
Identification and Validation of Cuproptosis-Related LncRNA Signatures in the Prognosis and Immunotherapy of Clear Cell Renal Cell Carcinoma Using Machine Learning

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
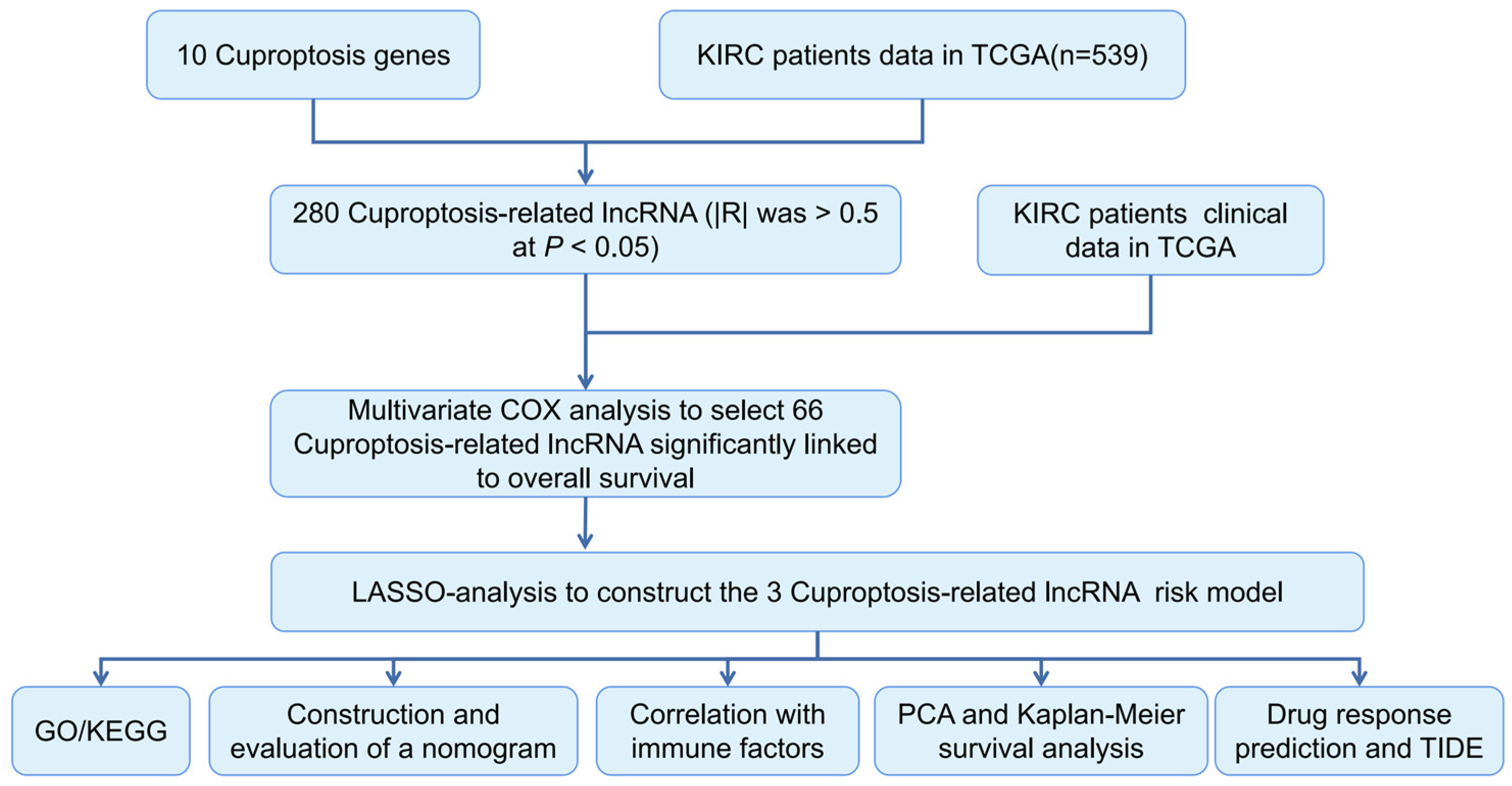
2.1. ccRCC Data Collection
2.2. Identification of CRLRs
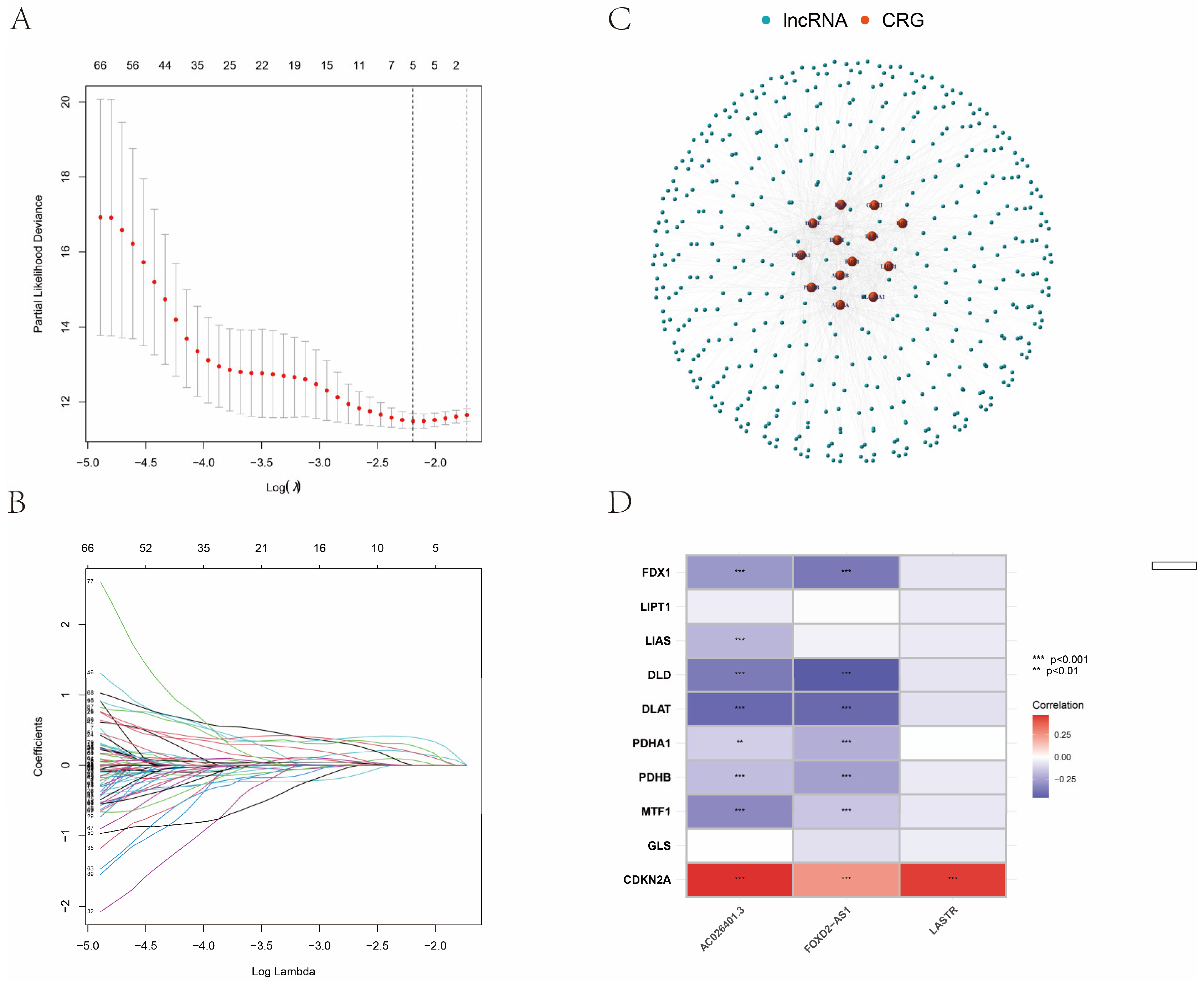
2.3. Construction of the CRLR Prognostic Model by a Machine Learning Algorithm
2.4. Kaplan–Meier (K–M) Survival Analysis and Principal Component Analysis (PCA)
2.5. Cell Culture
2.6. qPCR and RNA Isolation
2.7. Statistical Analysis
3. Results
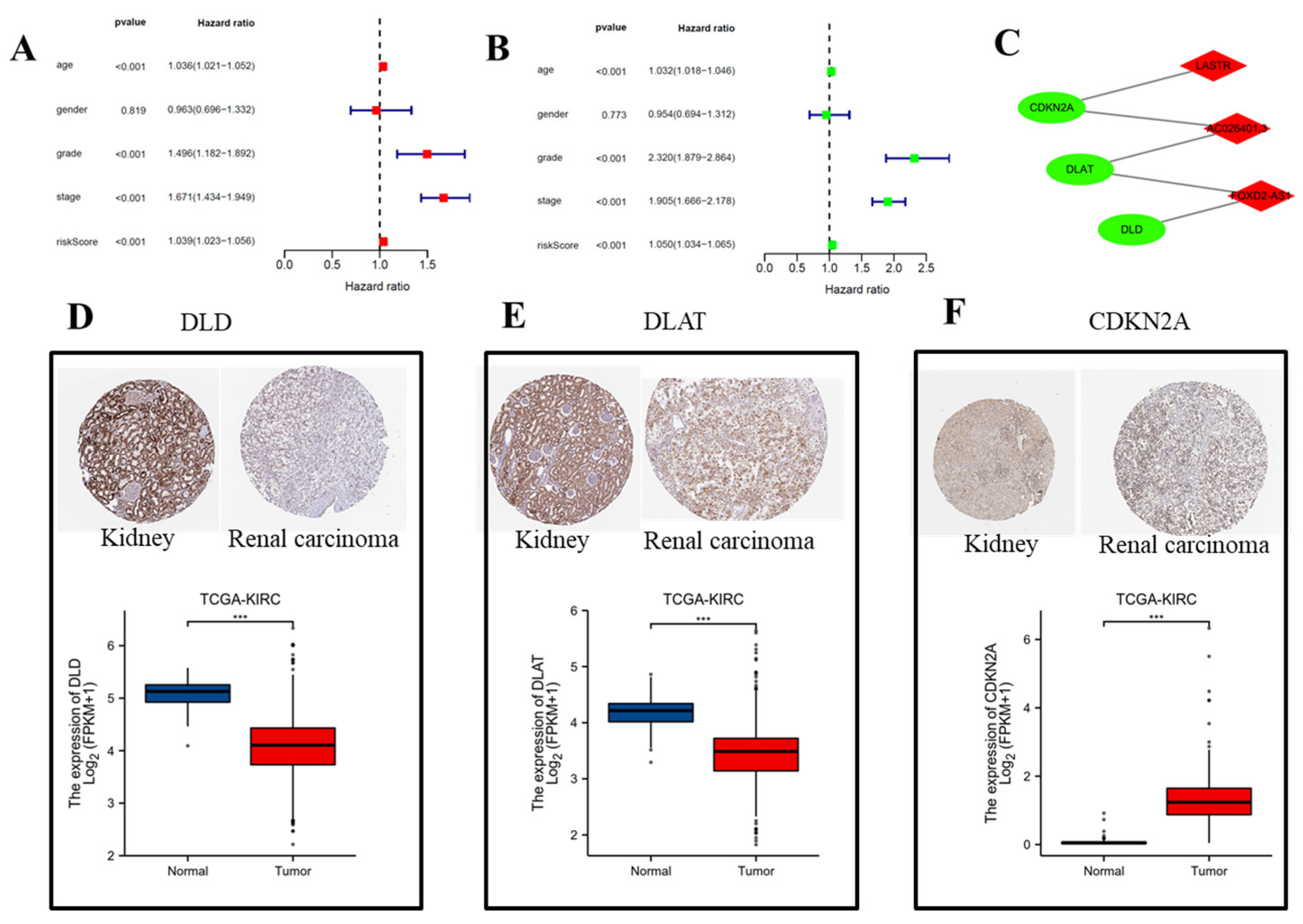
3.1. Identification of the Prognostic CRLR Signature in ccRCC
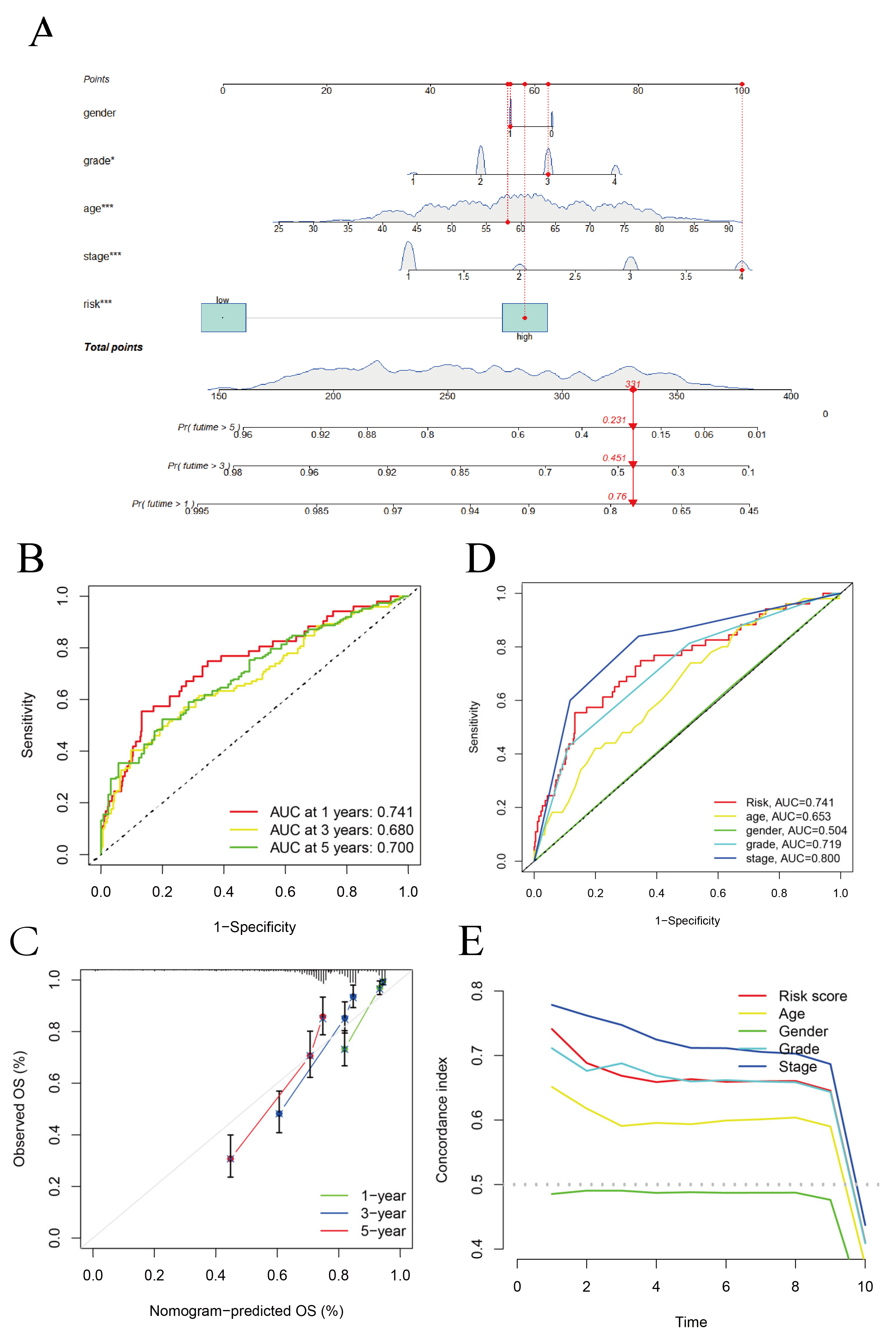
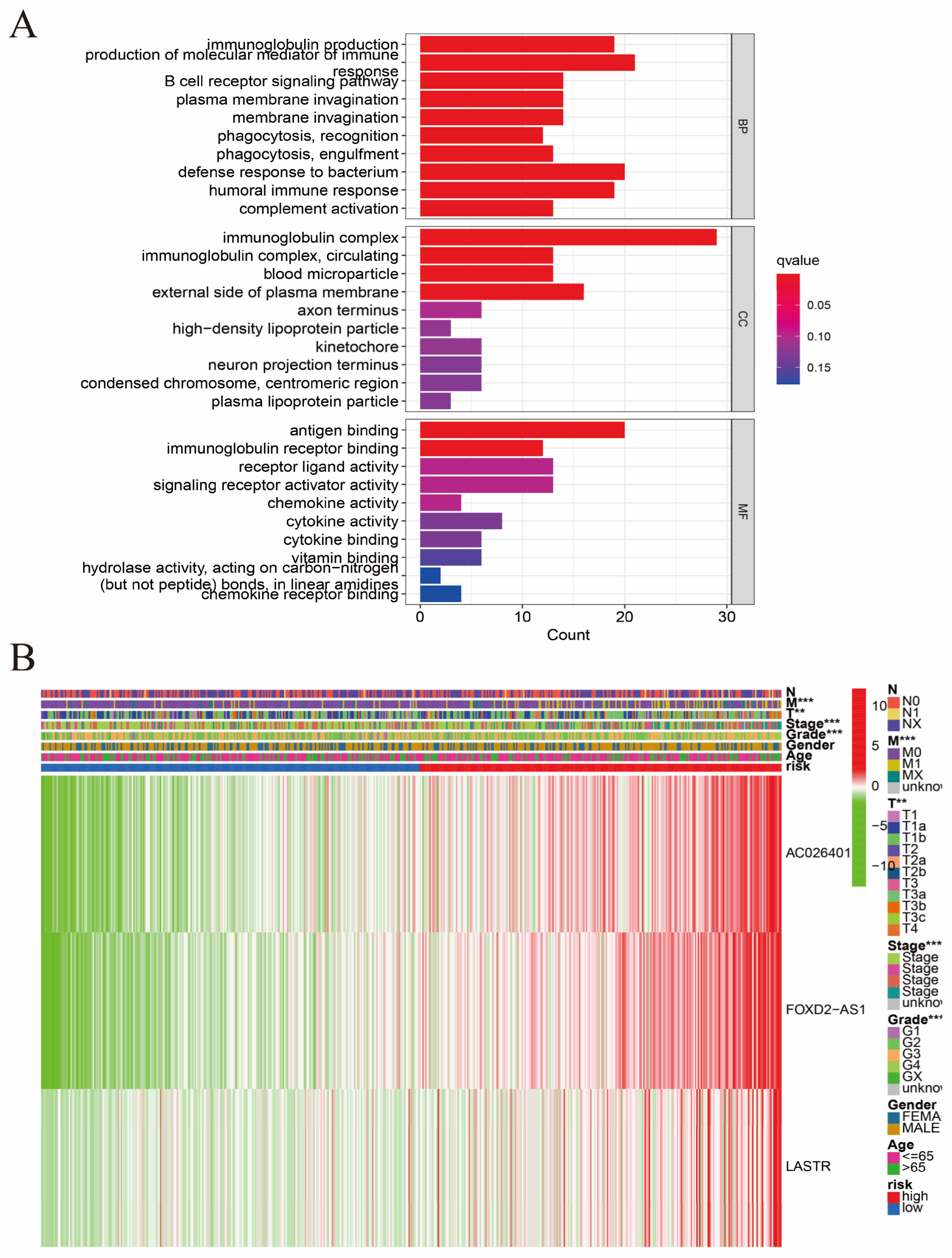
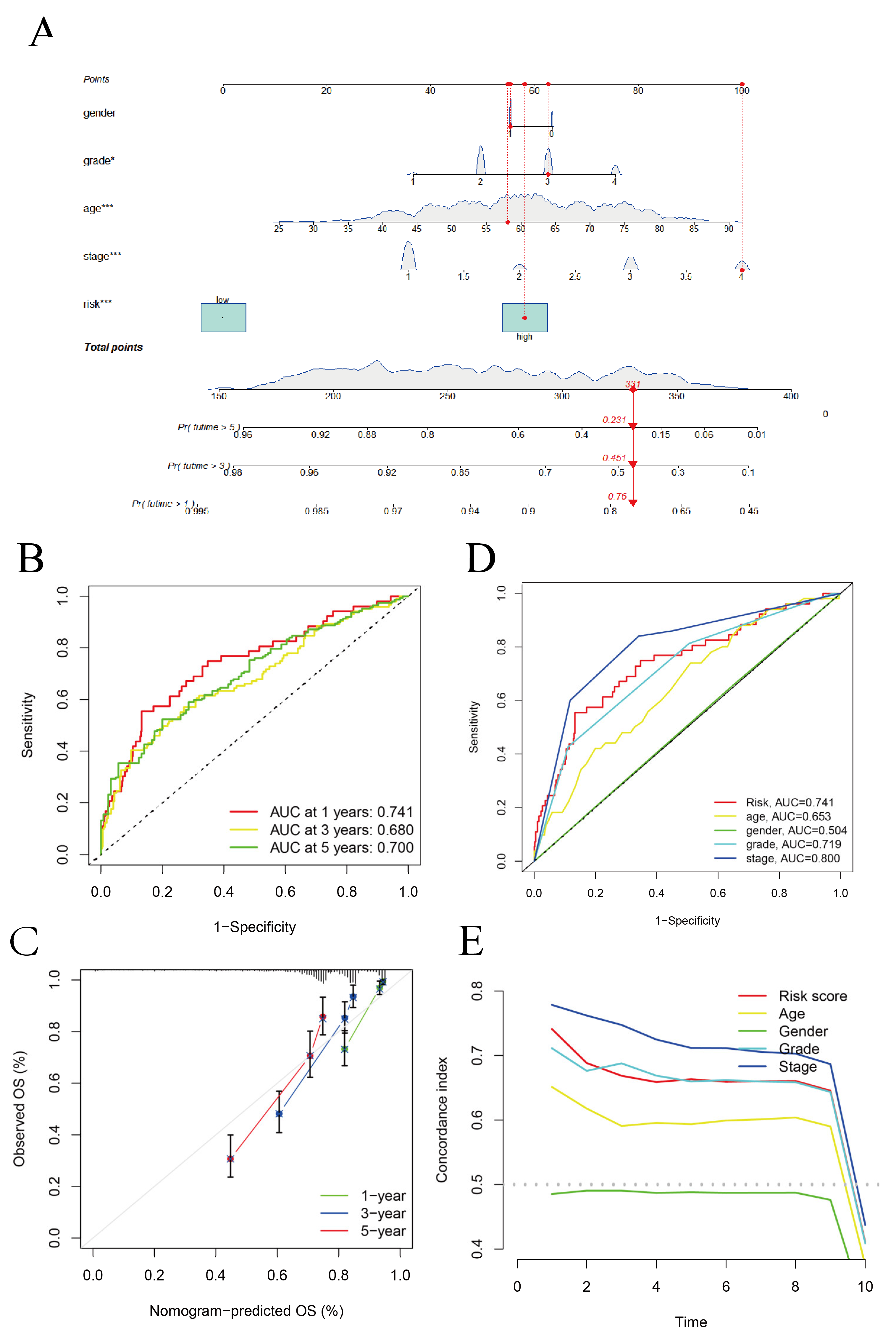
3.2. Construction of the Hybrid Nomogram and GO Analysis
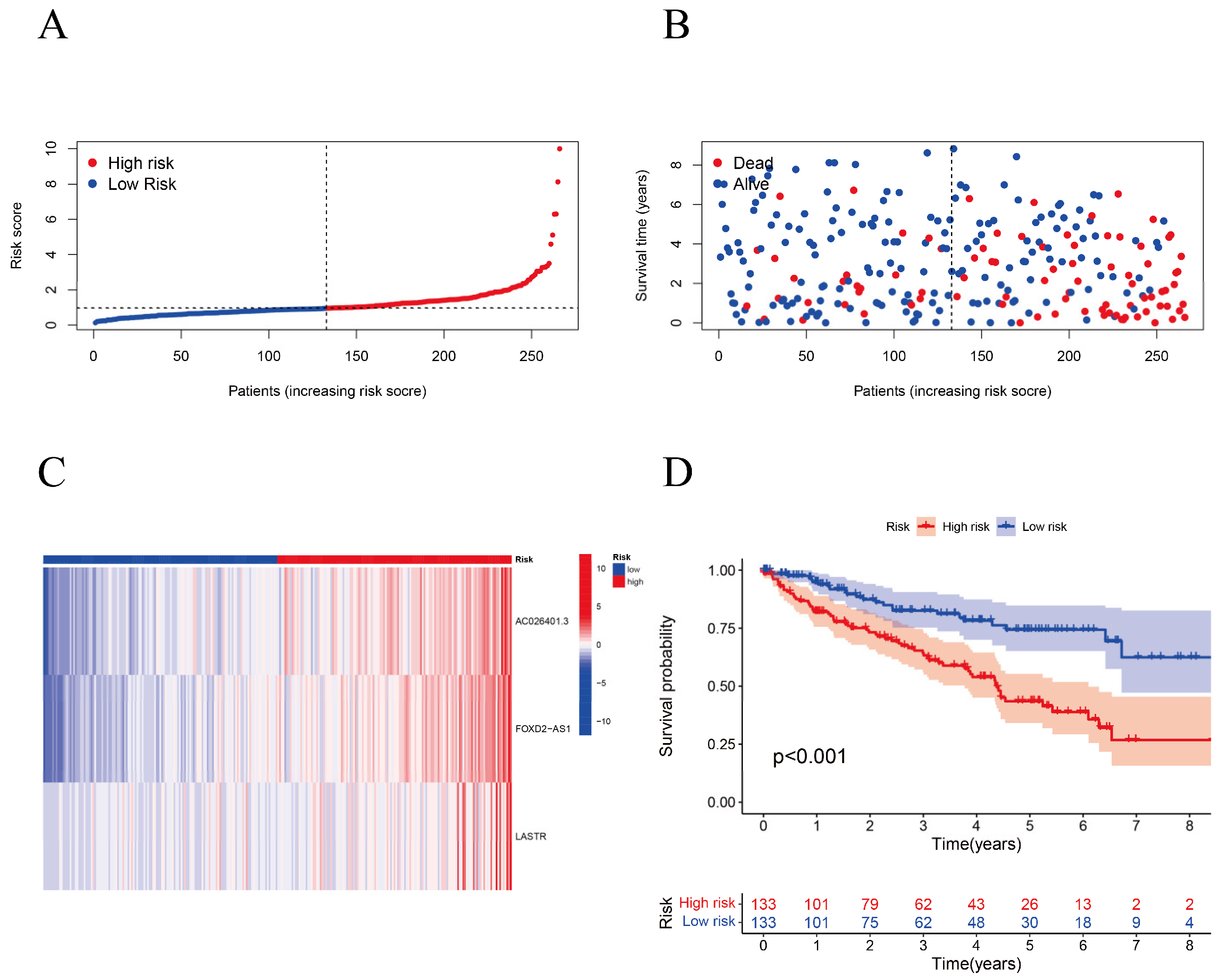
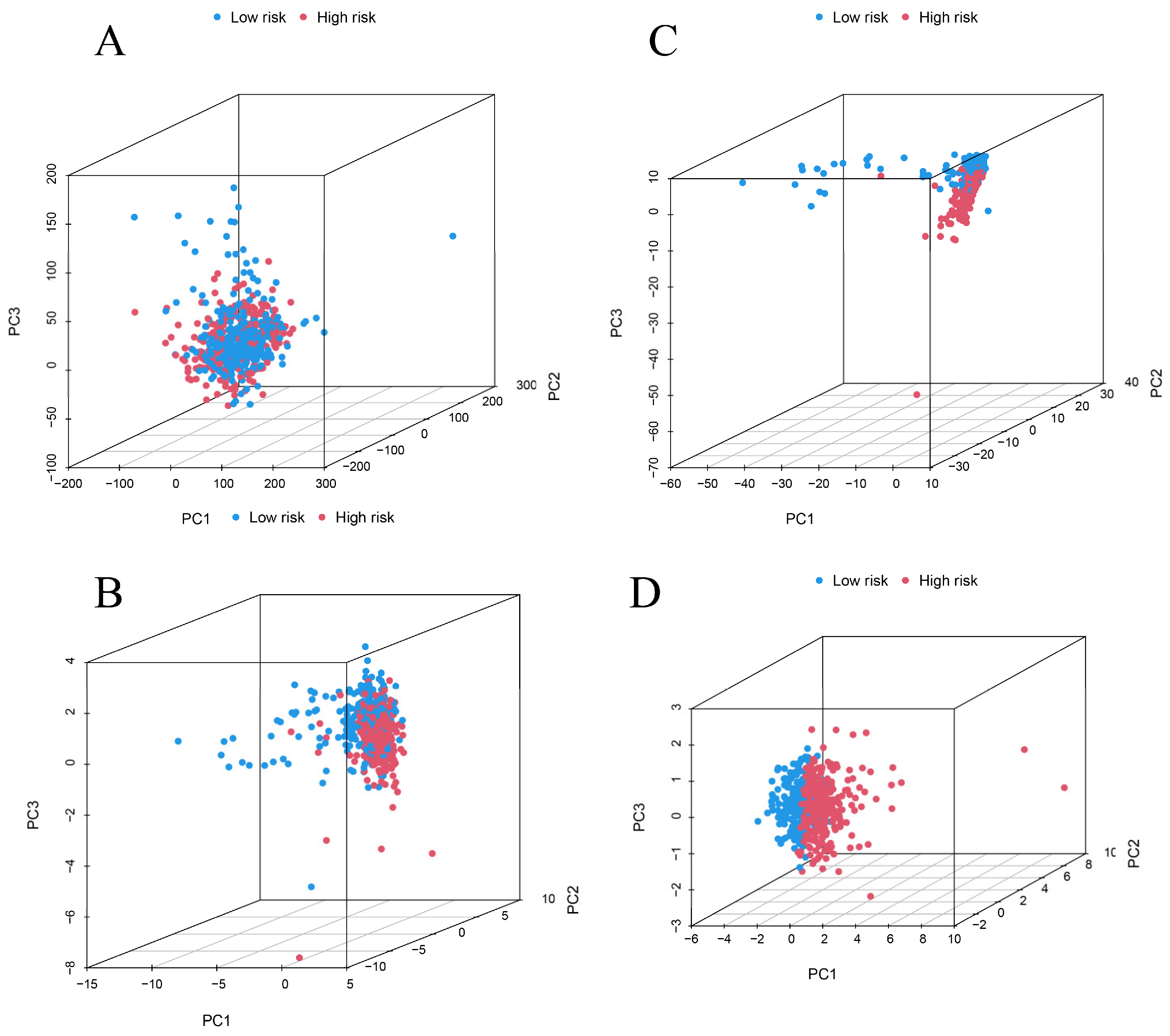
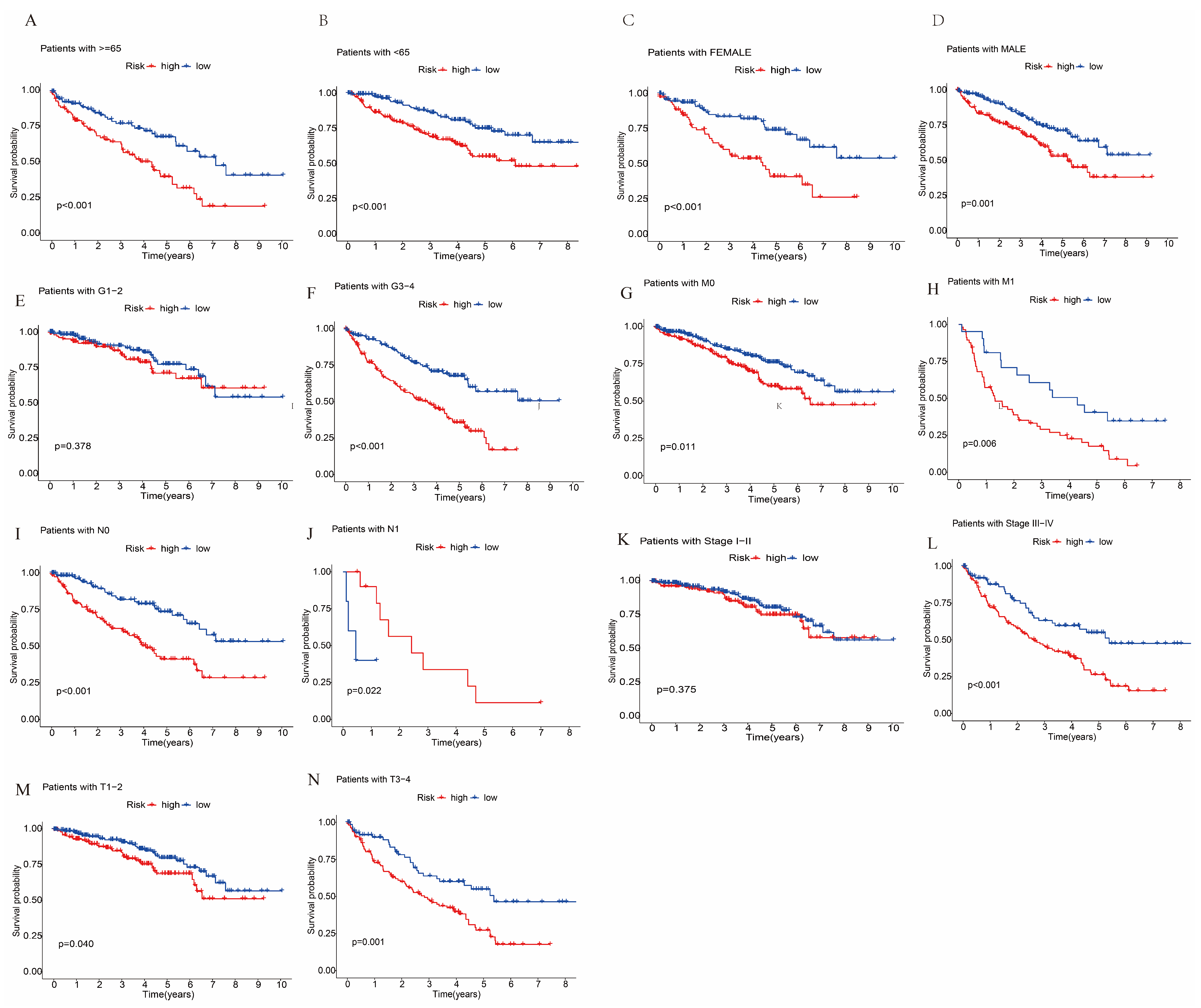
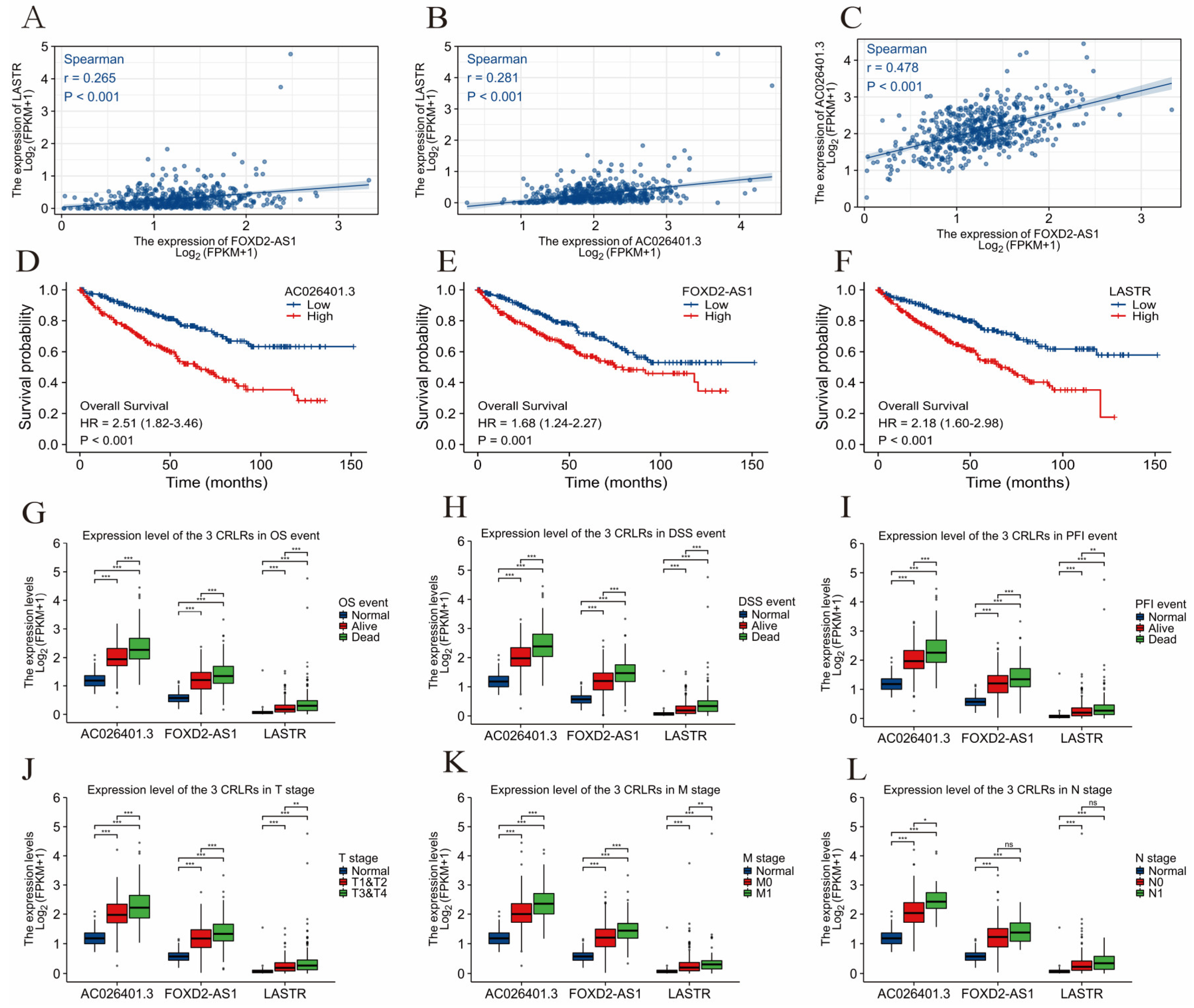
3.3. Survival Analysis and Principal Component Analysis
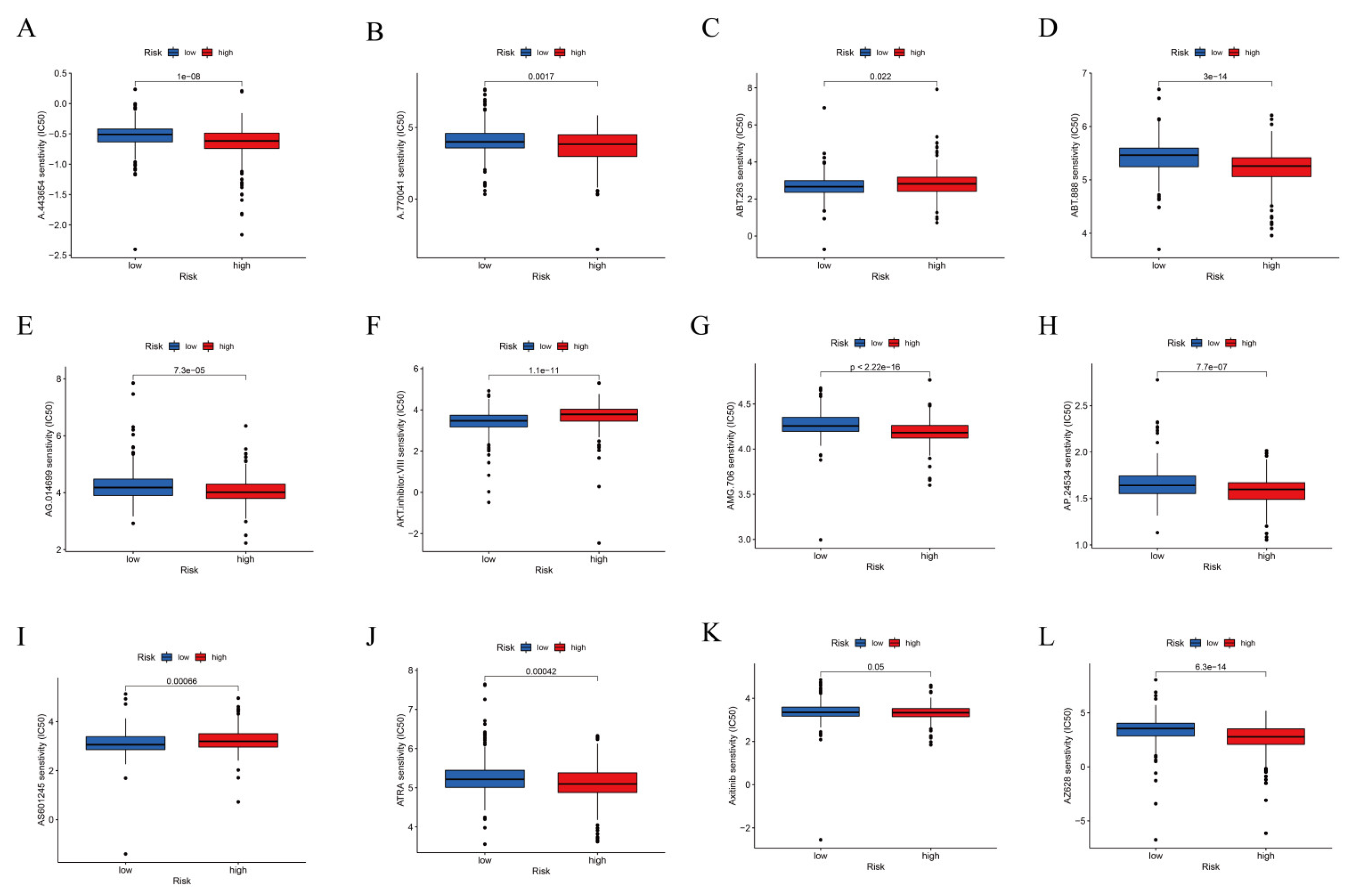
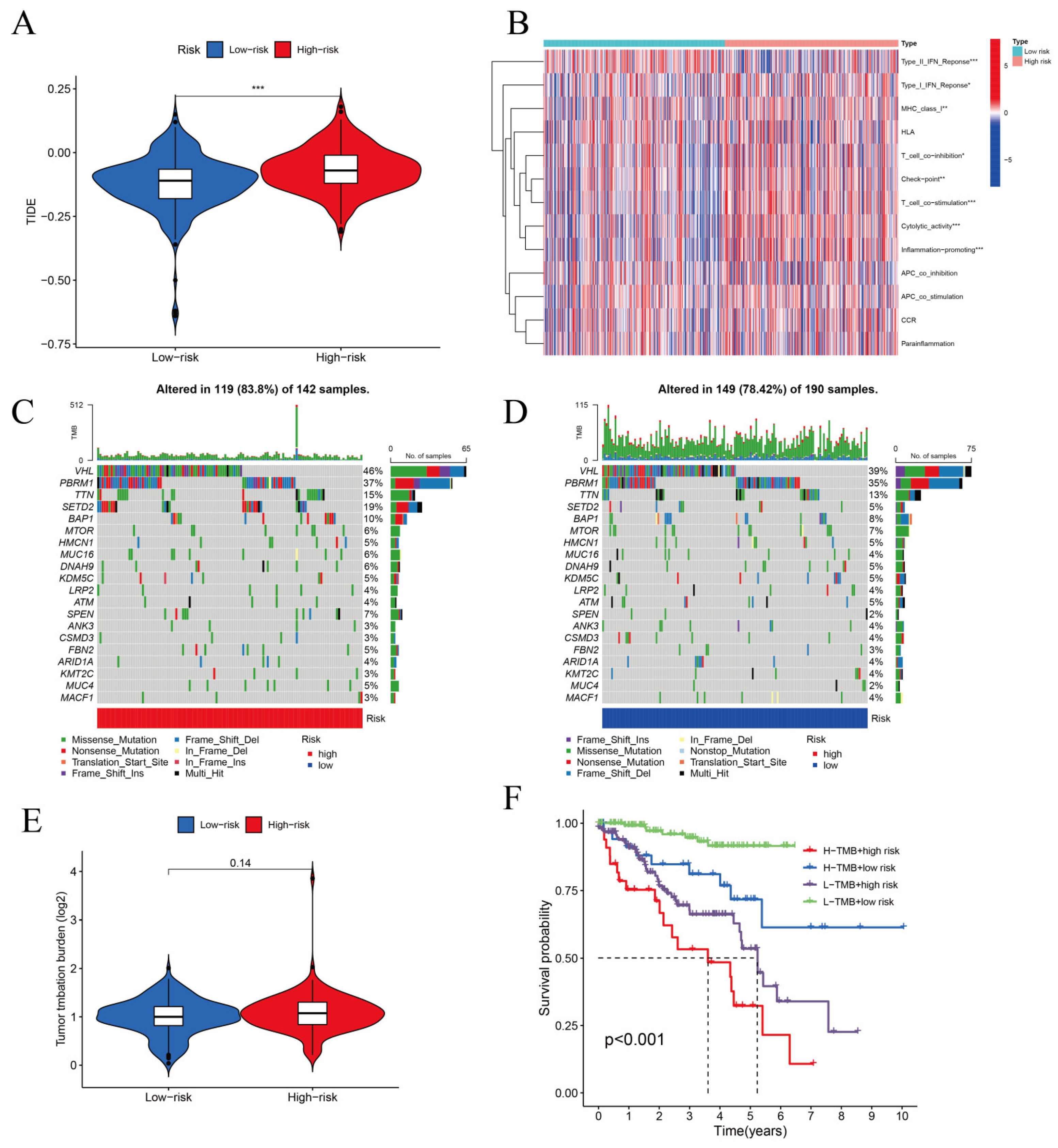
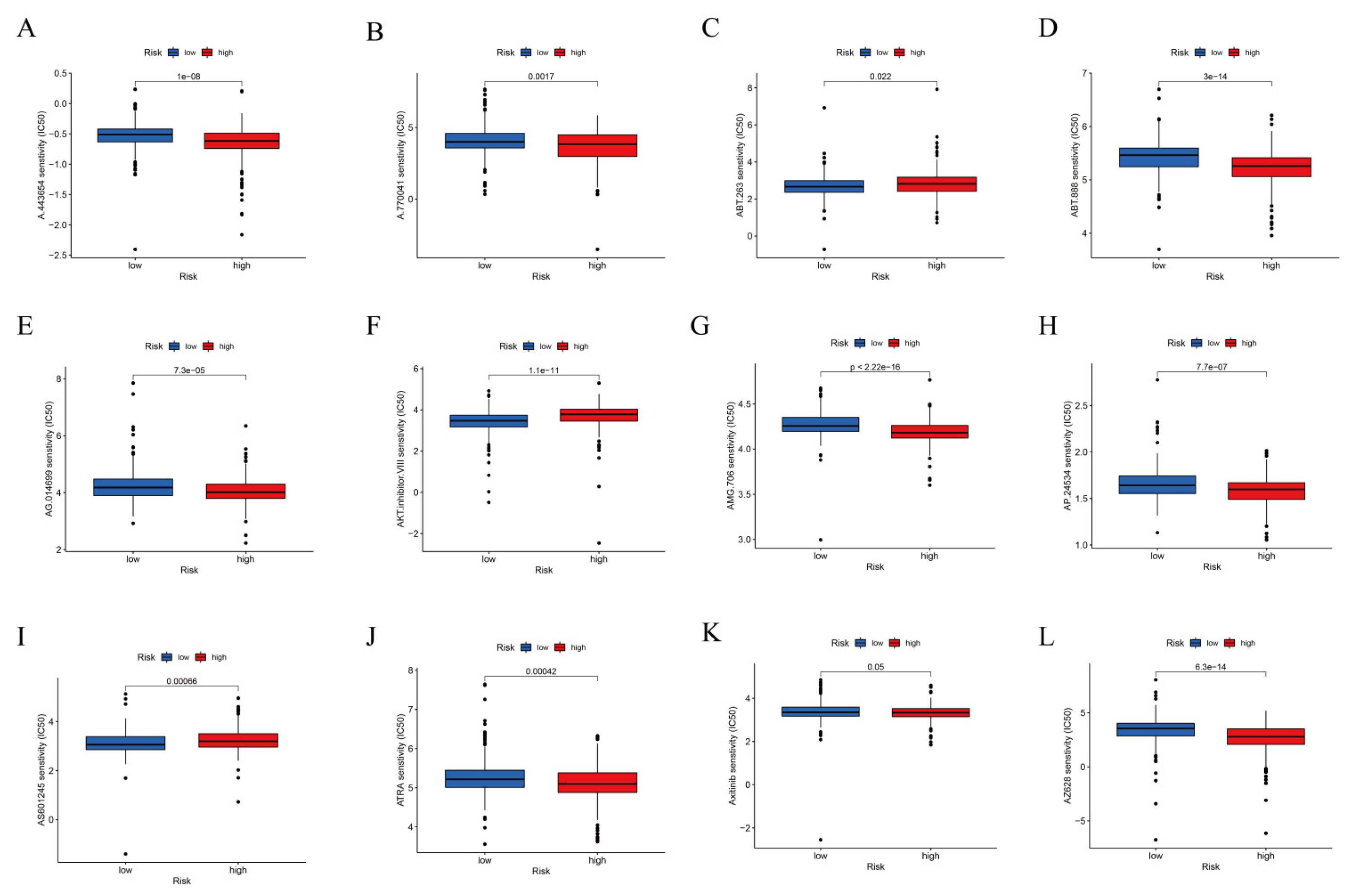
3.4. TIDE Algorithm and IC50 for Assessing Therapeutic Response
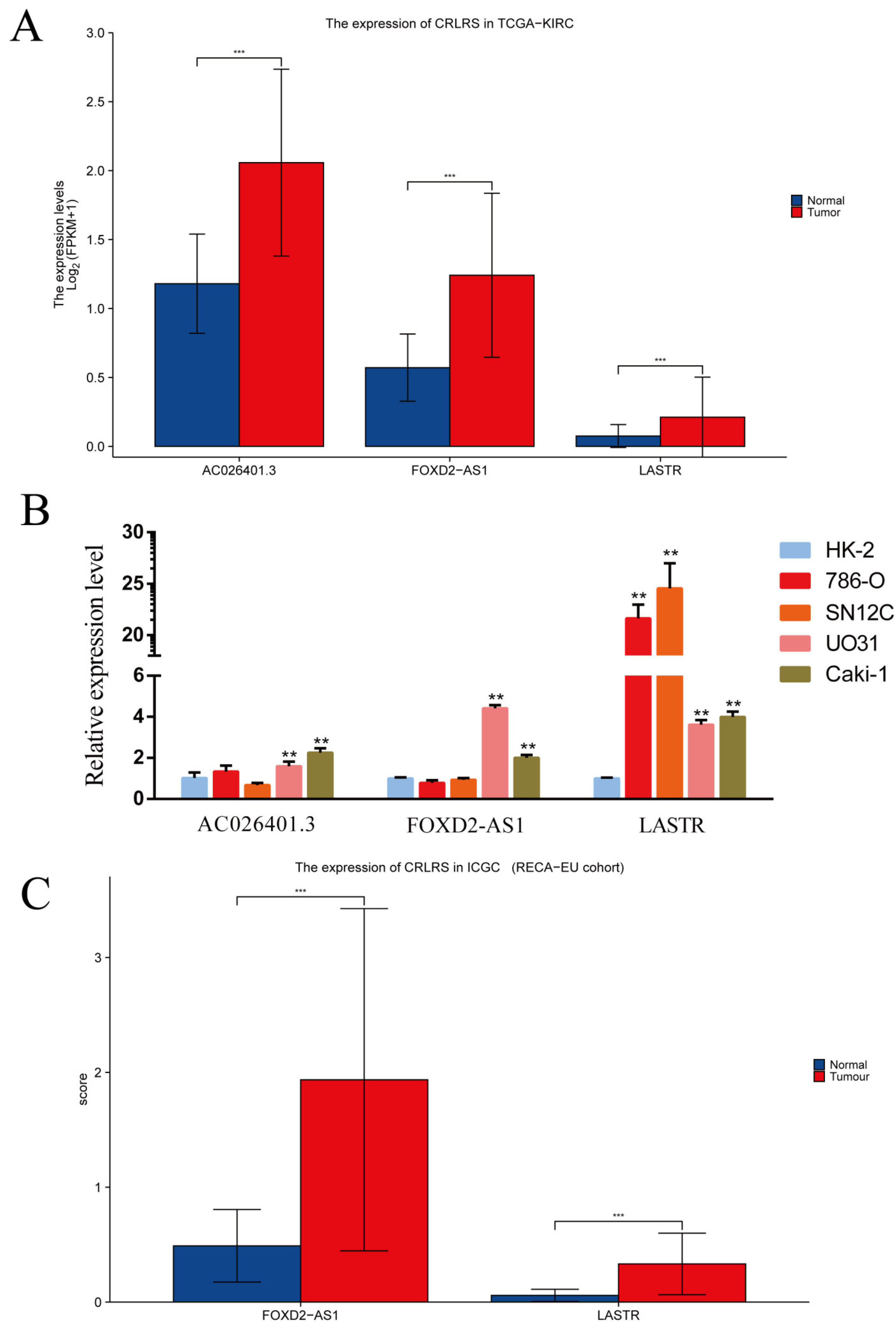
3.5. Validation of CRLRs by qPCR and ICGC Database
4. Discussion
5. Conclusions
Availability of Data and Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| lncRNAs | Long noncoding RNA |
| ICGC | International Cancer Genome Consortium |
| TCGA | The Cancer Genome Atlas |
| LASSO | Least absolute shrinkage and selection operator |
| ML | Machine learning |
| IC50 | Half maximal inhibitory concentration |
| ccRCC | Clear cell renal cell carcinoma |
| CRLR | Cuproptosis-related long noncoding RNA |
| TIDE | Tumor immune dysfunction and exclusion |
| TMB | Tumor mutational burden |
| CRGs | Cuproptosis-related genes |
| PCA | Principal component analysis |
| OS | Overall survival |
| AUC | Area under the curve |
References
- Hyuna, S.; Jacques, F.; Siegel, R.L.; Mathieu, L.; Isabelle, S.; Ahmedin, J.; Freddie, B. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Signoretti, S.; Flaifel, A.; Chen, Y.; Reuter, V.E. Renal Cell Carcinoma in the Era of Precision Medicine: From Molecular Pathology to Tissue-Based Biomarkers. J. Clin. Oncol. 2018, 36, 3553–3559. [Google Scholar] [CrossRef] [PubMed]
- Peter, T.; Shannon, C.; Boryana, P.; Margaret, D.; Ana, V.; Mai, A.; Jordan, R.; Lena, J.; Ranad, H.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Daolin, T.; Xin, C.; Guido, K. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, L.; Chen, J.; Liu, L.; Yang, X.; Cui, H.; Yue, G. Role of Metabolic Reprogramming of Long non-coding RNA in Clear Cell Renal Cell Carcinoma. J. Cancer 2022, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Braga, E.A.; Fridman, M.V.; Filippova, E.A.; Loginov, V.I.; Pronina, I.V.; Burdennyy, A.M.; Karpukhin, A.V.; Dmitriev, A.A.; Morozov, S.G. LncRNAs in the Regulation of Genes and Signaling Pathways through miRNA-Mediated and Other Mechanisms in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2021, 22, 11193. [Google Scholar] [CrossRef]
- Shen, H.; Luo, G.; Chen, Q. Long noncoding RNAs as tumorigenic factors and therapeutic targets for renal cell carcinoma. Cancer Cell Int. 2021, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.B.; Falkai, P.; Koutsouleris, N. Machine Learning Approaches for Clinical Psychology and Psychiatry. Annu. Rev. Clin. Psychol. 2018, 14, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, Z.; Luo, Y.; Yu, H.; Wu, S.; Ren, X.; Zheng, C.; Huang, X. Classifying 2-year recurrence in patients with dlbcl using clinical variables with imbalanced data and machine learning methods. Comput. Methods Programs Biomed. 2020, 196, 105567. [Google Scholar] [CrossRef]
- Mceligot, A.J.; Poynor, V.; Sharma, R.; Panangadan, A. Logistic LASSO Regression for Dietary Intakes and Breast Cancer. Nutrients 2020, 12, 2652. [Google Scholar] [CrossRef]
- Poldrack, R.A.; Huckins, G.; Varoquaux, G. Establishment of Best Practices for Evidence for Prediction: A Review. JAMA Psychiat. 2020, 77, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhang, S.; Jiao, Y.; Shi, Y.; Zhang, H.; Wang, F.; Wang, L.; Zhu, T.; Miao, Y.; Sang, W.; et al. LASSO Model Better Predicted the Prognosis of DLBCL than Random Forest Model: A Retrospective Multicenter Analysis of HHLWG. J. Oncol. 2022, 2022, 1618272. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Du, H.; Xie, S.; Guo, W.; Che, J.; Zhu, L.; Hang, J.; Li, H. Development and validation of an autophagy-related prognostic signature in esophageal cancer. Ann. Transl. Med. 2021, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Zhang, C.; Li, J.; Zhang, B.; Dong, Y.; Cui, X. Establishment and Validation of a Nomogram Prognostic Model for Epithelioid Hemangioendothelioma. J. Oncol. 2022, 2022, 6254563. [Google Scholar] [CrossRef]
- Qi, J.; Yin, J.; Ding, G. A Connexin-Based Biomarker Model Applicable for Prognosis and Immune Landscape Assessment in Lung Adenocarcinoma. J. Oncol. 2022, 2022, 9261339. [Google Scholar] [CrossRef]
- Yu, C.; Li, D.; Fan, F.; Li, C. Bioinformatics analysis of potential hub genes and miRNAs for hepatocellular carcinoma. J. Zunyi Med. Univ. 2022, 45, 37–45. [Google Scholar] [CrossRef]
- Liu, M.; Zheng, X.; Xu, Y.; Wang, J.; Feng, S.; Zhang, Z.; Wu, M.; Li, X. Prognostic value of ERGIC3 and its role on immune microenvironment in lung adenocarcinoma based on TCGA database. J. Zunyi Med. Univ. 2022, 45, 450–456. [Google Scholar] [CrossRef]
- Ye, J.; Liu, J.; Tang, T.; Xin, L.; Bao, X.; Yan, Y. miR-4306 inhibits the malignant behaviors of colorectal cancer by regulating lncRNA FoxD2-AS1. Mol. Med. Rep. 2021, 24, 723. [Google Scholar] [CrossRef]
- Ahmad, S.; Abbas, M.; Ullah, M.F.; Aziz, M.H.; Beylerli, O.; Alam, M.A.; Syed, M.A.; Uddin, S.; Ahmad, A. Long non-coding RNAs regulated NF-κB signaling in cancer metastasis: Micromanaging by not so small non-coding RNAs. Semin. Cancer Biol. 2022, 85, 155–163. [Google Scholar] [CrossRef]
- Zhao, Q.S.; Ying, J.B.; Jing, J.J.; Wang, S.S. LncRNA FOXD2-AS1 stimulates glioma progression through inhibiting P53. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4382–4388. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; He, W.; Chen, C.; Liu, M.; Liu, H.; Xue, F.; Bi, J.; Xu, D.; Zhao, Y.; Huang, J.; et al. The long non-coding RNA FOXD2-AS1 promotes bladder cancer progression and recurrence through a positive feedback loop with Akt and E2F1. Cell Death Dis. 2018, 9, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Jiang, X.; Jiang, S.; Guo, Z.; Zhou, Q.; He, J. LncRNA FOXD2-AS1 Regulates miR-25-3p/Sema4c Axis To Promote The Invasion And Migration Of Colorectal Cancer Cells. Cancer Manag. Res. 2019, 11, 10633–10639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, N.; Wang, X.; Di, Z.; Xiong, J.; Ma, Y.; Yan, Y.; Qian, Y.; Zhang, Q.; Yu, J. Silencing lncRNA FOXD2-AS1 inhibits proliferation, migration, invasion and drug resistance of drug-resistant glioma cells and promotes their apoptosis via microRNA-98-5p/CPEB4 axis. Aging 2019, 11, 10266–10283. [Google Scholar] [CrossRef]
- Ni, W.; Xia, Y.; Bi, Y.; Wen, F.; Hu, D.; Luo, L. FoxD2-AS1 promotes glioma progression by regulating miR-185-5P/HMGA2 axis and PI3K/AKT signaling pathway. Aging 2019, 11, 1427–1439. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.; Luo, X.; Zeng, M.; Xu, L.; Zhang, Q.; Liu, H.; Guo, J.; Xu, L. Overexpression of the Long Noncoding RNA FOXD2-AS1 Promotes Cisplatin Resistance in Esophageal Squamous Cell Carcinoma Through the miR-195/Akt/mTOR Axis. Oncol. Res. 2020, 28, 65–73. [Google Scholar] [CrossRef]
- Sui, C.; Dong, Z.; Yang, C.; Zhang, M.; Dai, B.; Geng, L.; Lu, J.; Yang, J.; Xu, M. LncRNA FOXD2-AS1 as a competitive endogenous RNA against miR-150-5p reverses resistance to sorafenib in hepatocellular carcinoma. J. Cell. Mol. Med. 2019, 23, 6024–6033. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhao, F.; Liu, C.; Xu, T.; Song, K. LncRNA FOXD2-AS1 promotes cell proliferation and invasion of fibroblast-like synoviocytes by regulation of miR-331-3p/PIAS3 pathway in rheumatoid arthritis. Autoimmunity 2021, 54, 254–263. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, W.; Lin, C.; Wang, X.; Zhang, X.; Zhang, Y.; Yang, R.; Chen, W.; Cao, W. Dysregulation of FOXD2-AS1 promotes cell proliferation and migration and predicts poor prognosis in oral squamous cell carcinoma: A study based on TCGA data. Aging 2020, 13, 2379–2396. [Google Scholar] [CrossRef]
- De Troyer, L.; Zhao, P.; Pastor, T.; Baietti, M.F.; Barra, J.; Vendramin, R.; Dok, R.; Lechat, B.; Najm, P.; Van Haver, D.; et al. Stress-induced lncRNA LASTR fosters cancer cell fitness by regulating the activity of the U4/U6 recycling factor SART3. Nucleic Acids Res. 2020, 48, 2502–2517. [Google Scholar] [CrossRef]
- Xia, M.; Zhu, W.; Tao, C.; Lu, Y.; Gao, F. LncRNA LASTR promote lung cancer progression through the miR-137/TGFA/PI3K/AKT axis through integration analysis. J. Cancer 2022, 13, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sun, L.; Wang, S.; Guo, J.; Xiao, R.; Li, W.; Qi, W.; Qiu, W. Ferroptosis-related long non-coding RNAs and the roles of LASTR in stomach adenocarcinoma. Mol. Med. Rep. 2022, 25, 118. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Tong, H.; Zhu, J.; Xie, C.; Qin, Z.; Li, T.; Liu, X.; He, W. A Glycolysis-Based Long Non-coding RNA Signature Accurately Predicts Prognosis in Renal Carcinoma Patients. Front. Genet. 2021, 12, 638980. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Zhang, J.; Cao, P.B.; Zhou, G.Q. Prognostic and predictive value of the hypoxia-associated long non-coding RNA signature in hepatocellular carcinoma. Yi Chuan 2022, 44, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fu, X.L.; Wang, J.J.; Guan, R.; Tang, X.J. Novel Strategies to Discover Effective Drug Targets in Metabolic and Immune Therapy for Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 17–39. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| LASTR | 3′-GCAAGAGAGAAGACAGTGGGTGAAG-5′ | 3′-CCAGTGAAGGGCTGAAGGGTTTAG-5′ |
| FOXD2−AS1 | 3′-TGGGTTGAGGGTCTGTGACTGTAG-5′ | 3′-GCTGCCGCTGGAGTATTCTTGG-5′ |
| AC026401.3 | 3′-AGTGGGAAATCTGACCTCTTTTGGC-5′ | 3′-TCCTGTTCTTAGTGGCTGCATTACC-5′ |
| β-Actin | 3′-CGGGAAATCGTGCGTGAC-5′ | 3′-CAGGAAGGAAGGCTGGAAG-5′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Z.; Lu, J.; Chen, A.; Zheng, X.; Wu, M.; Tan, Z.; Xie, J. Identification and Validation of Cuproptosis-Related LncRNA Signatures in the Prognosis and Immunotherapy of Clear Cell Renal Cell Carcinoma Using Machine Learning. Biomolecules 2022, 12, 1890. https://doi.org/10.3390/biom12121890
Bai Z, Lu J, Chen A, Zheng X, Wu M, Tan Z, Xie J. Identification and Validation of Cuproptosis-Related LncRNA Signatures in the Prognosis and Immunotherapy of Clear Cell Renal Cell Carcinoma Using Machine Learning. Biomolecules. 2022; 12(12):1890. https://doi.org/10.3390/biom12121890
Chicago/Turabian StyleBai, Zhixun, Jing Lu, Anjian Chen, Xiang Zheng, Mingsong Wu, Zhouke Tan, and Jian Xie. 2022. "Identification and Validation of Cuproptosis-Related LncRNA Signatures in the Prognosis and Immunotherapy of Clear Cell Renal Cell Carcinoma Using Machine Learning" Biomolecules 12, no. 12: 1890. https://doi.org/10.3390/biom12121890