
In Silico Protein Folding Prediction of COVID-19 Mutations and Variants



Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Identification
2.2. De Novo Protein Folding and Structural Analysis
2.3. Visualizing and Preliminary Analysis
2.4. Docking Analysis
2.5. In Vitro Test
- SARS-CoV-2 Spike protein (Sp) expression constructs: Spike protein (Sp) constructs were used in the experiments: mRFP-Spike Protein (Sp) Original (mRFP-Sp WT, EX-NV219-M55) and Kappa (B.1.617.1, mRFP-Sp Kappa, EX-NV248-M55) were purchased from Genecopoeia, Inc. Rockville, MD, U.S.A. Kappa includes the following eight mutations: K19R, G142D, E154K, L452R, E484Q, D614G, P681R, and Q1031H. Human ACE-2 conjugated mono-GFP (ACE-2-GFP, EX-U1285-M98) was also acquired from Genecopoeia, Inc.
- (1)
- Based on the kappa plasmid, the delta (B.1.617.2, mRFP-Sp Delta) was generated in the lab with the following nine mutations: T19R, 156&157del, R158G, L452R, T478K, D614G, P681R, and Q1031H.
- (2)
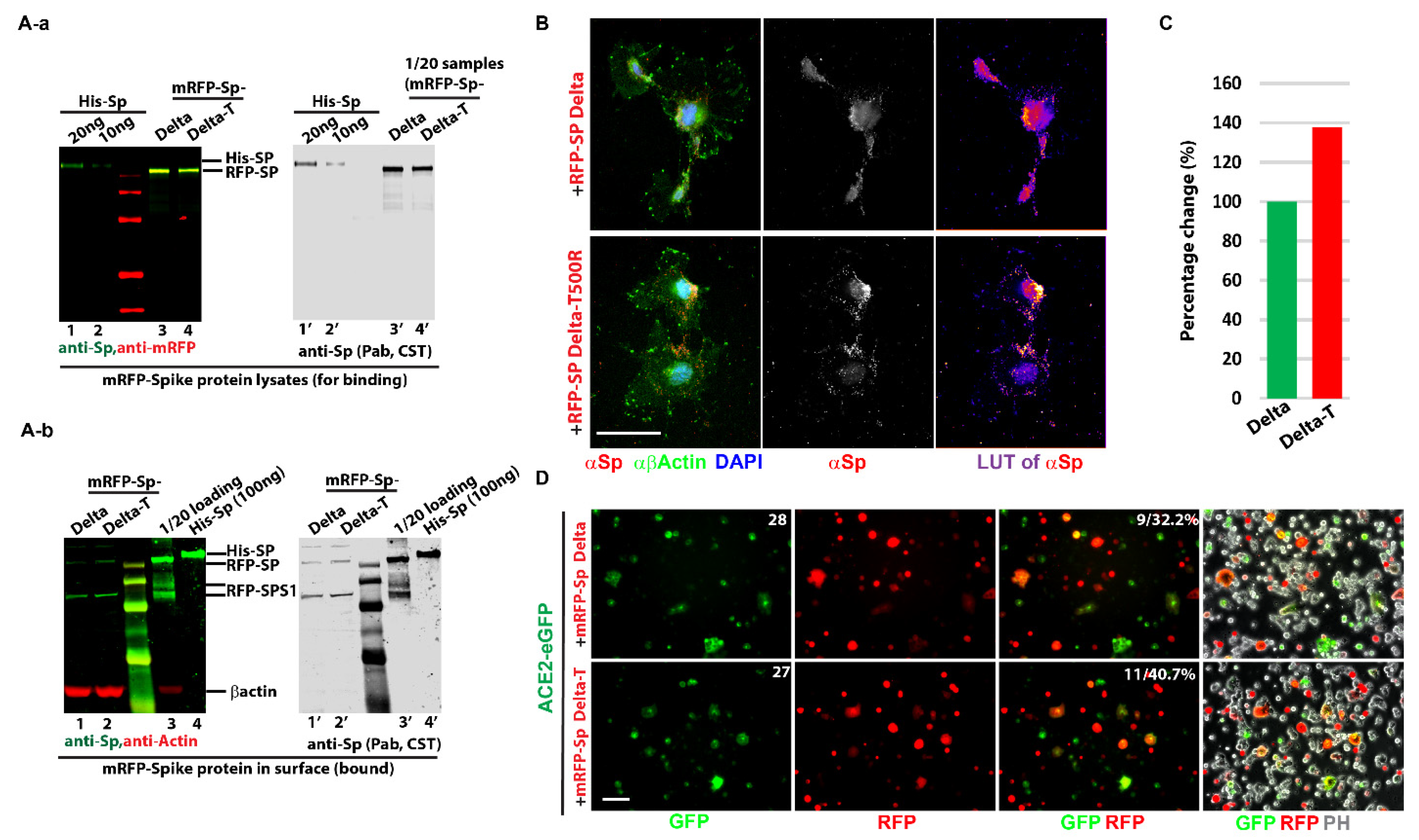
- As predicted, Threonine (Thr) at residue 500 of the delta variant was mutated to Arginine (Arg) to obtain the T500R mutations (mRFP-Sp Delta-T).
- Antibodies: Primary antibodies used were SARS-COV-2 Spike Protein (E7B3E) (Cell Signaling Technology, Inc. Danvers, MA, U.S.A. #63847, rabbit monoclonal, 1:2000 for WB, 1:200 for IF). SARS-COV-2 Spike Protein (2B3E5) (Cell Signaling Technology, Inc. #52342S, mouse monoclonal, 1:2000 for WB, 1:200 for IF). SARS-CoV-2 Spike RBD (R&D Systems, Inc. Minneapolis, MN, U.S.A. AB#105401, mouse monoclonal, 1:2000 for WB, 1:200 for IF). Beta-actin (13E5) (Cell Signaling Technology, Inc. #4970, rabbit monoclonal, 1:2000 for WB, 1:200 for IF). Beta-actin (C4) (Santa Cruz Bio, Santa Cruz, CA, USA, SC-47778, mouse monoclonal, 1:1000 for WB, 1:100 for IF). Secondary antibodies for immunofluorescence were Alexa Fluora® 488- and Alexa Fluor® 594 conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) and IRDye 800CW Goat anti-Rabbit (Li Cor Biosciences, Lincoln, NE, USA, 926-32211, 1:20,000) and IRDye 680LT Goat anti-Mouse IgG (Li Cor Biosciences, 926-68020, 1:20,000) for Western blotting.
- Cells lines and culture: HEK 293T cell line (CRL-11268TM, ATCC) and COS7 cells (CRL-1651TM, ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FBS and incubated in a humidified incubator at 5% CO2 and 37 °C.
- Soluble Spike proteins preparation from cells (Freeze & Thaw lysis): 293T cells were set up in p100 dishes and transfected with 8 μg of different Sp plasmids with Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA) for 2 days. Cells were then scraped and transferred to 1.5 mL microfuge tube with 1 mL ice-cold PBS. After centrifuge, the pellet was resuspended in 500 μL lysis buffer (600 mM KCl, 20 M Tric-Cl (pH 7.8), and 20% Glycerol) with protease inhibitor (Roche Applied Sciences, Penzberg, Germany). The tube was sealed and frozen using liquid nitrogen, then thawed on ice. After three rounds of the “Freeze–Thaw” process, 250U of Benzonase was added to digest DNA for 10 min at RT (or alternatively via sonication). After a quick vortex, the samples were centrifuged with maximum speed for 30 min in a cold room, and 100 μL per tube of supernatants was aliquoted and saved as ligand samples.
- Ligand bound assay and Western blot analysis: COS7 cells were set up in p60 dishes. After overnight plating, the total amount of Sp proteins (around 300 ng) was added to the cells. Then, 2 h after incubation, the cell dishes were washed twice with PBS for 5 min to remove unbound ligands. A total of 125 μL SDS sample loading buffer was used to lysis cells and collected into the 1.5 mL micro-centrifuge tubes. The cell lysates in the tube were heated at 100 °C for 7 min and centrifuged for 5 min. The standard purified His-Sp (R & D System) was used as control for quantification. One-third of the total lysates were loaded to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to a PVDF membrane (Immobilon-FL membrane, 0.45 μm, IPFL00010). The membranes were blocked in Intercept Blocking buffer (Li Cor Biosciences, 927-60001) for 30 min at room temperature and then incubated overnight at 4 °C with primary antibodies. The next day, membranes were incubated at room temperature for 1 h with secondary antibodies. The fluorescent signal was assayed using an Odyssey Infrared Imaging system (Li Cor Biosciences).
- Immunofluorescence: COS 7 Cells cultured on coverslips after spike protein adding were fixed in 4% paraformaldehyde for 12 min at room temperature (RT), then permeabilized TBS with 0.1% Triton X-100 for 5 min. After blocking with 5% BSA/TBS for 30 min at RT, the coverslips were incubated with either primary individual antibodies or combined antibodies from different species in at cold room overnight, followed by incubation of primary antibodies overnight, secondary antibodies for 1 h and DAPI-Fluoromount-G® (0100-20; South-ern Biotech, Birmingham, AL, USA) stain. Images were analyzed using a fluorescence microscope (Nikon Image system).
- Ligand-receptor endosomal assay: COS7 cells were cultured on coverslips overnight and added with ligand (mRFP-Sp) for 2 h, then cells on a coverslip were quickly treated with 0.025% Trypsin for 1 min after being washed with PBS. Next, cells were stained following the immunofluorescence procedures. Heatmap was considered for the intensity comparison (LUT).
- Cell in-fusion assay: 293T cells were set up in p60 dishes and individually transfected with 3 µg of different mRFP-Sp plasmids (Sp Delta and Sp Delta T500R), mRFP, eGFP, and ACE-2-GFP with Lipofectamine 3000 for overnight. All transfected cells were trypsinized and resuspended in a 4 mL culture medium. Different combinations were set (eGFP + mRFP, eGFP + mRFP-Sps, ACE-2-eGFP + mRFP, and ACE-2-eGFP + mRFP-Sps), an example like: 0.5 mL of ACE-2-eGFP transfected 293T cells was mixed with 0.5 mL of 293T cells with mRFP-Sp delta, in 15mL falcon tubes for 15 min at 37 °C cell incubator, then diluted in the 3mL culture medium and settled down on the coverslips for live imaging and were monitored for 4 h, 8 h and overnight to check cell in fusion statues. In-fusion percentages of ACE-2-eGFP were compared among different spike protein variants or mutations.
- Statistical Analysis: The data were analyzed by Student’s t-test or two-tailed t-tests (two-way ANOVA) with the Prism 8 (GraphPad) or Excel (Microsoft) software. p-values < 0.05 were considered statistically significant.
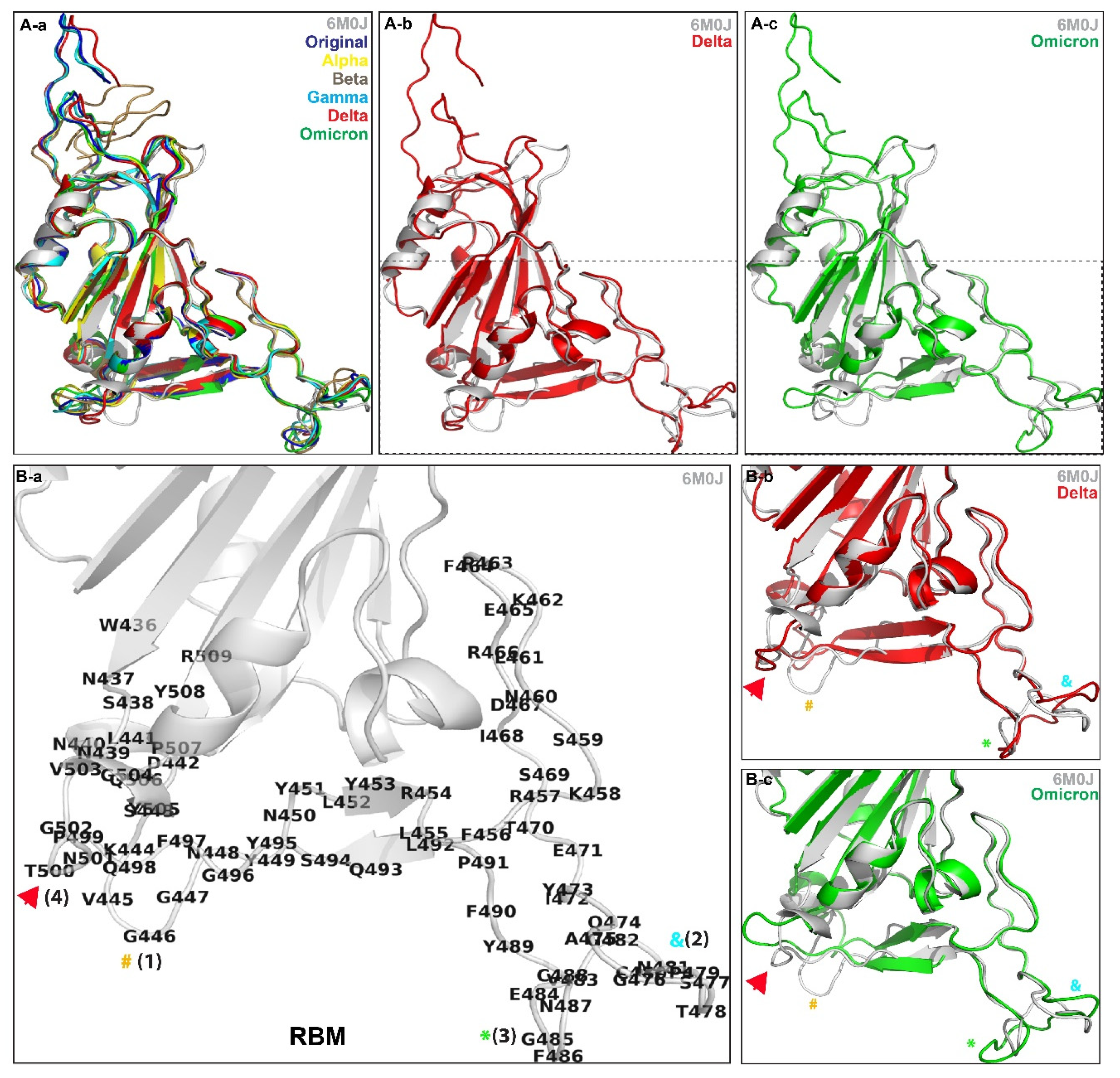
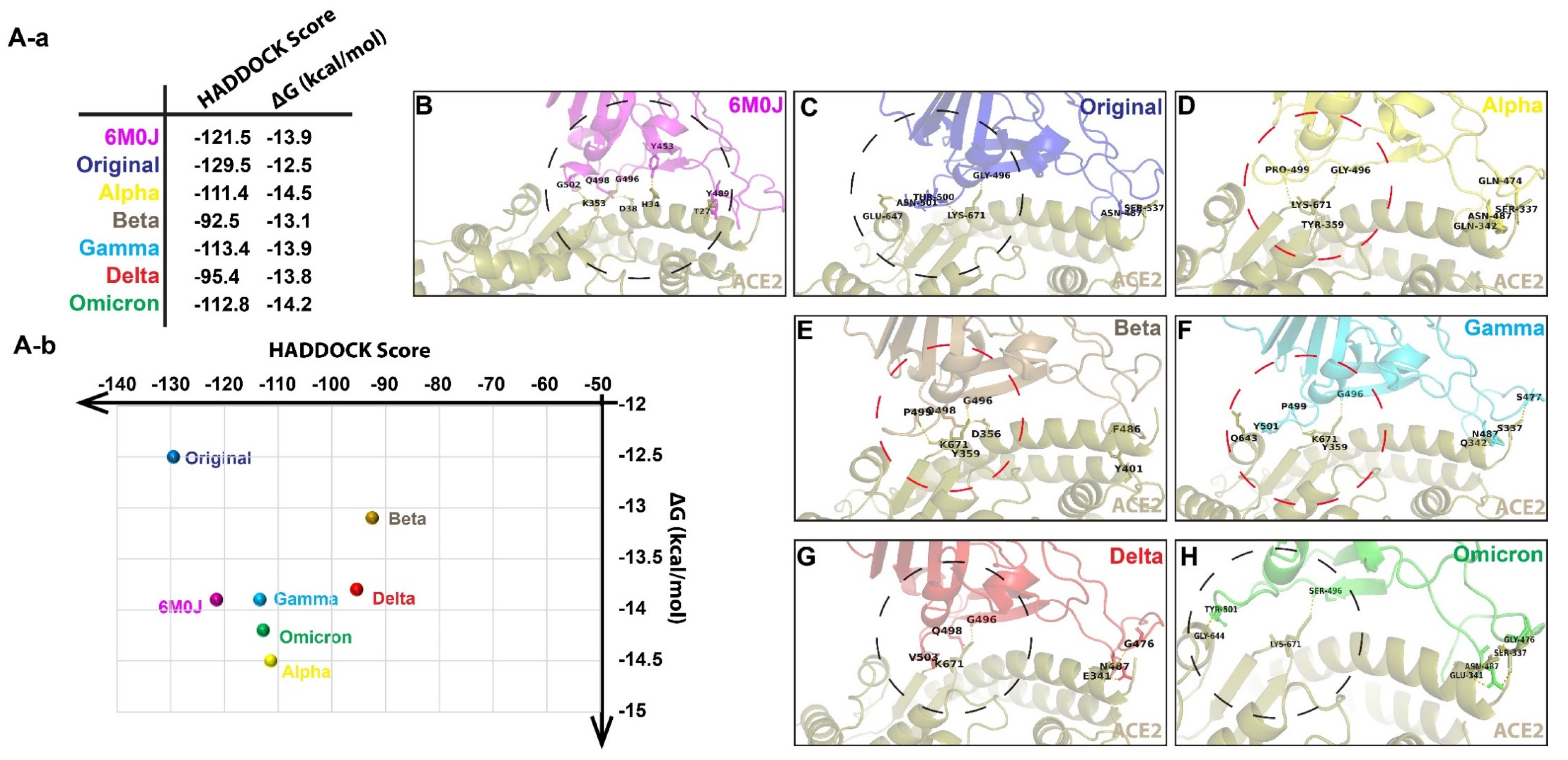
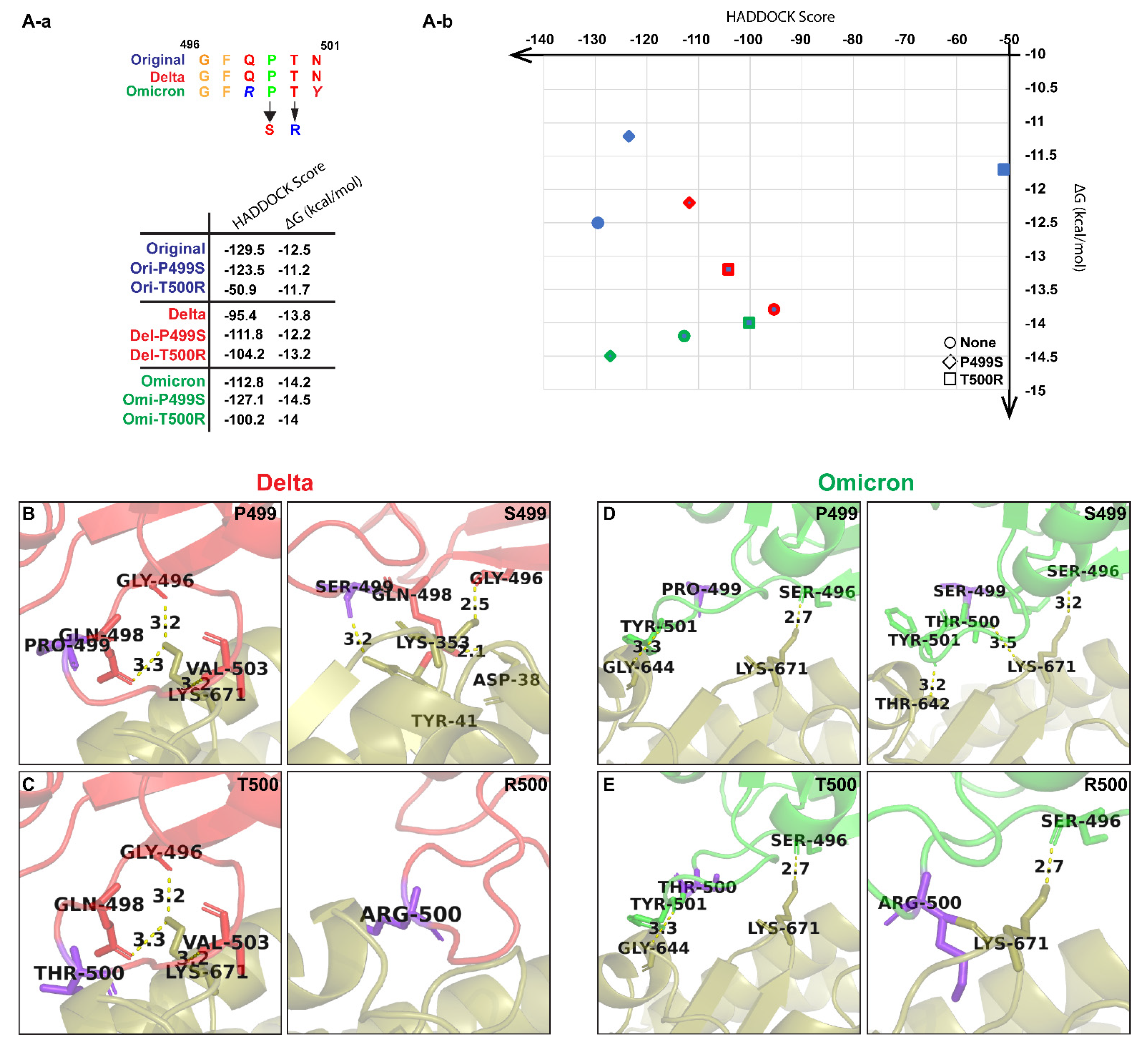
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilcox, G.L.; Poliac, M.; Liebman, M.N. Neural Network Analysis of Protein Tertiary Structure. Tetrahedron Comput. Methodol. 1990, 3, 191–204. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C. Combining Evolutionary Information and Neural Networks to Predict Protein Secondary Structure. Proteins Struct. Funct. Bioinf. 1994, 19, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.T.; Kooperberg, C.; Huang, E.; Baker, D. Assembly of Protein Tertiary Structures from Fragments with Similar Local Sequences Using Simulated Annealing and Bayesian Scoring Functions. J. Mol. Biol. 1997, 268, 209–225. [Google Scholar] [CrossRef]
- Bohr, H.; Bohr, J.; Brunak, S.; Cotterill, R.M.J.; Fredholm, H.; Lautrupt, B.; Petersen, S.B. A Novel Approach to Prediction of the 3-Dimensional Structures of Protein Backbones by Neural Networks. FEBS Lett. 1990, 261, 43–46. [Google Scholar] [CrossRef]
- Fariselli, P.; Casadio, R. A Neural Network Based Predictor of Residue Contacts in Proteins. Protein Eng. 1999, 12, 15–21. [Google Scholar] [CrossRef]
- Xue, B.; Faraggi, E.; Zhou, Y. Predicting Residue-Residue Contact Maps by a Two-Layer, Integrated Neural-Network Method. Proteins Struct. Funct. Bioinf. 2009, 76, 176–183. [Google Scholar] [CrossRef]
- Wu, S.; Szilagyi, A.; Zhang, Y. Improving Protein Structure Prediction Using Multiple Sequence-Based Contact Predictions. Structure 2011, 19, 1182–1191. [Google Scholar] [CrossRef]
- Zhang, G.Z.; Huang, D.S.; Quan, Z.H. Combining a Binary Input Encoding Scheme with RBFNN for Globulin Protein Inter-Residue Contact Map Prediction. Pattern Recognit. Lett. 2005, 26, 1543–1553. [Google Scholar] [CrossRef]
- Vullo, A.; Walsh, I.; Pollastri, G. A Two-Stage Approach for Improved Prediction of Residue Contact Maps. BMC Bioinf. 2006, 7, 180. [Google Scholar] [CrossRef]
- Ovchinnikov, S.; Kim, D.E.; Wang, R.Y.R.; Liu, Y.; Dimaio, F.; Baker, D. Improved de Novo Structure Prediction in CASP11 by Incorporating Coevolution Information into Rosetta. Proteins Struct. Funct. Bioinf. 2016, 84, 67–75. [Google Scholar] [CrossRef]
- Sun, H.P.; Huang, Y.; Wang, X.F.; Zhang, Y.; Shen, H. Bin Improving Accuracy of Protein Contact Prediction Using Balanced Network Deconvolution. Proteins Struct. Funct. Bioinf. 2015, 83, 485–496. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XII. Proteins Struct. Funct. Bioinf. 2018, 86, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)-Round XIII. Proteins Struct. Funct. Bioinf. 2019, 87, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)-Round XIV. Proteins Struct. Funct. Bioinf. 2021, 89, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Y.; Vakser, I.A. DOCKGROUND Protein-Protein Docking Decoy Set. Bioinf. Appl. NOTE 2008, 24, 2634–2635. [Google Scholar] [CrossRef]
- Koehler Leman, J.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. General Overview and Challenges Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct Project-IntAct as a Common Curation Platform for 11 Molecular Interaction Databases. Nucleic Acids Res. 2013, 42, D358–D363. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Jensen, L.J.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2018, 47, 607–613. [Google Scholar] [CrossRef]
- Cong, Q.; Anishchenko, I.; Ovchinnikov, S.; Baker, D. Protein Interaction Networks Revealed by Proteome Coevolution. Science 2019, 365, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The TrRosetta Server for Fast and Accurate Protein Structure Prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Singh, A. Deep Learning 3D Structures. Nat. Methods 2020, 17, 249. [Google Scholar] [CrossRef] [PubMed]
- Jianyi, Y.; Ivan, A.; Hahnbeom, P.; Zhenling, P.; Sergey, O.; David, B. Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Norn, C.; Wicky, B.I.M.; Juergens, D.; Liu, S.; Kim, D.; Tischer, D.; Koepnick, B.; Anishchenko, I.; Players, F.; Baker, D.; et al. Protein Sequence Design by Conformational Landscape Optimization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017228118. [Google Scholar] [CrossRef]
- Pleiner, T.; Tomaleri, G.P.; Januszyk, K.; Inglis, A.J.; Hazu, M.; Voorhees, R.M. Structural Basis for Membrane Insertion by the Human ER Membrane Protein Complex. Science 2020, 369, 433–436. [Google Scholar] [CrossRef]
- O’donnell, J.P.; Phillips, B.P.; Yagita, Y.; Juszkiewicz, S.; Wagner, A.; Malinverni, D.; Keenan, R.J.; Miller, E.A.; Hegde, R.S. The Architecture of EMC Reveals a Path for Membrane Protein Insertion. eLife 2020, 9, e57887. [Google Scholar] [CrossRef]
- Melia, T.J.; Reinisch, K.M. A Possible Role for VPS13-Family Proteins in Bulk Lipid Transfer, Membrane Expansion and Organelle Biogenesis. J. Cell Sci. 2022, 135, jcs259357. [Google Scholar] [CrossRef]
- Yu, T.; Hu, Y.; Zhang, Y.; Zhao, R.; Yan, X.; Dayananda, B.; Wang, J.; Jiao, Y.; Li, J.; Yi, X. Whole-Genome Sequencing of Acer Catalpifolium Reveals Evolutionary History of Endangered Species GBE. Genome Biol. Evol. 2021, 13, evab271. [Google Scholar] [CrossRef]
- Seong, K.; Krasileva, K. V Computational Structural Genomics Unravels Common Folds and Novel Families in the Secretome of Fungal Phytopathogen Magnaporthe Oryzae. Mol. Plant-Microbe Interact. 2021, 34, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Alici, H.; Uversky, V.N.; Kang, D.E.; Woo, A.; Coskuner-Weber, O. Structures of the Wild-Type and S59L Mutant CHCHD10 Proteins Important in Amyotrophic Lateral Sclerosis−Frontotemporal Dementia. ACS Chem. Neurosci. 2022, 13, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, P.; Melarkode Vattekatte, A.; Shinada, N.; Diharce, J.; Martins, C.; Cadet, F.; Gardebien, F.; Etchebest, C.; Nadaradjane, A.A.; De Brevern, A.G. VHH Structural Modelling Approaches: A Critical Review. Int. J. Mol. Sci. 2022, 23, 3721. [Google Scholar] [CrossRef]
- Jaan, S.; Zaman, A.; Ahmed, S.; Shah, M.; Chandra Ojha, S. MRNA Vaccine Designing Using Chikungunya Virus E Glycoprotein through Immunoinformatics-Guided Approaches. Vaccines 2022, 10, 1476. [Google Scholar] [CrossRef]
- Callaway, E. “It Will Change Everything”: DeepMind’s AI Makes Gigantic Leap in Solving Protein Structures. Nature 2020, 588. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Nagesha, S.N.; Ramesh, B.N.; Pradeep, C.; Shashidhara, K.S.; Ramakrishnappa, T.; Krishnaprasad, B.T.; Jnanashree, S.M.; Manohar, M.; Arunkumar, N.; Yallappa; et al. SARS-CoV 2 Spike Protein S1 Subunit as an Ideal Target for Stable Vaccines: A Bioinformatic Study. Mater. Today Proc. 2021, 49, 904–912. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Saha, P.; Kumar Banerjee, A.; Tripathi, P.P.; Srivastava, A.K.; Ray, U.; Banerjee, A.K. A Virus That Has Gone Viral: Amino Acid Mutation in S Protein of Indian Isolate of Coronavirus COVID-19 Might Impact Receptor Binding, and Thus, Infectivity. Biosci. Rep. 2020, 40, 20201312. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Schoeder, C.T.; Brown, J.A.; Smart, C.D.; Moth, C.; Wikswo, J.P.; Capra, J.A.; Meiler, J.; Chen, W.; Madhur, M.S. Which Animals Are at Risk? Predicting Species Susceptibility to COVID-19. bioRxiv 2020, bioRxiv:2020.07.09.194563. [Google Scholar]
- Alenquer, M.; Ferreira, F.; Lousa, D.; Valério, M.; Medina-Lopes, M.; Bergman, M.L.; Gonçalves, J.; Demengeot, J.; Leite, R.B.; Lilue, J.; et al. Signatures in SARS-CoV-2 Spike Protein Conferring Escape to Neutralizing Antibodies. PLoS Pathog. 2021, 17, e1009772. [Google Scholar] [CrossRef] [PubMed]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 Infects Cells after Viral Entry via Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, J.; Nguyen, L.N.T.; Adkins, J.L.; Schank, M.; Khanal, S.; Nguyen, L.N.; Dang, X.; Cao, D.; Krishna, B.; et al. Blockade of SARS-CoV-2 Spike Protein-Mediated Cell-Cell Fusion Using COVID-19 Convalescent Plasma. Sci. Rep. 2021, 11, 5558. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Modeling of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Proteins by Machine Learning and Physics-Based Refinement. bioRxiv 2020, bioRxiv:2020.03.25.008904. [Google Scholar] [CrossRef]
- Kalita, P.; Padhi, A.K.; Zhang, K.Y.J.; Tripathi, T. Design of a Peptide-Based Subunit Vaccine against Novel Coronavirus SARS-CoV-2. Microb. Pathog. 2020, 145, 104236. [Google Scholar] [CrossRef]
- Tripathy, S.P.; Ponnapati, M.; Jacobson, J.M.; Chatterjee, P. Sub-Picomolar Detection of SARS-CoV-2 RBD via Computationally-Optimized Peptide Beacons. bioRxiv 2021, bioRxiv:2021.06.04.447114. [Google Scholar]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and Bat RaTG13 Spike Glycoprotein Structures Inform on Virus Evolution and Furin-Cleavage Effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. Distinguishing Features of Current COVID-19 Vaccines: Knowns and Unknowns of Antigen Presentation and Modes of Action. NPJ Vaccines 2021, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Wurtzer, S.; Levert, M.; Dhenain, E.; Accrombessi, H.; Manco, S.; Fagour, N.; Goulet, M.; Boudaud, N.; Gaillard, L.; Bertrand, I.; et al. From Alpha to Omicron BA.2: New Digital RT-PCR Approach and Challenges for SARS-CoV-2 VOC Monitoring and Normalization of Variant Dynamics in Wastewater. Sci. Total Environ. 2022, 848, 157740. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Root-Mean-Square Deviation (RMSD) for trRosetta-Generated Models (Å) | Root-Mean-Square Deviation (RMSD) for AlphaFold v2.1.0-Generated Models (Å) |
|---|---|---|
| Original Variant | 0.705 for 1434 atoms | 1.257 for 1442 atoms |
| Alpha Variant | 0.694 for 1446 atoms | 1.596 for 1444 atoms |
| Beta Variant | 0.777 for 1434 atoms | 0.969 for 1505 atoms |
| Gamma Variant | 0.762 for 1474 atoms | 1.199 for 1488 atoms |
| Delta Variant | 0.785 for 1466 atoms | 1.407 for 1370 atoms |
| Omicron Variant | 0.732 for 1421 atoms | 0.828 for 1482 atoms |
| Type of Variants | Residues Mutated |
|---|---|
| Variant 1 | Mutations from both the Alpha and Delta variants |
| Variant 2 | Mutation only at N501Y residue in the Original variant |
| Variant 3 | Mutation only at S494P residue in the Original variant |
| Variant 4 | Mutation only at S494P residue in the Delta variant |
| Variant 5 | Mutation only at S494P residue in the Omicron variant |
| Variant 6 | Mutation only at P499S residue in the Original variant |
| Variant 7 | Mutation only at P499S residue in the Delta variant |
| Variant 8 | Mutation only at P499S residue in the Omicron variant |
| Variant 9 | Mutation only at T500R residue in the Original variant |
| Variant 10 | Mutation only at T500R residue in the Delta variant |
| Variant 11 | Mutation only at T500R residue in the Omicron variant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhowmick, S.; Jing, T.; Wang, W.; Zhang, E.Y.; Zhang, F.; Yang, Y. In Silico Protein Folding Prediction of COVID-19 Mutations and Variants. Biomolecules 2022, 12, 1665. https://doi.org/10.3390/biom12111665
Bhowmick S, Jing T, Wang W, Zhang EY, Zhang F, Yang Y. In Silico Protein Folding Prediction of COVID-19 Mutations and Variants. Biomolecules. 2022; 12(11):1665. https://doi.org/10.3390/biom12111665
Chicago/Turabian StyleBhowmick, Sumana, Tim Jing, Wei Wang, Elena Y. Zhang, Frank Zhang, and Yanmin Yang. 2022. "In Silico Protein Folding Prediction of COVID-19 Mutations and Variants" Biomolecules 12, no. 11: 1665. https://doi.org/10.3390/biom12111665
APA StyleBhowmick, S., Jing, T., Wang, W., Zhang, E. Y., Zhang, F., & Yang, Y. (2022). In Silico Protein Folding Prediction of COVID-19 Mutations and Variants. Biomolecules, 12(11), 1665. https://doi.org/10.3390/biom12111665