastasis to occur via the decreased integrity of TJs. The effect of HAVcR-1 on paracellular permeability was also assessed due to TJs being the primary determinant of epithelial permeability, with Claudin expression patterns in particular being responsible for pore selectivity [
24]. However, HAVcR-1 appeared to have no effect on PC-3 permeability and, thus, further suggesting that HAVcR-1 expression has no bearing on the integrity of TJ within PC-3 cells or on the composition of TJs within PC-3 cells. The compositional stability of PC-3 TJs with manipulated HAVcR-1 expression was further validated with the gene expression of all TJ proteins investigated remaining constant. Preliminary investigations into TJ protein expression showed minute changes in the expression and localisation of occludin and ZO-1. Decreased occludin staining in PC-3 HAVcR-1EXP and increased staining in PC-3 HAVcR-1KD suggests that the overexpression of HAVcR-1 would decrease TJ integrity and that targeting HAVcR-1 could, therefore, be a potential therapeutic target for prostate cancer. However, contradictory to this, there was also an increase in occludin membranous staining with HAVcR-1 overexpression, suggesting an increase in TJs. Furthermore, in PC-3 cells that overexpressed HAVcR-1, there was a decreased nuclear staining of ZO-1. ZO-1 contains both NLS and NES; thus, can shuttle between TJs and the cell nucleus [
25]. Nuclear levels are generally associated with a decreased TJ integrity being found in proliferating low confluent cells [
26]. Therefore, low ZO-1 nuclear staining further suggests an increased junctional stability with HAVcR-1 overexpression. There was, however, a decrease in the barrier resistance with HAVcR-1 overexpression and knockdown during cell adhesion and spreading, indicating a decrease in the cell–cell junction integrity. TER results suggested that the junctional integrity remained constant regardless of HAVcR-1 expression; it is a possibility that HAVcR-1 levels affect the initiation of junction assembly, but do not affect the integrity of junctions once formed. The cell adhesion assay as well as the ECIS initial attachment experiment showed no change in cell adhesion with a manipulated HAVcR-1 expression. However, there was an increased constraint under cells with decreased HAVcR-1 expression, suggesting a decreased focal adhesion. To validate the changes to focal adhesion, further analyses are required, such as conducting a dynamic culture cell adhesion assay [
27]. There has been some research to indicate that HGF is important in HAVcR-1 activation and, therefore, it would be interesting to investigate the effect of a combination of HGF and HAVcR-1 on cell behaviours and TJ integrity [
14]
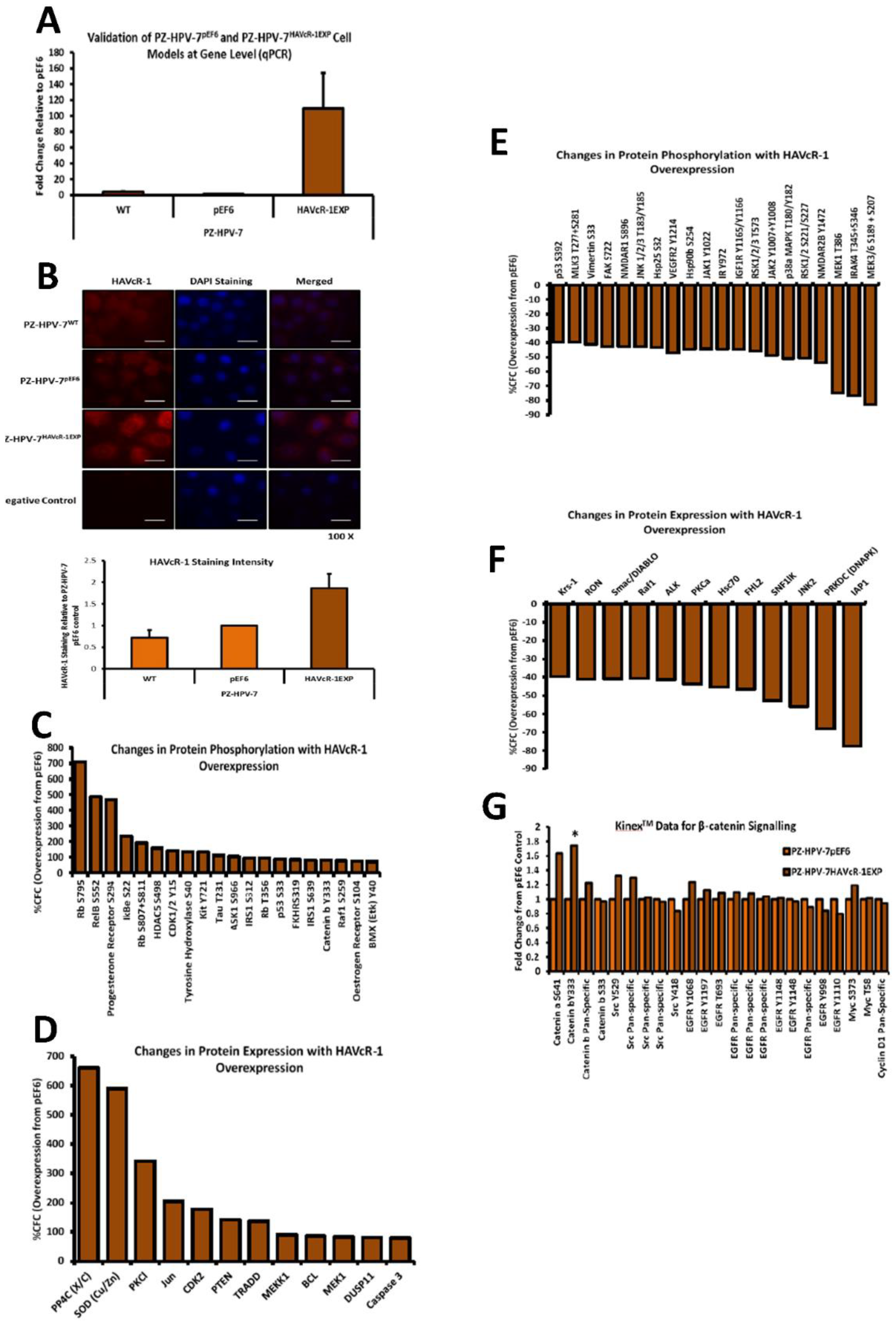
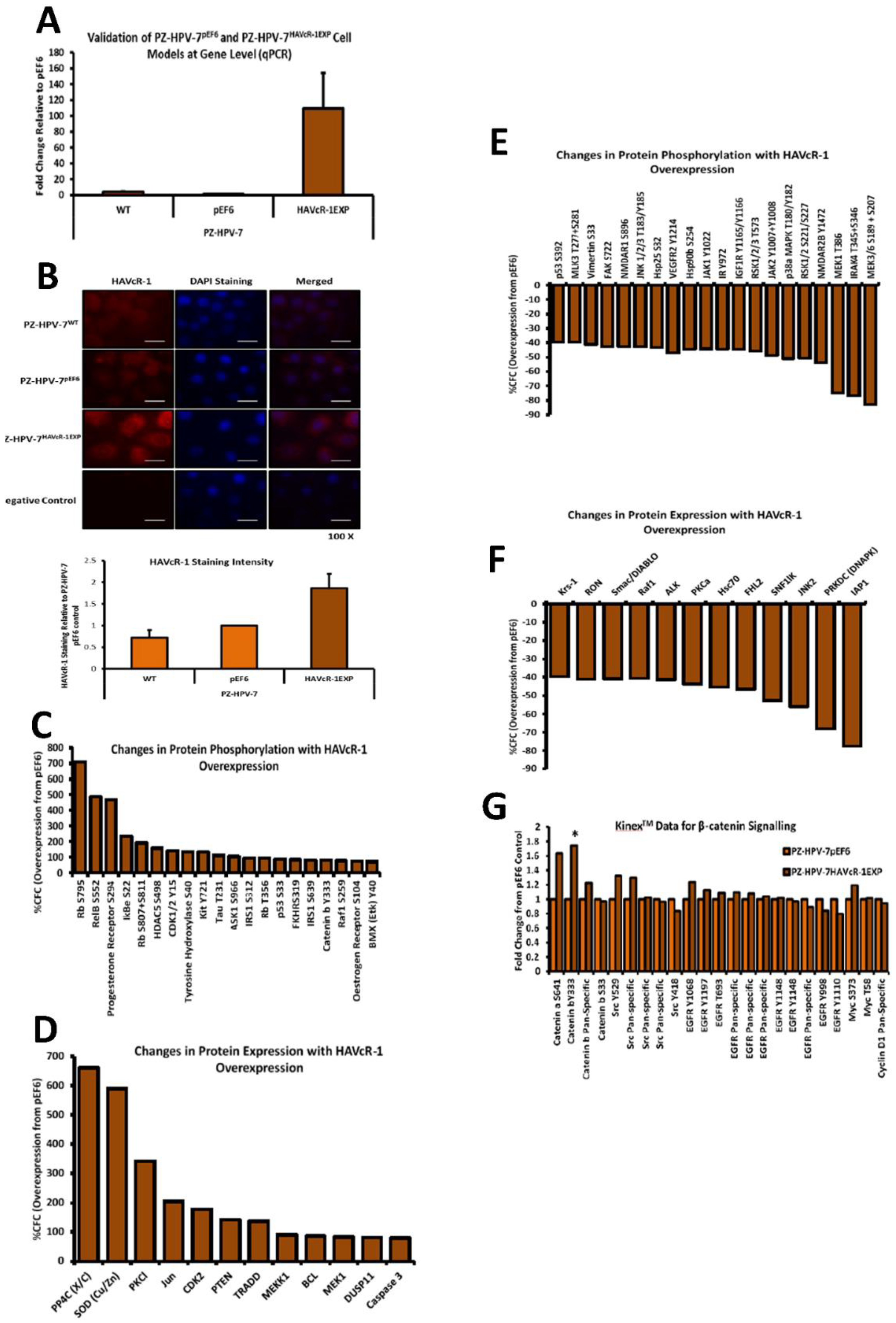
The Kinex™ KAM880 protein array analysis indicated 64 proteins as being statistically altered, either in expression or phosphorylation. Although there were numerous proteins that would be of interest for further study, β-catenin was chosen. β-catenin Y333 showed a significantly increased phosphorylation with HAVcR-1 overexpression. As well as β-catenin being an important structure component of AJs, it also has a role in the de-differentiation process [
28]. β-catenin has been shown to play a role in the tumorigenesis of numerous cancers, with dysregulation being associated with prostate cancer progression; however, studies have generally focused on the Wnt/wingless cascade and activation mutations [
29]. However, the phosphorylation of β-catenin at residue Y333 was due to EGFR signalling; thus, identifying a novel area of interest in the study of prostate cancer research [
30]. HAVcR-1 had no direct effect on the total gene or protein expression of β-catenin in PZ-HPV-7 cells, although HAVcR-1 overexpression altered the subcellular localisation of β-catenin, which is an important indicator of signalling [
29]. We found an increase in the β-catenin nuclear staining, and since nuclear staining is indicative of β-catenin activation, this, therefore, supports the Kinex™ KAM880 protein array data and the theory that HAVcR-1 is involved in β-catenin signalling [
31]. Interestingly, the nuclear accumulation of β-catenin has been associated with poorly differentiated and highly proliferative tumours, with an increased vascular invasion [
29,
30]. Since invasion is a hallmark of malignancy and a prerequisite for cancer metastasis, this proposes HAVcR-1 as a potential anti-metastatic target [
32]. The Kinex™ KAM880 protein array also revealed an increase in the α-catenin S641 phosphorylation, again, as a result of EGFR activation and, subsequently, resulted in the dissociation of β-catenin from the membrane and its nuclear translocation [
33]. The gene and protein analysis of α-catenin expression showed that HAVcR-1 had no effect on the total expression levels, although immunofluorescence showed a discontinuous staining of α-catenin at the cell membrane; thus, suggesting a breakdown of AJs, which was further shown by the discontinuous membrane staining of E-cadherin with HAVcR-1 overexpression. These results, therefore, support the Kinex™ KAM880 protein array and the theory that HAVcR-1 could lead to the phosphorylation of α-catenin, which, subsequently, results in the disassociation of β-catenin from AJs and nuclear accumulation.
The junctional integrity was also shown to decrease with HAVcR-1 overexpression, with a decreased barrier resistance during wound healing and an increased PCP. Thus, it is plausible that HAVcR-1 results in a decreased junctional stability via the activation and, thus, the dissociation of β-catenin from AJs. Once accumulated in the nucleus, β-catenin binds PKM2 and this complex can be recruited to the CCND1 promoter leading to targeted gene transcription, including cyclin D1 [
30,
34]. HAVcR-1 had no direct effect of PKM2 gene expression; however, this was unsurprising due to the localisation of PKM2 being important in β-catenin signalling. The cyclin-D1 gene expression remained constant with HAVcR-1 overexpression; however, the protein expression significantly increased. It would be expected that activated β-catenin signalling would increase cyclin D1 transcription; therefore, increasing the cyclin D1 gene expression and protein expression [
35]. Cyclin-D1 is a cell cycle control protein and has been linked to the development and progression of cancer. Cyclin D1 is a regulator of the progression of cells into the proliferation stage of the cell cycle, in LNCaP cells cyclin D1 overexpression enhancing S-phase entry, increasing colony formation and the tumour growth rate [
35,
36,
37]. EMT is a multi-step process involving the decreased integrity of junctional complexes [
38]. As previously discussed, immunofluorescence showed as decreased integrity of AJs with E-cadherin, α-catenin and β-catenin membrane staining.
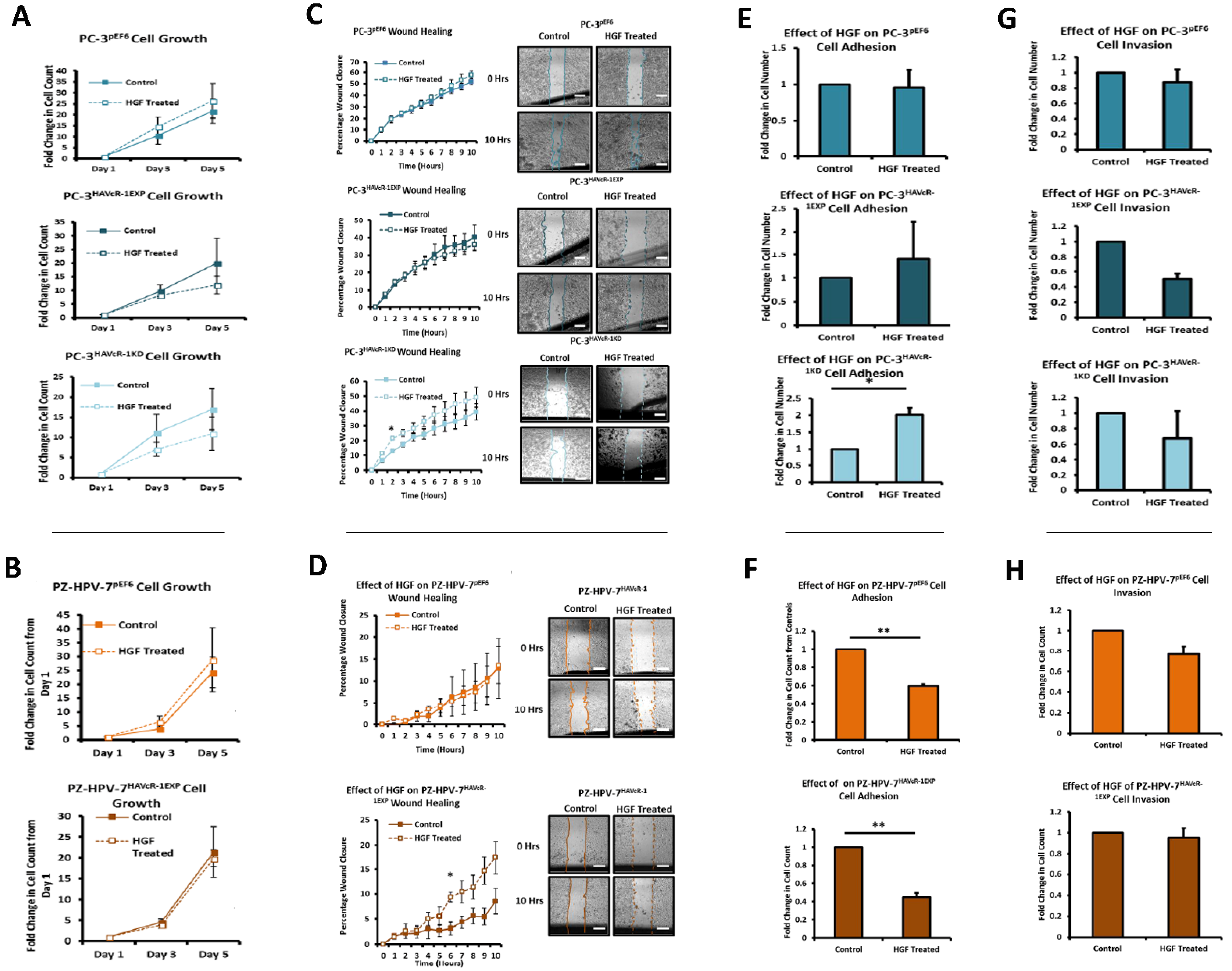
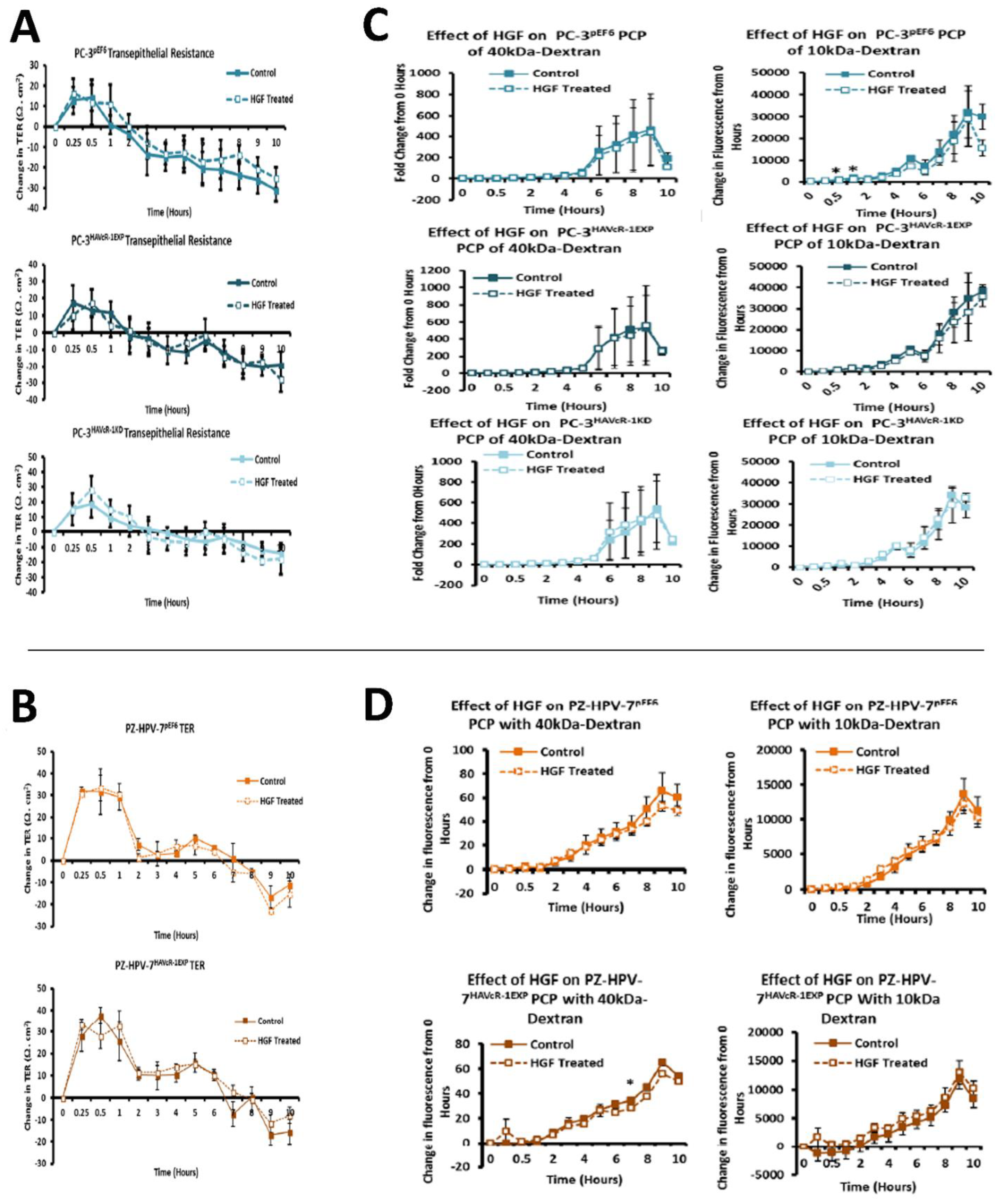
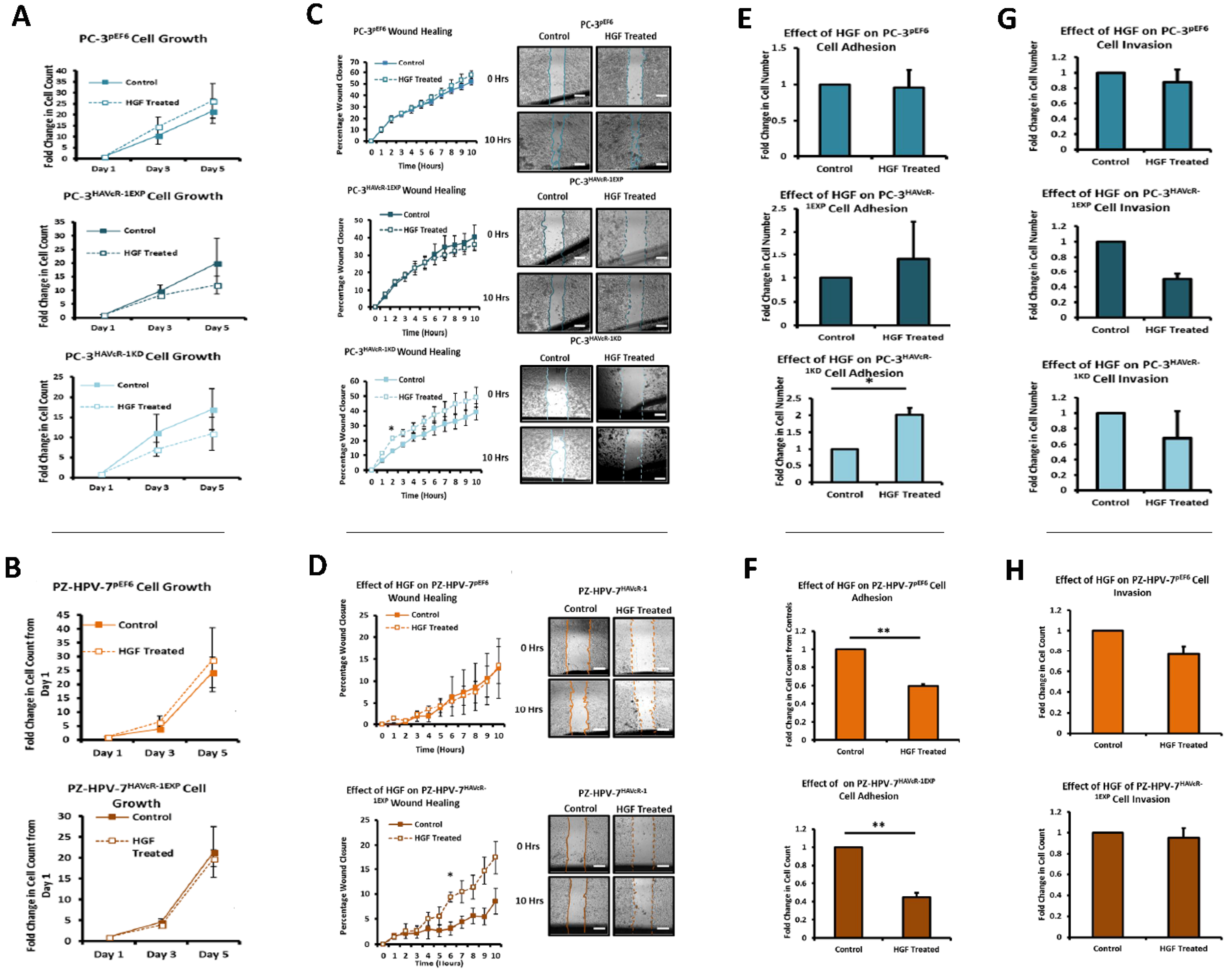
The HGF treatment has been shown to increase the cell growth of KGN and HO8910 ovarian cancer cell lines and the PC-3 cell line [
39,
40,
41]. HAVcR-1 overexpression and knockdown had no effect on this with no change in the cell growth with the HGF treatment. HGF has also been shown to influence cell migration with the HGF treatment increasing the cell migration of HO8910 ovarian cells line, the MKN1, MKN7 and MKN28 gastric cell lines and the PC-3 cell line [
39,
40,
41,
42]. However, there was no change in the cell migration of PC-3 pEF6 or PZ-HPV-7 pEF6 cells. Although HAVcR-1 overexpression had no effect on this with no change in PC-3 cell migration with the HGF treatment, the HAVcR-1 knockdown increased the PC-3 cell migration when treated with HGF. This, therefore, suggests that HAVcR-1 inhibited the ability of HGF to influence cell migration. However, HAVcR-1 overexpression in PZ-HPV-7 cells resulted in an increase in cell migration with the HGF treatment, which suggests that HAVcR-1 is required for HGF to influence cell migration. It is, therefore, possible that the control of HAVcR-1 levels is essential with only a small range in expression allowing HGF signalling. HGF had no effect on the PC-3 pEF6 cell adhesion and an inhibitory effect on PZ-HPV-7 pEF6 cell adhesion; however, this contrasted a previous report, whereby HGF had been shown to increase PC-3 cell adhesion [
39]. HAVcR-1 knockdown, however, resulted in an increased PC-3 adhesion with the HGF treatment, which further suggests that HAVcR-1 has an inhibitory effect on HGF signalling in PC-3 cells due to HAVcR-1 overexpression also resulting in a decreased cell adhesion of PZ-HPV-7 cells, causing the HGF signalling in PC-3 and PZ-HPV-7 to be different. The effect of the HGF treatment on cell invasion has previously had conflicting results, with some showing the treatment resulting in an increased cell line invasion (HO8910 and PC-3) and others showing that the cell line (PC-3 and PZ-HPV-7) invasion was unaffected by the treatment [
39,
41,
43]. Similarly, to the latter, the HGF treatment had no effect on PC-3 pEF6or PZ-HPV-7 pEF6 cell invasion. HAVcR-1 overexpression and knockdown in PC-3 cells as well as HAVcR-1 overexpression in PZ-HPV-7 had no effect on this, with no difference in invasion with the HGF treatment. These results, therefore, suggest that change to cell invasion in prostate cancer cells is independent of HGF signalling. Previous studies had demonstrated a decreased TER and decreased TJ protein expression and membrane localisation and, therefore, a decreased junctional integrity with the HGF treatment [
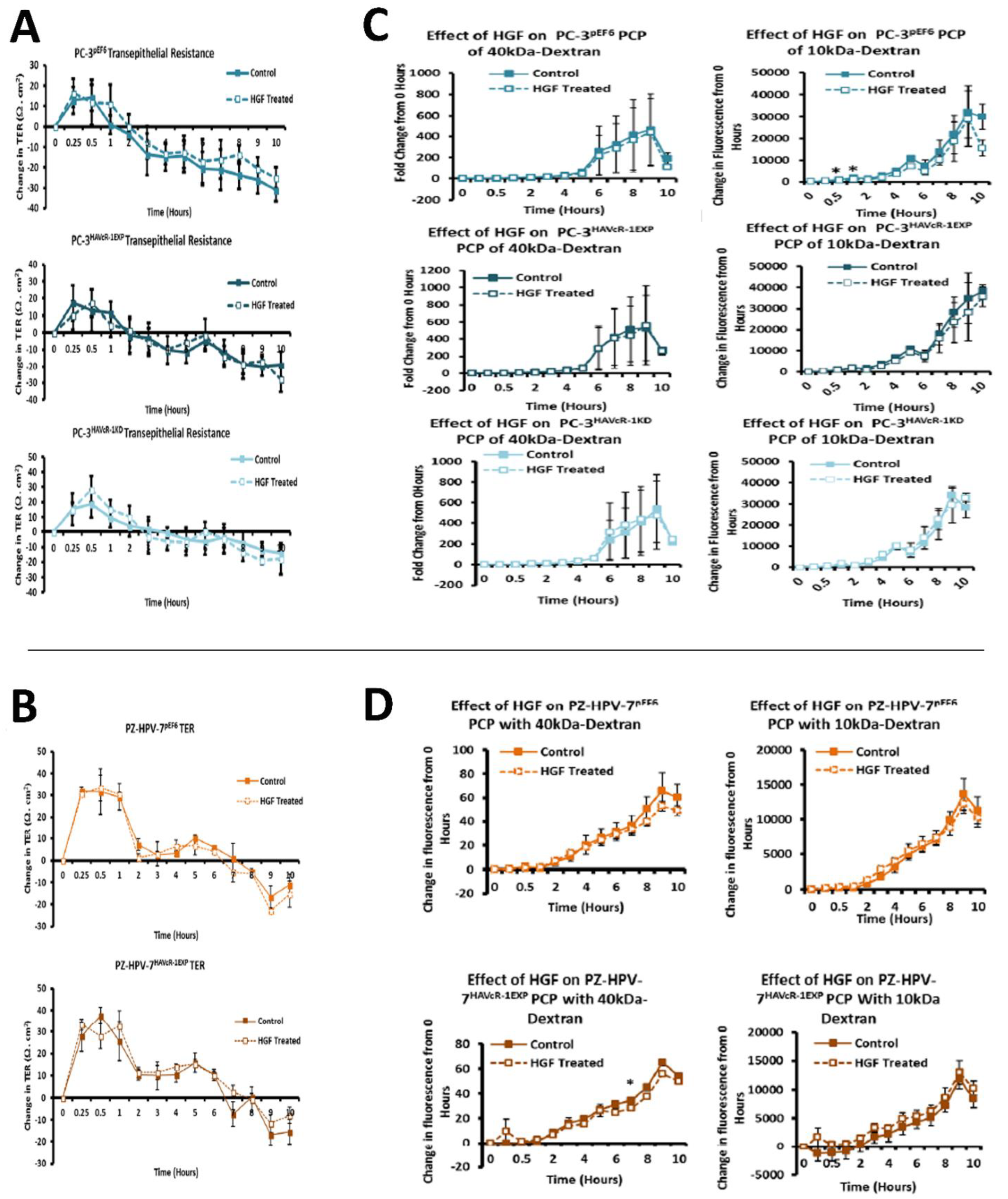
44,
45]. There was no effect with the HGF treatment when HAVcR-1 was overexpressed or knocked-down in PC-3 cells or when overexpressed in PZ-HPV-7 cells. This was contradictory to the literature, whereby the knockdown of HAVcR-1 resulted in HECV cells being resistant to an HGF-induced decreased TER [
14]. As HECV cells are endothelial and PC-3 and PZ-HPV-7 are epithelial derived, the regulation mechanisms of junctional integrity may differ between endothelial and epithelial cells. This is further supported by the potential initial decrease in the PCP of 10 kDa FITC–dextran conjugate of PC-3 pEF6 with the HGF treatment, which would suggest an increase.



 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}