
Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells




Abstract
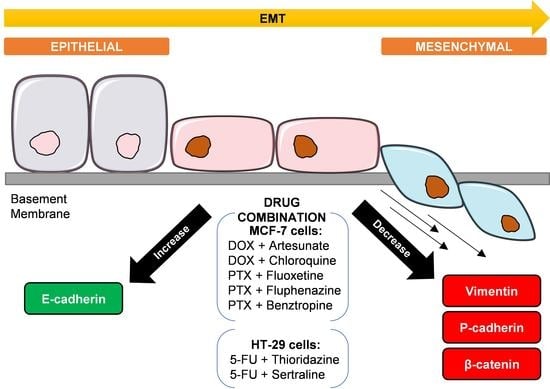
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Evaluation of the Cytotoxic Effect of CNS Drugs Combined with DOX in MCF-7 Cells
2.2.1. Cell Line and Cell Culture
2.2.2. Cell Treatment
2.2.3. Cell-Based Assays
2.2.4. Cell Morphology Visualization
2.2.5. Data Analysis
2.2.6. Evaluation of Synergism Using CompuSyn and SynergyFinder Software
2.2.7. Statistical Analysis
2.3. Evaluation of EMT Biomarkers in MCF-7 and HT-29 Cells Treated with Repurposed Drugs Combined with Antineoplastic Agents
2.3.1. Cell Line and Cell Culture
2.3.2. Preparation of Cells for Immunohistochemistry (IHC)
2.3.3. Immunohistochemistry Protocol
3. Results
3.1. Evaluation of the Cytotoxic Effect of CNS Drugs Combined with DOX in MCF-7 Cells
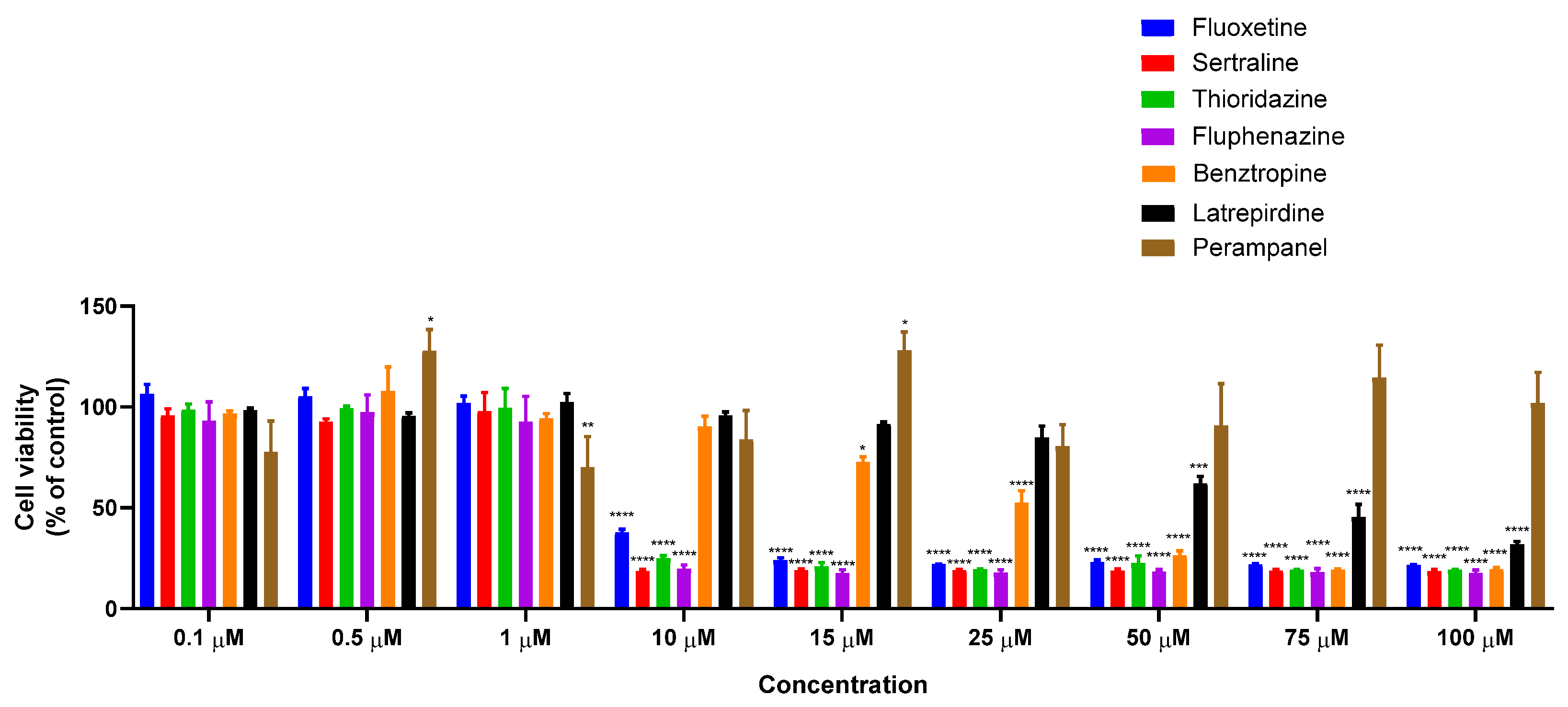
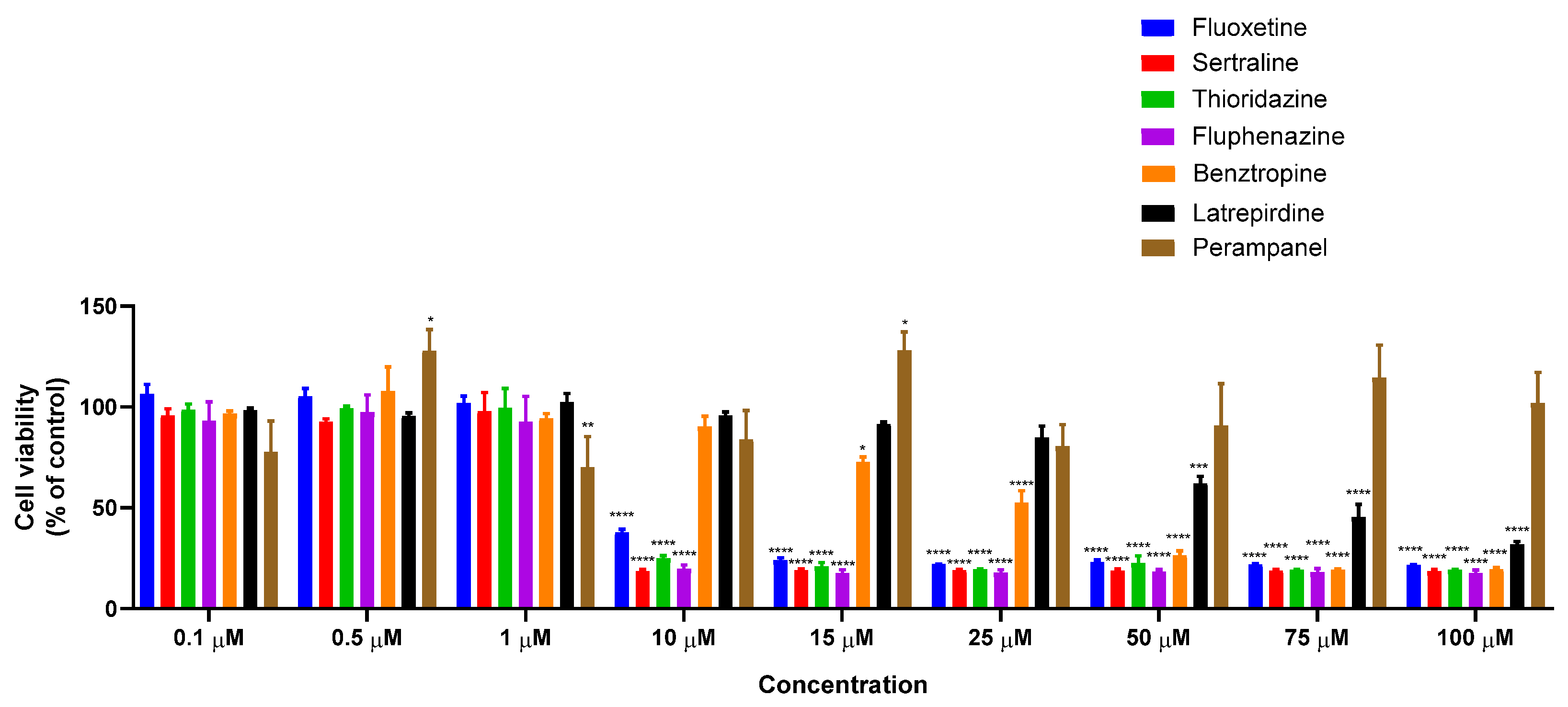
3.1.1. Single Treatment of MCF-7 Breast Cancer Cells with CNS Drugs
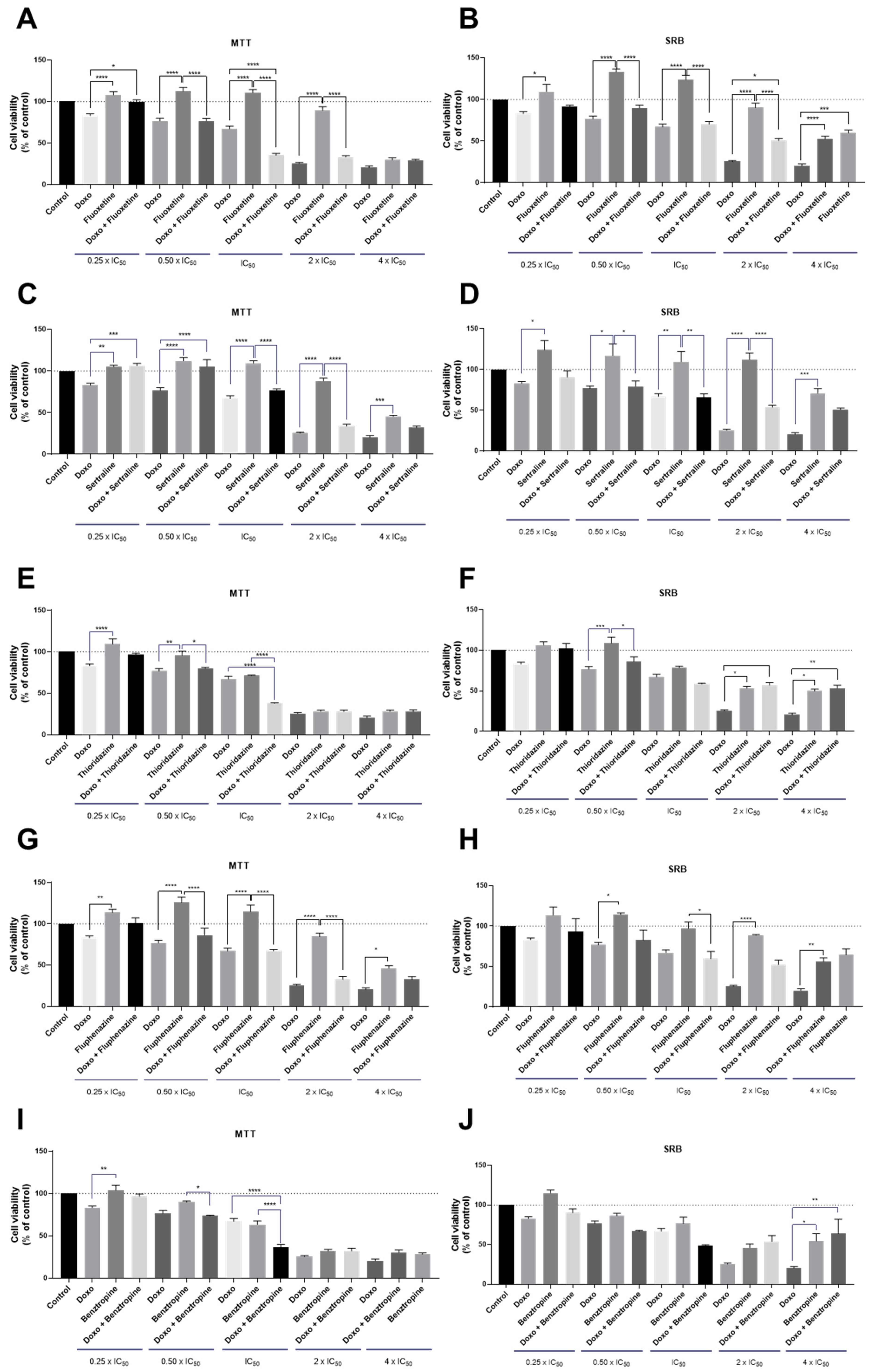
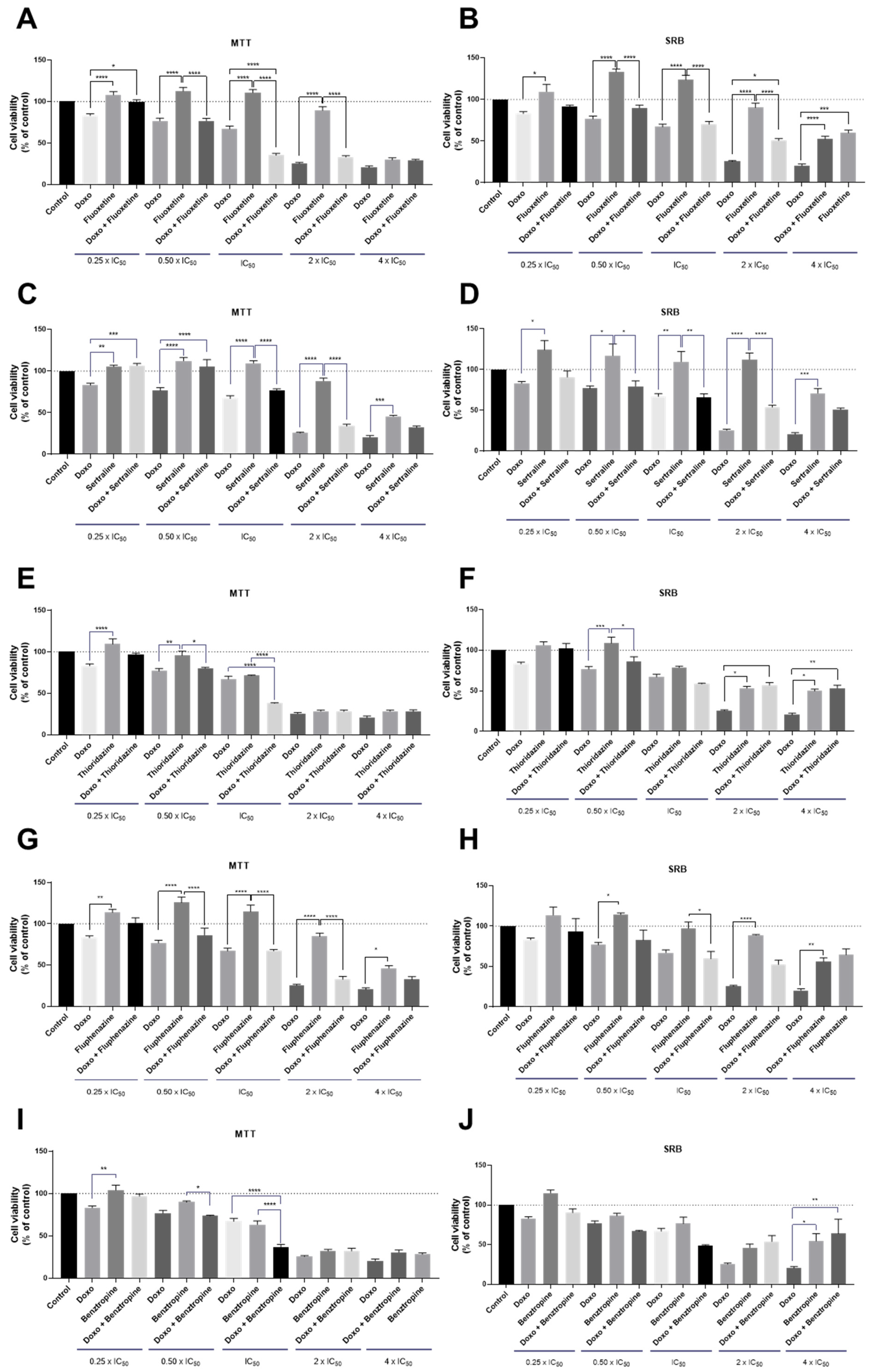
3.1.2. Co-Treatment of MCF-7 Breast Cancer Cells with DOX and CNS Drugs
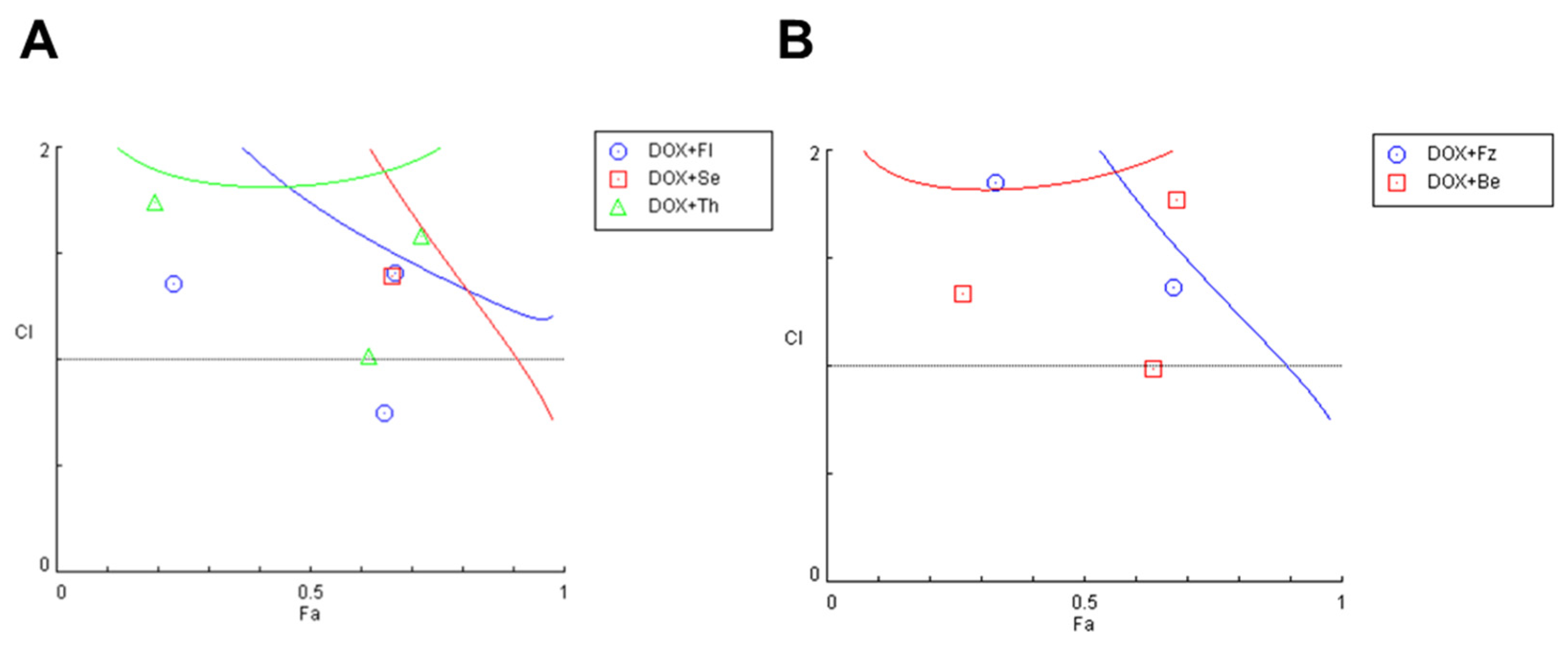
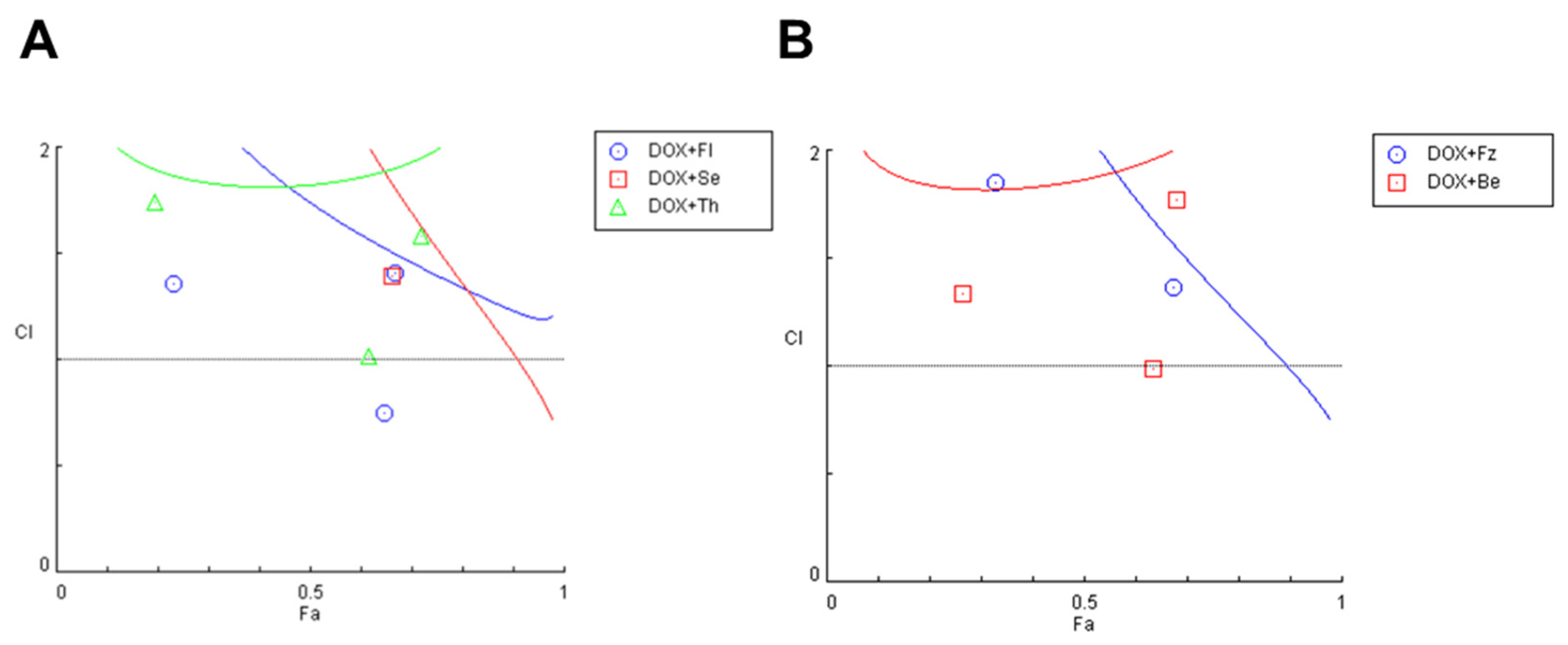
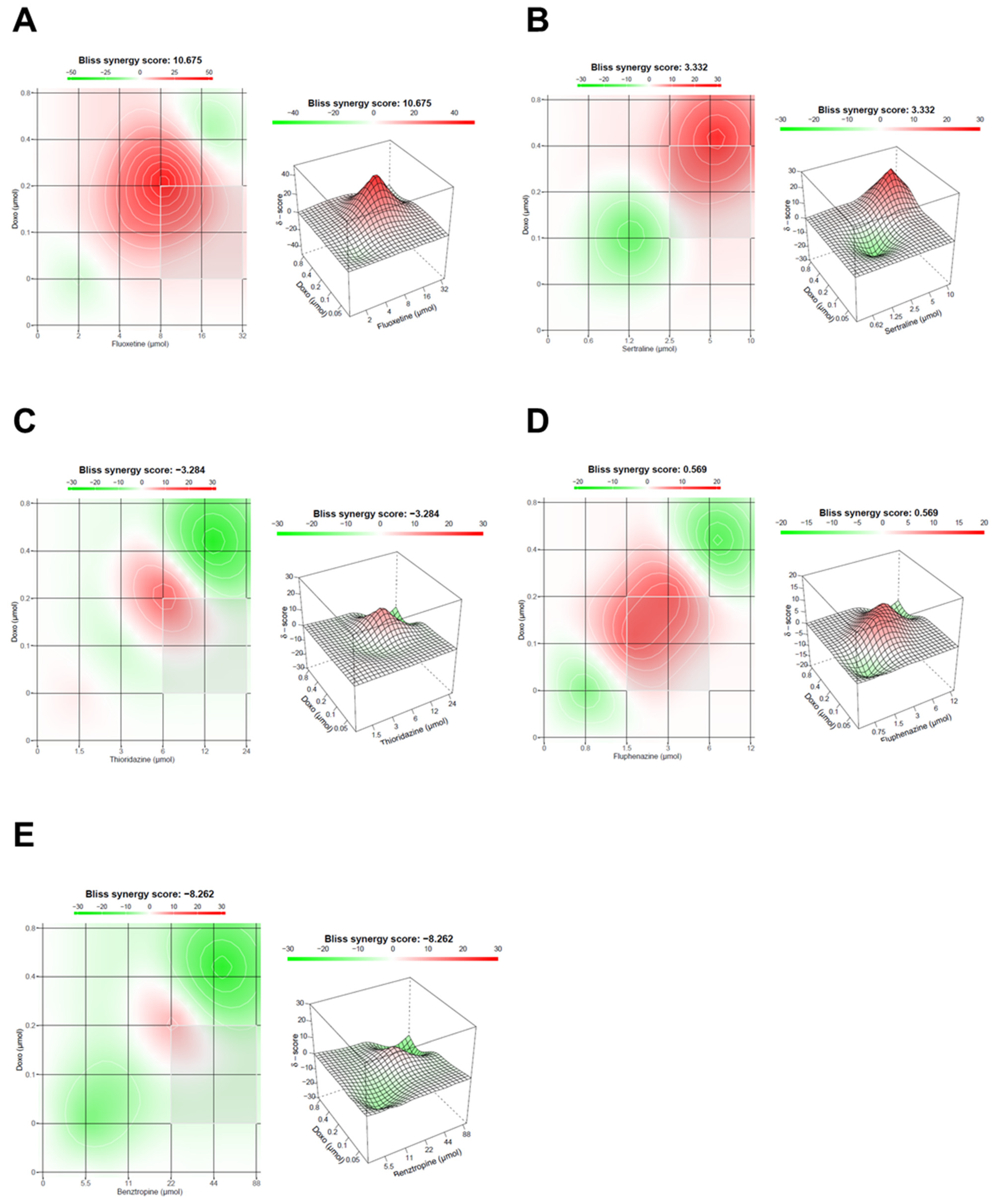
3.1.3. Evaluation of Synergism in MCF-7 Cells Co-Treated with DOX and Different CNS Drugs
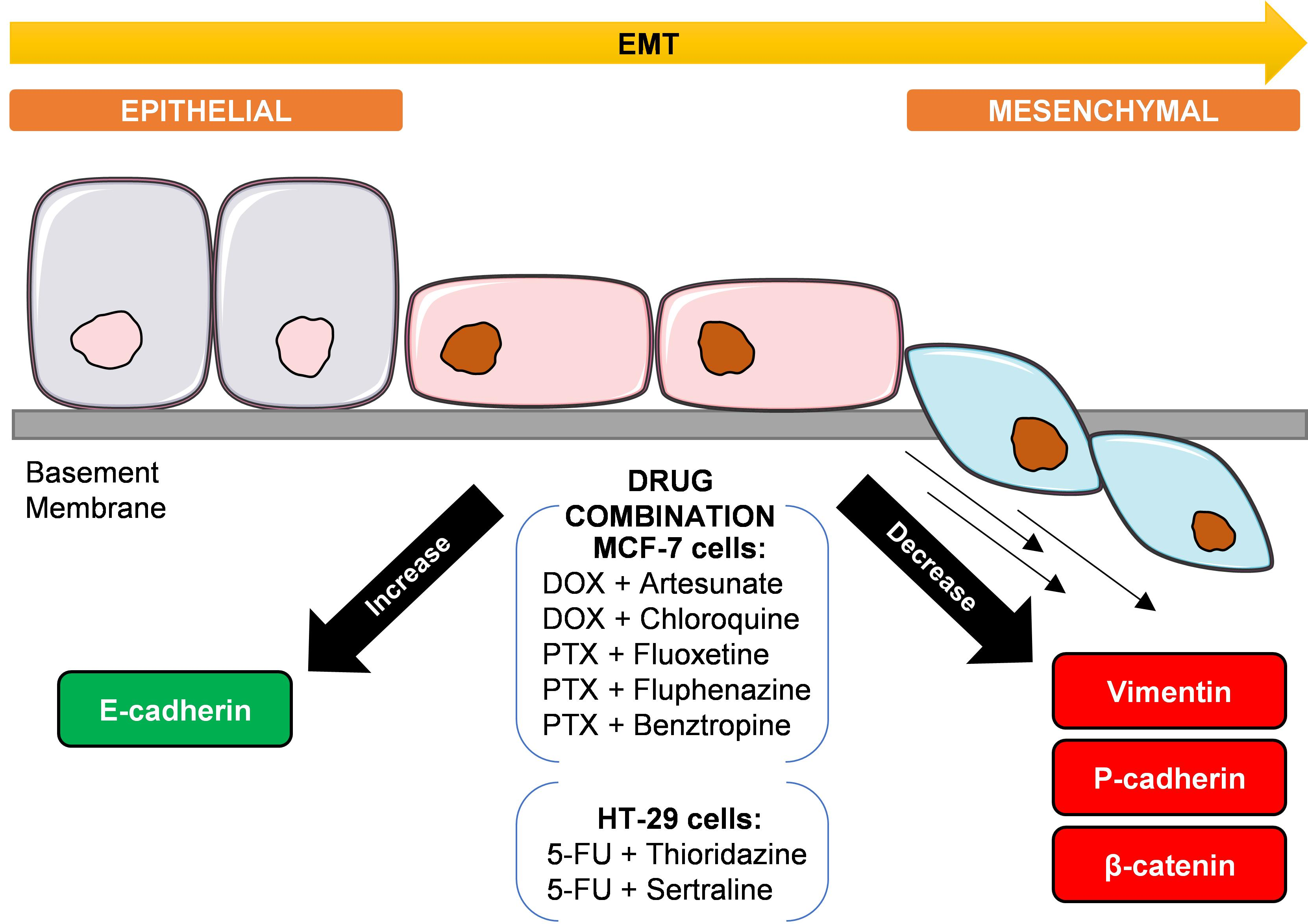
3.2. Evaluation of EMT Biomarkers in MCF-7 and HT-29 Cells Treated with Repurposed Drugs Combined with Antineoplastic Agents
3.2.1. EMT Biomarkers in MCF-7 Cells Treated with Antimalarial Drugs Combined with Antineoplastic Agents
3.2.2. EMT Biomarkers in MCF-7 Cells Treated with CNS Drugs Combined with Antineoplastic Agents
3.2.3. EMT Biomarkers in HT-29 Cells Treated with CNS Drugs Combined with Antineoplastic Agents
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramos, F.S.; Wons, L.; Cavalli, I.J.; Ribeiro, E.M. Epithelial-mesenchymal transition in cancer: An overview. Integr. Cancer Sci. Ther. 2017, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Kerrigan, B.C.P.; Yang, D.; Hu, L.; Shmulevich, I.; Sood, A.K.; Xue, F.; Zhang, W. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J. Hematol. Oncol. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinestel, K.; Eder, S.; Schrader, A.J.; Steinestel, J. Clinical significance of epithelial-mesenchymal transition. Clin. Transl. Med. 2014, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, B.P. Epithelial-Mesenchymal Transition—A Hallmark of Breast Cancer Metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micalizzi, D.S.; Ford, H.L. Epithelial–mesenchymal transition in development and cancer. Futur. Oncol. 2009, 5, 1129–1143. [Google Scholar] [CrossRef]
- Thompson, E.W.; Newgreen, D.F. Carcinoma invasion and metastasis: A role for epithelial-mesenchymal transition? Cancer Res. 2005, 65, 5991–5995. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.F.; Paredes, J. P-cadherin and the journey to cancer metastasis. Mol. Cancer 2015, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin. J. Cancer 2011, 30, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.A.; Patel, P.S.; Vora, H.H. Role of PRL-3, Snail, Cytokeratin and Vimentin expression in epithelial mesenchymal transition in breast carcinoma. Breast Dis. 2015, 35, 113–127. [Google Scholar] [CrossRef]
- Lehtinen, L.; Ketola, K.; Mäkelä, R.; Mpindi, J.-P.; Viitala, M.; Kallioniemi, O.; Iljin, K. High-throughput RNAi screening for novel modulators of vimentin expression identifies MTHFD2 as a regulator of breast cancer cell migration and invasion. Oncotarget 2012, 4, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Antoszczak, M.; Markowska, A.; Huczyński, A. Old wine in new bottles: Drug repurposing in oncology. Eur. J. Pharmacol. 2020, 866, 172784. [Google Scholar] [CrossRef]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Capistrano, I.R.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The repurposing drugs in oncology database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology—patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691. [Google Scholar] [CrossRef] [Green Version]
- Talib, W.H.; Odeh, L.H.; A Basheti, I. Synergistic effect of thymoquinone and melatonin against breast cancer implanted in mice. J. Cancer Res. Ther. 2018, 14, S324–S330. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Ponce-Cusi, R.; Carrión, F. Curcumin and paclitaxel induce cell death in breast cancer cell lines. Oncol. Rep. 2018, 40, 2381–2388. [Google Scholar] [CrossRef] [PubMed]
- Mirzapur, P.; Khazaei, M.R.; Moradi, M.T.; Khazaei, M. Apoptosis induction in human breast cancer cell lines by synergic effect of raloxifene and resveratrol through increasing proapoptotic genes. Life Sci. 2018, 205, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Yuan, B.; Wang, X.; Sato, A.; Sakuma, K.; Kaneko, K.; Komuro, H.; Okazaki, A.; Hayashi, H.; Toyoda, H.; et al. Synergistic cytotoxic effects of arsenite and tetrandrine in human breast cancer cell line MCF-7. Int. J. Oncol. 2017, 51, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.-X.; Du, Y.-Y.; Zhang, Y.; Pan, Y.-Y. Synergistic effects of exemestane and aspirin on MCF-7 human breast cancer cells. Asian Pac. J. Cancer Prev. 2012, 13, 5903–5908. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Cardoso, A.; Vale, N. Synergistic Growth Inhibition of HT-29 Colon and MCF-7 Breast Cancer Cells with Simultaneous and Sequential Combinations of Antineoplastics and CNS Drugs. Int. J. Mol. Sci. 2021, 22, 7408. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells. Biomol. 2020, 10, 1623. [Google Scholar] [CrossRef]
- Munson, J.M.; Fried, L.; Rowson, S.A.; Bonner, M.Y.; Karumbaiah, L.; Diaz, B.; Courtneidge, S.A.; Knaus, U.G.; Brat, D.J.; Arbiser, J.L.; et al. Anti-Invasive Adjuvant Therapy with Imipramine Blue Enhances Chemotherapeutic Efficacy Against Glioma. Sci. Transl. Med. 2012, 4, 127ra36. [Google Scholar] [CrossRef]
- Rajamanickam, S.; Panneerdoss, S.; Gorthi, A.; Timilsina, S.; Onyeagucha, B.; Kovalskyy, D.; Ivanov, D.; Hanes, M.A.; Vadlamudi, R.K.; Chen, Y.; et al. Inhibition of FoxM1-Mediated DNA Repair by Imipramine Blue Suppresses Breast Cancer Growth and Metastasis. Clin. Cancer Res. 2016, 22, 3524–3536. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-H.; Su, Y.-H.; Hsu, W.-H.; Wang, C.-C.; Arbiser, J.L.; Yang, M.-H. Imipramine blue halts head and neck cancer invasion through promoting F-box and leucine-rich repeat protein 14-mediated Twist1 degradation. Oncogene 2016, 35, 2287–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metts, J.; Bradley, H.L.; Wang, Z.; Shah, N.P.; Kapur, R.; Arbiser, J.L.; Bunting, K.D. Imipramine blue sensitively and selectively targets FLT3-ITD positive acute myeloid leukemia cells. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, K.M.E.; Berhan, S.; Liu, S.; Silvestri, G.; Holyoake, T.L.; Frank, D.A.; Aggarwal, B.; Bonner, M.Y.; Perrotti, D.; Jørgensen, H.G.; et al. Cooperation of imipramine blue and tyrosine kinase blockade demonstrates activity against chronic myeloid leukemia. Oncotarget 2016, 7, 51651–51664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorab, M.M.; Alsaid, M.S.; Samir, N.; Abdel-Latif, G.A.; Soliman, A.M.; Ragab, F.A.; El Ella, D.A.A. Aromatase inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and molecular modeling studies of novel phenothiazine derivatives carrying sulfonamide moiety as hybrid molecules. Eur. J. Med. Chem. 2017, 134, 304–315. [Google Scholar] [CrossRef]
- Zong, D.; Zielinska-Chomej, K.; Juntti, T.; Mork, B.; Lewensohn, R.; Haag, P.; Viktorsson, K. Harnessing the lysosome-dependent antitumor activity of phenothiazines in human small cell lung cancer. Cell Death Dis. 2014, 5, e1111. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Bai, L.-Y.; Tsai, M.-H.; Chu, P.-C.; Chiu, C.-F.; Chen, M.Y.; Chiu, S.-J.; Chiang, J.-H.; Weng, J.-R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents–A drug repurposing strategy. Sci. Rep. 2016, 6, 27540. [Google Scholar] [CrossRef]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.-T.; Wu, A.; Chang, P.M.-H.; Chen, K.-Y.; Yang, C.-N.; Yang, S.-C.; Ho, C.-C.; Chen, C.-C.; Kuo, Y.-L.; Lee, P.-Y.; et al. Trifluoperazine, an Antipsychotic Agent, Inhibits Cancer Stem Cell Growth and Overcomes Drug Resistance of Lung Cancer. Am. J. Respir. Crit. Care Med. 2012, 186, 1180–1188. [Google Scholar] [CrossRef]
- Bertolesi, G.E.; Shi, C.; Elbaum, L.; Jollimore, C.; Rozenberg, G.; Barnes, S.; Kelly, M.E.M. The Ca2+Channel Antagonists Mibefradil and Pimozide Inhibit Cell Growth via Different Cytotoxic Mechanisms. Mol. Pharmacol. 2002, 62, 210–219. [Google Scholar] [CrossRef]
- Chou, Y.-W.; Chaturvedi, N.K.; Ouyang, S.; Lin, F.-F.; Kaushik, D.; Wang, J.; Kim, I.; Lin, M.-F. Histone deacetylase inhibitor valproic acid suppresses the growth and increases the androgen responsiveness of prostate cancer cells. Cancer Lett. 2011, 311, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Terranova-Barberio, M.; Roca, M.S.; Zotti, A.I.; Leone, A.; Bruzzese, F.; Vitagliano, C.; Scognamiglio, G.; Russo, D.; D’Angelo, G.; Franco, R.; et al. Valproic acid potentiates the anticancer activity of capecitabine in vitro and in vivo in breast cancer models via induction of thymidine phosphorylase expression. Oncotarget 2015, 7, 7715–7731. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liang, B.; Jia, H.; Jiao, Y.; Pang, Z.; Huang, Y. Evaluation of cell death pathways initiated by antitumor drugs melatonin and valproic acid in bladder cancer cells. FEBS Open Bio 2017, 7, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roell, K.R.; Reif, D.; Motsinger-Reif, A.A. An Introduction to Terminology and Methodology of Chemical Synergy—Perspectives from Across Disciplines. Front. Pharmacol. 2017, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Stonier, P.D. Repurposing old drugs in oncology: Opportunities with clinical and regulatory challenges ahead. J. Clin. Pharm. Ther. 2019, 44, 6–22. [Google Scholar] [CrossRef] [Green Version]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 2020, 15, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Chen, Z.; Chi, J.; Sun, Y.; Sun, Y. Recent progress in synergistic chemotherapy and phototherapy by targeted drug delivery systems for cancer treatment. Artif. Cells Nanomed. Biotechnol. 2018, 46, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nat. 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Böttcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Shen, Y.; Hong, J.; Xia, Q.; Zhou, F.; Liu, X. The contribution of TGF-β in Epithelial–Mesenchymal Transition (EMT): Down-regulation of E-cadherin via snail. Neoplasma 2015, 62, 1–15. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Paredes, J. P-Cadherin Linking Breast Cancer Stem Cells and Invasion: A Promising Marker to Identify an “Intermediate/Metastable” EMT State. Front. Oncol. 2015, 4, 371. [Google Scholar] [CrossRef]
- Nan, J.N.; Kim, O.R.; Lee, M.A. β-Catenin expression is associated with cell invasiveness in pancreatic cancer. Korean J. Intern. Med. 2019, 34, 618–625. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | HT-29 ‡ | MCF-7 | |
|---|---|---|---|
| IC50 (µM) | IC50 (µM) | ||
| Doxorubicin | N.D. | 0.17 | |
| Paclitaxel | 0.44 (nM) | ||
| 5-FU | 3 | N.D. | |
| Central Nervous System | Rivastigmine | >100 | N.D. |
| m-chlorophenilbiguanide | >100 | ||
| Safinamide | >100 | ||
| Fluoxetine | 6.12 | 7.78 * | |
| Benztropine | 18.23 | 21.71 * | |
| Thioridazine | 4.26 | 5.72 * | |
| Carbidopa | >100 | N.D. | |
| Bromocriptine | >100 | ||
| Nepicastat | 61.24 | ||
| Scopolamine | >100 | ||
| Carbamazepine | >100 | ||
| Sertraline | 2.45 | 2.22 * | |
| Selegiline | >100 | ND | |
| Entacapone | 40.89 | ND | |
| Tolcapone | 35.47 | ND | |
| Latrepirdine | 7.75 | 75.37 | |
| Fluphenazine | 1.86 | 2.68 * | |
| Perampanel | >100 | >100 | |
| Antimalarials ‡ | Artesunate | 17.88 | 11.60 |
| Chloroquine | 32.13 | N.D. | |
| Mefloquine | 11.49 | 1.24 | |
| 6-methoxy-8-nitroquinoline | N.D. | >100 | |
| Atovaquone | >100 | ||
| Cycloguanil | 20.30 | ||
| Dapsone | 53.99 | ||
| Ethosuximide | >100 | ||
| Lumefantrine | >100 | ||
| Natamycin | >100 | ||
| Piperazine | 3.24 | ||
| Primaquine | 29.90 | ||
| Primidone | >100 | ||
| Pyronaridine | 1.39 | ||
| Quinidine | >100 | ||
| Rufinamide | >100 | ||
| Sitamaquine | >100 | ||
| Sulfamethoxazole | >100 | ||
| Tafenoquine | 2.60 | ||
| Tobramycin | >100 | ||
| Tunicamycin | N.D. |
| Drug A | Drug B | HT-29 * | MCF-7 |
|---|---|---|---|
| Number of Synergic Interactions | Number of Synergic Interactions | ||
| 5-FU | Mefloquine | 1 | N.D. |
| Artesunate | 0 | ||
| Latrepirdine | 1 | ||
| Fluphenazine | 0 | ||
| Fluoxetine | 1 | ||
| Benztropine | 1 | ||
| Thioridazine | 3 | ||
| Sertraline | 5 | ||
| DOX | Artesunate | N.D. | 4 |
| Chloroquine | 3 | ||
| Mefloquine | 1 | ||
| Pyronaridine | 2 | ||
| Tafenoquine | 0 | ||
| Fluoxetine | 1 | ||
| Sertraline | 0 | ||
| Thioridazine | 0 | ||
| Benztropine | 0 | ||
| Fluphenazine | 0 | ||
| PTX * | Artesunate | N.D. | 2 |
| Chloroquine | 2 | ||
| Mefloquine | 1 | ||
| Pyronaridine | 3 | ||
| Tafenoquine | 0 | ||
| Fluoxetine | 3 | ||
| Sertraline | 2 | ||
| Thioridazine | 3 | ||
| Benztropine | 3 | ||
| Fluphenazine | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, D.; Rêma, A.; Amorim, I.; Vale, N. Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells. Biomolecules 2022, 12, 190. https://doi.org/10.3390/biom12020190
Duarte D, Rêma A, Amorim I, Vale N. Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells. Biomolecules. 2022; 12(2):190. https://doi.org/10.3390/biom12020190
Chicago/Turabian StyleDuarte, Diana, Alexandra Rêma, Irina Amorim, and Nuno Vale. 2022. "Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells" Biomolecules 12, no. 2: 190. https://doi.org/10.3390/biom12020190
APA StyleDuarte, D., Rêma, A., Amorim, I., & Vale, N. (2022). Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells. Biomolecules, 12(2), 190. https://doi.org/10.3390/biom12020190