
Endoplasmic Reticulum Stress in Chronic Obstructive Pulmonary Disease: Mechanisms and Future Perspectives


Abstract
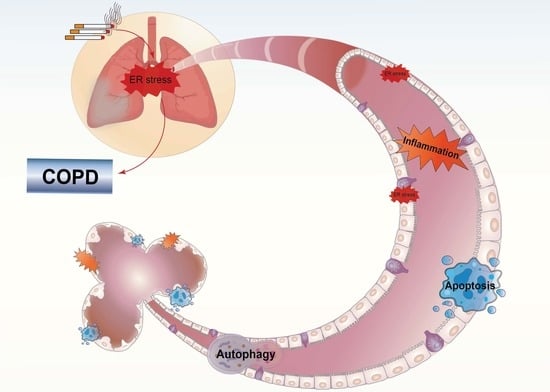
1. Introduction
2. The Function of the ER
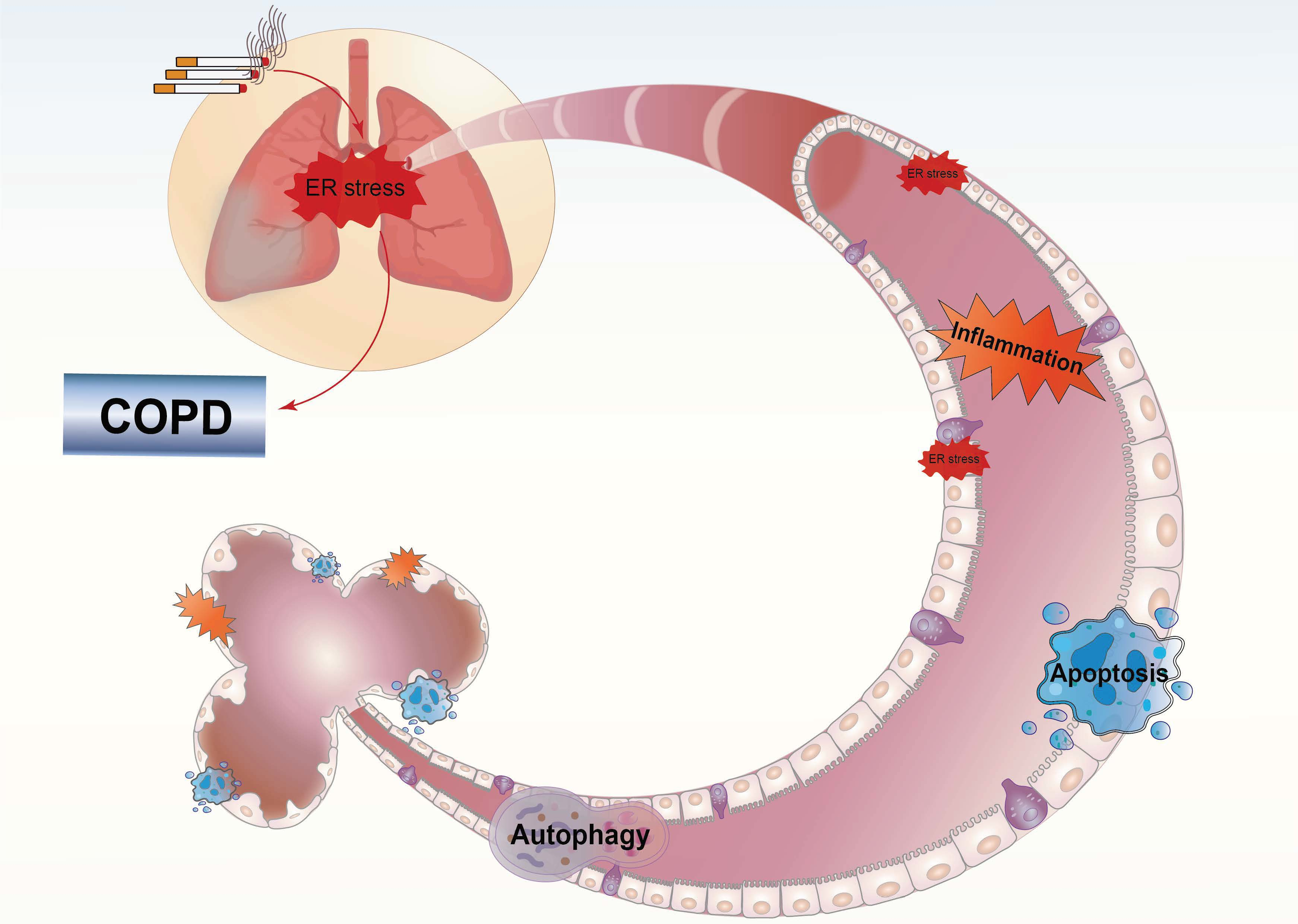
3. UPR Signaling
3.1. UPR Pathway Proteins
3.1.1. IRE1
3.1.2. PERK
3.1.3. ATF6
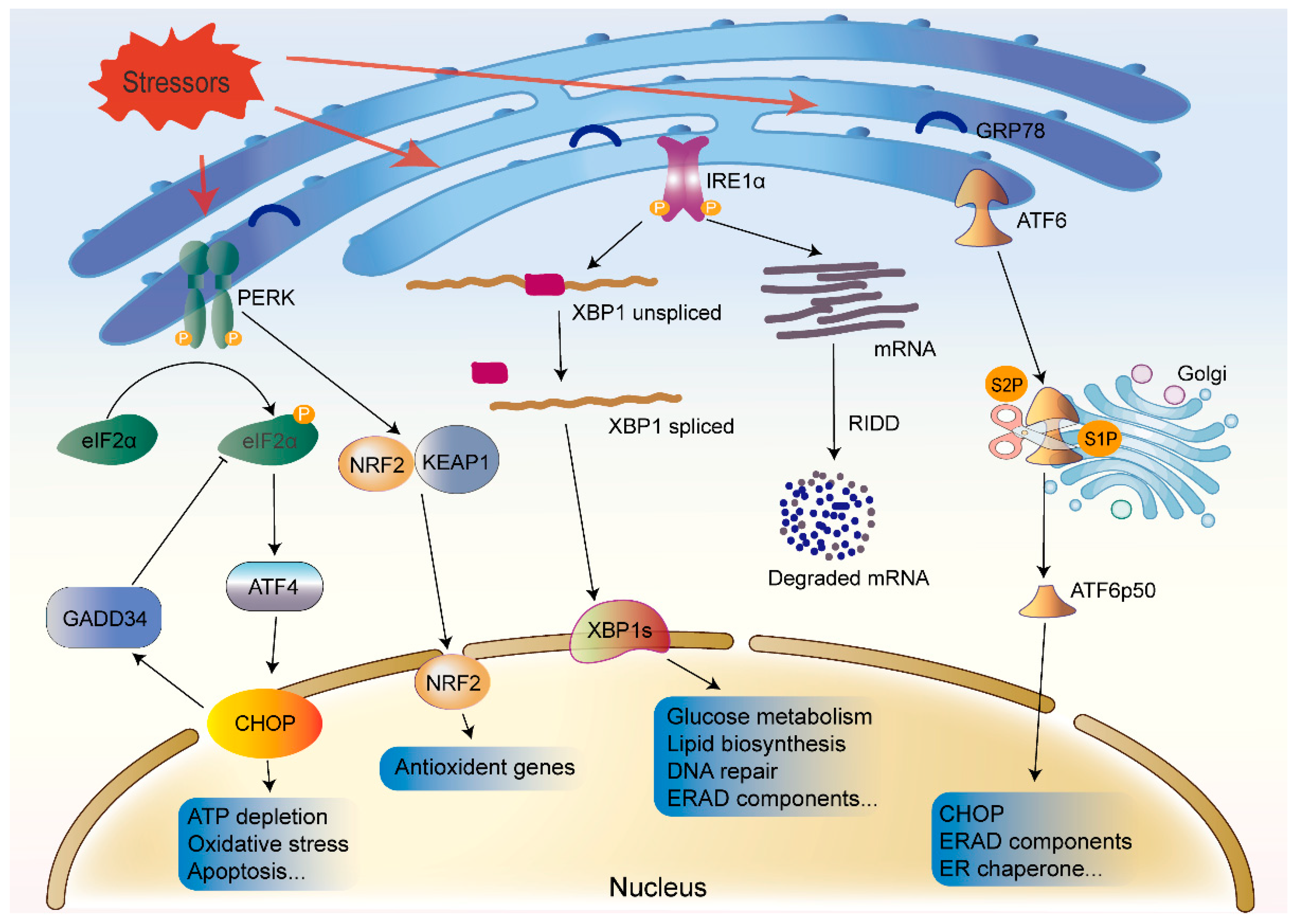
3.2. The UPR and Other Cellular Processes
3.2.1. Inflammation
3.2.2. ERAD
3.2.3. Autophagy
3.2.4. Cellular Senescence
3.2.5. Apoptosis
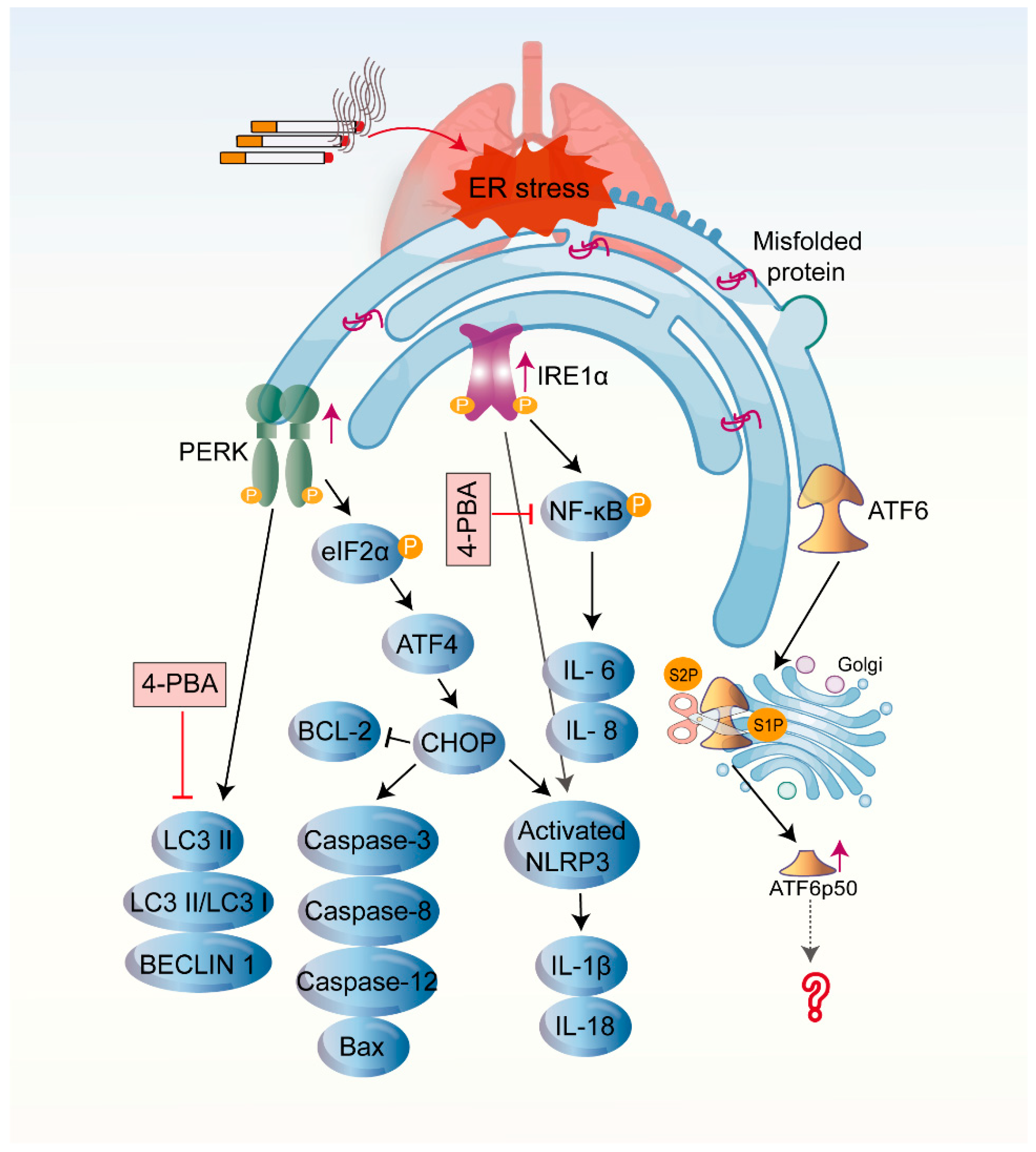
4. COPD and ER Stress
4.1. CS-Induced UPR in COPD
4.2. CS-Induced UPR and Other Cellular Processes in COPD
4.2.1. Inflammation
4.2.2. Cellular Senescence
4.2.3. Apoptosis
4.2.4. Autophagy
4.3. The Influence of Three Branches of UPR on the Pathogenesis of COPD
5. The UPR and Potential Therapeutic Interventions in COPD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol. Res. 2017, 119, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Dufey, E.; Sepulveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, H.; Yang, Y.; Lan, T.; Wang, H.; Wu, D. Hydrogen sulfide plays an important role by regulating endoplasmic reticulum stress in Diabetes-Related diseases. Int. J. Mol. Sci. 2022, 23, 7170. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, S.G. The unfolded protein response in chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 2), S138–S145. [Google Scholar] [CrossRef]
- Mccaffrey, K.; Braakman, I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235. [Google Scholar] [CrossRef]
- Naiel, S.; Tat, V.; Padwal, M.; Vierhout, M.; Mekhael, O.; Yousof, T.; Ayoub, A.; Abed, S.; Dvorkin-Gheva, A.; Ask, K. Protein misfolding and endoplasmic reticulum stress in chronic lung disease: Will Cell-Specific targeting be the key to the cure? Chest 2020, 157, 1207–1220. [Google Scholar] [CrossRef]
- Wang, F.; Ma, J.; Wang, J.; Chen, M.; Xia, H.; Yao, S.; Zhang, D. SIRT1 ameliorated septic associated-lung injury and macrophages apoptosis via inhibiting endoplasmic reticulum stress. Cell. Signal. 2022, 97, 110398. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Bhardwaj, A.; Dhawan, D.K.; Tandon, C.; Kaur, T. 4-PBA rescues hyperoxaluria induced nephrolithiasis by modulating urinary glycoproteins: Cross talk between endoplasmic reticulum, calcium homeostasis and mitochondria. Life Sci. 2022, 305, 120786. [Google Scholar] [CrossRef]
- Jiang, M.; Li, Z.; Zhu, G. The role of endoplasmic reticulum stress in the pathophysiology of periodontal disease. J. Periodontal Res. 2022, 57, 915–932. [Google Scholar] [CrossRef]
- Barreiro, E.; Salazar-Degracia, A.; Sancho-Munoz, A.; Aguilo, R.; Rodriguez-Fuster, A.; Gea, J. Endoplasmic reticulum stress and unfolded protein response in diaphragm muscle dysfunction of patients with stable chronic obstructive pulmonary disease. J. Appl. Physiol. (1985) 2019, 126, 1572–1586. [Google Scholar] [CrossRef]
- Wu, G.; Yuan, T.; Zhu, H.; Zhang, H.; Su, J.; Guo, L.; Zhou, Q.; Xiong, F.; Yu, Q.; Yang, P.; et al. Chrysophanol protects human bronchial epithelial cells from cigarette smoke extract (CSE)-induced apoptosis. Int. J. Mol. Epidemiol. Genet. 2020, 11, 39–45. [Google Scholar] [PubMed]
- Wang, H.L.; Chen, F.Q.; Wu, L.J. Ephedrine ameliorates chronic obstructive pulmonary disease (COPD) through restraining endoplasmic reticulum (ER) stress in vitro and in vivo. Int. Immunopharmacol. 2022, 103, 107842. [Google Scholar] [CrossRef] [PubMed]
- Weidner, J.; Jarenback, L.; Aberg, I.; Westergren-Thorsson, G.; Ankerst, J.; Bjermer, L.; Tufvesson, E. Endoplasmic reticulum, Golgi, and lysosomes are disorganized in lung fibroblasts from chronic obstructive pulmonary disease patients. Physiol. Rep. 2018, 6, e13584. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Sirish, P.; Diloretto, D.A.; Thai, P.N.; Chiamvimonvat, N. The critical roles of proteostasis and endoplasmic reticulum stress in atrial fibrillation. Front. Physiol. 2021, 12, 793171. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef]
- van Anken, E.; Braakman, I. Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 191–228. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Moon, H.W.; Han, H.G.; Jeon, Y.J. Protein quality control in the endoplasmic reticulum and cancer. Int. J. Mol. Sci. 2018, 19, 3020. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome system (UPS) and autophagy two main protein degradation machineries in response to cell stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef]
- Ozgur, R.; Uzilday, B.; Iwata, Y.; Koizumi, N.; Turkan, I. Interplay between the unfolded protein response and reactive oxygen species: A dynamic duo. J. Exp. Bot. 2018, 69, 3333–3345. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef] [PubMed]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Chaudhary, M.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum (ER) stress response failure in diseases. Trends Cell Biol. 2020, 30, 672–675. [Google Scholar] [CrossRef]
- Elfiky, A.A.; Baghdady, A.M.; Ali, S.A.; Ahmed, M.I. GRP78 targeting: Hitting two birds with a stone. Life Sci. 2020, 260, 118317. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Sims, S.G.; Cisney, R.N.; Lipscomb, M.M.; Meares, G.P. The role of endoplasmic reticulum stress in astrocytes. Glia 2022, 70, 5–19. [Google Scholar] [CrossRef]
- Raymundo, D.P.; Doultsinos, D.; Guillory, X.; Carlesso, A.; Eriksson, L.A.; Chevet, E. Pharmacological targeting of IRE1 in cancer. Trends Cancer 2020, 6, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Doultsinos, D.; Avril, T.; Lhomond, S.; Dejeans, N.; Guedat, P.; Chevet, E. Control of the unfolded protein response in health and disease. SLAS Discov. 2017, 22, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; Mccarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Karagoz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. eLife 2017, 6, e30700. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Liu, J.; Ibi, D.; Taniguchi, K.; Lee, J.; Herrema, H.; Akosman, B.; Mucka, P.; Salazar, H.M.; Uyar, M.F.; Park, S.W.; et al. Inflammation improves glucose homeostasis through IKKbeta-XBP1s interaction. Cell 2016, 167, 1052–1066. [Google Scholar] [CrossRef]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Adachi, M.; Zhao, S.; Hareyama, M.; Koong, A.C.; Luo, D.; Rando, T.A.; Imai, K.; Shinomura, Y. Preventing oxidative stress: A new role for XBP1. Cell Death Differ. 2009, 16, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Liew, C.W.; Lu, S.; Hu, J.; Martinez, R.; Hambro, B.; Kennedy, R.T.; Kulkarni, R.N. X-box binding protein 1 is essential for insulin regulation of pancreatic alpha-cell function. Diabetes 2013, 62, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Sone, M.; Zeng, X.; Larese, J.; Ryoo, H.D. A modified UPR stress sensing system reveals a novel tissue distribution of IRE1/XBP1 activity during normal Drosophila development. Cell Stress Chaperones 2013, 18, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Tak, J.; Kim, Y.S.; Kim, T.H.; Park, G.C.; Hwang, S.; Kim, S.G. Galpha12 overexpression in hepatocytes by ER stress exacerbates acute liver injury via ROCK1-mediated miR-15a and ALOX12 dysregulation. Theranostics 2022, 12, 1570–1588. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.R.; Chae, H.J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Wiese, W.; Siwecka, N.; Wawrzynkiewicz, A.; Rozpedek-Kaminska, W.; Kucharska, E.; Majsterek, I. IRE1alpha inhibitors as a promising therapeutic strategy in blood malignancies. Cancers 2022, 14, 2526. [Google Scholar] [CrossRef]
- Wodrich, A.; Scott, A.W.; Shukla, A.K.; Harris, B.T.; Giniger, E. The unfolded protein responses in health, aging, and neurodegeneration: Recent advances and future considerations. Front. Mol. Neurosci. 2022, 15, 831116. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, Y.; Lu, Y.; Xiang, M. ROS and endoplasmic reticulum stress in pulmonary disease. Front. Pharmacol. 2022, 13, 879204. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Wortel, I.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving stress: Modulation of ATF4-Mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Wang, J.; Wang, X.; Wang, Y.; Mao, H.; Liu, X. The PERK/Nrf2 pathway mediates endoplasmic reticulum stress-induced injury by upregulating endoplasmic reticulophagy in H9c2 cardiomyoblasts. Life Sci. 2019, 237, 116944. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, W.T.; Arrieta, A.; Blackwood, E.A.; Glembotski, C.C. Sledgehammer to scalpel: Broad challenges to the heart and other tissues yield specific cellular responses via transcriptional regulation of the ER-Stress master regulator ATF6alpha. Int. J. Mol. Sci. 2020, 21, 1134. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell. 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Li, Y.; Schwabe, R.F.; Devries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Choi, H.J.; Yang, H.; Do, K.H.; Kim, J.; Lee, D.W.; Moon, Y. Endoplasmic reticulum stress-activated C/EBP homologous protein enhances nuclear factor-kappaB signals via repression of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2010, 285, 35330–35339. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Zhu, J.; Zhang, M.; Li, C.; Zhou, X.; Zhou, M.; Ke, M.; Busuttil, R.W.; Ying, Q.L.; Kupiec-Weglinski, J.W.; et al. The myeloid heat shock transcription factor 1/beta-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology 2016, 64, 1683–1698. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nunez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and Caspase-2-Driven mitochondrial damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef]
- Qu, J.; Zou, T.; Lin, Z. The roles of the Ubiquitin-Proteasome system in the endoplasmic reticulum stress pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. EMBO J. 2019, 38, e100999. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, J.; Sun, H.; Jiang, C.; Dong, Y.; Shan, Q.; Su, S.; Xie, Y.; Xu, N.; Lou, X.; et al. Ubiquitination of inositol-requiring enzyme 1 (IRE1) by the E3 ligase CHIP mediates the IRE1/TRAF2/JNK pathway. J. Biol. Chem. 2014, 289, 30567–30577. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Ishiguro, M.; Niinuma, Y.; Uesugi, M.; Nomura, Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett. 2002, 532, 147–152. [Google Scholar] [CrossRef]
- Xu, Y.; Melo-Cardenas, J.; Zhang, Y.; Gau, I.; Wei, J.; Montauti, E.; Zhang, Y.; Gao, B.; Jin, H.; Sun, Z.; et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight 2019, 4, e121887. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2020, 39, e105826. [Google Scholar] [CrossRef]
- Huang, Y.; Sun, Y.; Cao, Y.; Sun, H.; Li, M.; You, H.; Su, D.; Li, Y.; Liang, X. HRD1 prevents apoptosis in renal tubular epithelial cells by mediating eIF2alpha ubiquitylation and degradation. Cell Death Dis. 2017, 8, 3202. [Google Scholar] [CrossRef]
- Yamamoto, K.; Suzuki, N.; Wada, T.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Mori, K. Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J. Biochem. 2008, 144, 477–486. [Google Scholar] [CrossRef]
- Baird, L.; Tsujita, T.; Kobayashi, E.H.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yamamoto, M. A homeostatic shift facilitates endoplasmic reticulum proteostasis through transcriptional integration of proteostatic stress response pathways. Mol. Cell. Biol. 2017, 37, e00439-16. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, Z.; Luo, W.; Wang, L.; Zhou, H.; Nie, C. JNK initiates Beclin-1 dependent autophagic cell death against Akt activation. Exp. Cell Res. 2022, 414, 113105. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Q.; Bai, Q.; Tong, T.; Zhou, Y.; Li, Z.; Xiao, C.; Chen, L. Chlamydia psittaci Induces Autophagy in Human Bronchial Epithelial Cells via PERK and IRE1alpha, but Not ATF6 Pathway. Infect. Immun. 2022, 90, e7922. [Google Scholar] [CrossRef] [PubMed]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Joe, Y.; Kim, S.K.; Park, S.U.; Park, J.; Chen, Y.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.J.; et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2alpha-ATF4 pathway. Free Radic. Biol. Med. 2017, 110, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef]
- Sun, X.; Li, W.; Deng, Y.; Dong, B.; Sun, Y.; Xue, Y.; Wang, Y. Hepatic conditional knockout of ATF6 exacerbates liver metabolic damage by repressing autophage through MTOR pathway. Biochem. Biophys. Res. Commun. 2018, 505, 45–50. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Dai, C.; Tang, S.; Yang, X.; Li, D.; Zhao, K.; Xiao, X. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol. Toxicol. 2016, 32, 141–152. [Google Scholar] [CrossRef]
- L’Hote, V.; Courbeyrette, R.; Pinna, G.; Cintrat, J.C.; Le Pavec, G.; Delaunay-Moisan, A.; Mann, C.; Thuret, J.Y. Ouabain and chloroquine trigger senolysis of BRAF-V600E-induced senescent cells by targeting autophagy. Aging Cell 2021, 20, e13447. [Google Scholar] [CrossRef]
- Lee, D.; Hokinson, D.; Park, S.; Elvira, R.; Kusuma, F.; Lee, J.M.; Yun, M.; Lee, S.G.; Han, J. ER stress induces cell cycle arrest at the G2/M phase through eIF2alpha phosphorylation and GADD45alpha. Int. J. Mol. Sci. 2019, 20, 6309. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef] [PubMed]
- Cormenier, J.; Martin, N.; Desle, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6alpha arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Franco-Juarez, B.; Coronel-Cruz, C.; Hernandez-Ochoa, B.; Gomez-Manzo, S.; Cardenas-Rodriguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Avila, L.M.; Ortega-Cuellar, D. TFEB; Beyond its role as an autophagy and lysosomes regulator. Cells. 2022, 11, 3153. [Google Scholar] [CrossRef]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef]
- Rotem-Dai, N.; Muraleedharan, A.; Livneh, E. PKCeta promotes Stress-Induced autophagy and senescence in breast cancer cells, presenting a target for therapy. Pharmaceutics 2022, 14, 1704. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; Mcmanus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhang, P.; Xia, N.; Wu, S.; Liu, B.; Weng, L.; Shang, M. GPx8 regulates apoptosis and autophagy in esophageal squamous cell carcinoma through the IRE1/JNK pathway. Cell. Signal. 2022, 93, 110307. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Wu, H.; Liu, D.; Yang, J.; Yang, J.; Ding, J.W.; Zhou, G.; Zhang, J.; Zhang, D. CFLIPL alleviates myocardial Ischemia-Reperfusion injury by inhibiting endoplasmic reticulum stress. Cardiovasc. Drugs Ther. 2021, 1–14. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Shao, C.; Yu, Z.; Luo, T.; Zhou, B.; Song, Q.; Li, Z.; Yu, X.; Jiang, S.; Zhou, Y.; Dong, W.; et al. Chitosan-Coated selenium nanoparticles attenuate PRRSV replication and ROS/JNK-Mediated apoptosis in vitro. Int. J. Nanomed. 2022, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.Y.; Hsieh, M.J.; Lo, Y.S.; Lin, C.C.; Chuang, Y.C.; Chen, M.K.; Chou, M.C. Xanthohumol targets the JNK1/2 signaling pathway in apoptosis of human nasopharyngeal carcinoma cells. Environ. Toxicol. 2022, 37, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Lee, K.T.; Lim, M.C.; Choi, J.H. TRPV1 antagonist DWP05195 induces ER Stress-Dependent apoptosis through the ROS-p38-CHOP pathway in human ovarian cancer cells. Cancers 2020, 12, 1702. [Google Scholar] [CrossRef]
- Su, S.; Zhang, D.; Liu, J.; Zhao, H.; Tang, X.; Che, H.; Wang, Q.; Ren, W.; Zhen, D. Folate ameliorates homocysteine-induced osteoblast dysfunction by reducing endoplasmic reticulum stress-activated PERK/ATF-4/CHOP pathway in MC3T3-E1 cells. J. Bone Miner. Metab. 2022, 40, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Park, J.Y.; Pyo, J.; Lee, S.Y.; Kim, H.S. Induction of DR5-Dependent apoptosis by PGA2 through ATF4-CHOP pathway. Molecules 2022, 27, 3804. [Google Scholar] [CrossRef]
- Zhang, F.; Ni, Z.; Zhao, S.; Wang, Y.; Chang, X.; Zhou, Z. Flurochloridone induced cell apoptosis via ER stress and eIF2alpha-ATF4/ATF6-CHOP-Bim/Bax signaling pathways in mouse TM4 sertoli cells. Int. J. Environ. Res. Public Health. 2022, 19, 4564. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef]
- Chang, X.; Tian, M.; Zhang, Q.; Liu, F.; Gao, J.; Li, S.; Liu, H.; Hou, X.; Li, L.; Li, C.; et al. Grape seed proanthocyanidin extract ameliorates cisplatin-induced testicular apoptosis via PI3K/Akt/mTOR and endoplasmic reticulum stress pathways in rats. J. Food Biochem. 2021, 45, e13825. [Google Scholar] [CrossRef]
- Lugg, S.T.; Scott, A.; Parekh, D.; Naidu, B.; Thickett, D.R. Cigarette smoke exposure and alveolar macrophages: Mechanisms for lung disease. Thorax 2022, 77, 94–101. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Duan, X.; Ji, R.; Perez, O.; Liu, C.; Merali, S. Cigarette smoke induces an unfolded protein response in the human lung: A proteomic approach. Am. J. Respir. Cell Mol. Biol. 2008, 38, 541–550. [Google Scholar] [CrossRef]
- Merali, S.; Barrero, C.A.; Bowler, R.P.; Chen, D.E.; Criner, G.; Braverman, A.; Litwin, S.; Yeung, A.; Kelsen, S.G. Analysis of the plasma proteome in COPD: Novel low abundance proteins reflect the severity of lung remodeling. COPD 2014, 11, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, M.O.; Kim, V.; Cornwell, W.D.; Rogers, T.J.; Kosmider, B.; Bahmed, K.; Barrero, C.; Merali, S.; Shetty, N.; Kelsen, S.G. Secretion of the endoplasmic reticulum stress protein, GRP78, into the BALF is increased in cigarette smokers. Respir. Res. 2017, 18, 78. [Google Scholar] [CrossRef]
- Bon, J.; Kahloon, R.; Zhang, Y.; Xue, J.; Fuhrman, C.R.; Tan, J.; Burger, M.; Kass, D.J.; Csizmadia, E.; Otterbein, L.; et al. Autoreactivity to glucose regulated protein 78 links emphysema and osteoporosis in smokers. PLoS ONE 2014, 9, e105066. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crane, E.D.; Al-Hashimi, A.A.; Chen, J.; Lynn, E.G.; Won, K.D.; Lhotak, S.; Naeim, M.; Platko, K.; Lebeau, P.; Byun, J.H.; et al. Anti-GRP78 autoantibodies induce endothelial cell activation and accelerate the development of atherosclerotic lesions. JCI Insight 2018, 3, e99363. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef]
- Tang, F.; Ling, C. Curcumin ameliorates chronic obstructive pulmonary disease by modulating autophagy and endoplasmic reticulum stress through regulation of SIRT1 in a rat model. J. Int. Med. Res. 2019, 47, 4764–4774. [Google Scholar] [CrossRef]
- Wang, Y.; Su, N.X.; Pan, S.G.; Ge, X.P.; Dai, X.P. Fengbaisan suppresses endoplasmic reticulum stress by up-regulating SIRT1 expression to protect rats with chronic obstructive pulmonary diseases. Pharm. Biol. 2020, 58, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.; Hu, R.; Dai, A.; Tan, S.; Ouyang, Q.; Fu, D.; Jiang, D. The role of endoplasmic reticulum stress in emphysema results from cigarette smoke exposure. Cell. Physiol. Biochem. 2011, 28, 725–732. [Google Scholar] [CrossRef]
- Kenche, H.; Baty, C.J.; Vedagiri, K.; Shapiro, S.D.; Blumental-Perry, A. Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J. 2013, 27, 965–977. [Google Scholar] [CrossRef]
- Geraghty, P.; Wallace, A.; D’Armiento, J.M. Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4-C/EBP homologous protein mediated process. Int. J. Chron. Obstruct. Pulmon. Dis. 2011, 6, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, J.; Luo, Y. The effect of various durations of cigarette smoke exposure on muscle fibre remodeling in rat diaphragms. Biomed. Pharmacother. 2019, 117, 109053. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Morrow, J.; Owen, C.A.; Qiu, W.; Glass, K.; Lao, T.; Jiang, Z.; Perrella, M.A.; Silverman, E.K.; Zhou, X.; et al. Transcriptomic analysis of lung tissue from cigarette Smoke-Induced emphysema murine models and human chronic obstructive pulmonary disease show shared and distinct pathways. Am. J. Respir. Cell Mol. Biol. 2017, 57, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yamashita, Y.; Tanaka, T.; Takaki, M.; Le, M.N.; Yoshida, L.M.; Morimoto, K. Cigarette smoke induces endoplasmic reticulum stress and suppresses efferocytosis through the activation of RhoA. Sci. Rep. 2020, 10, 12620. [Google Scholar] [CrossRef]
- Yoo, Y.M.; Jung, E.M.; Jeon, B.H.; Tran, D.N.; Jeung, E.B. Cigarette smoke extraxt influences intracellular calcium concentration in A549 cells. J. Physiol. Pharmacol. 2020, 71, 679–687. [Google Scholar] [CrossRef]
- Kenche, H.; Ye, Z.W.; Vedagiri, K.; Richards, D.M.; Gao, X.H.; Tew, K.D.; Townsend, D.M.; Blumental-Perry, A. Adverse outcomes associated with cigarette smoke radicals related to damage to protein-disulfide isomerase. J. Biol. Chem. 2016, 291, 4763–4778. [Google Scholar] [CrossRef] [PubMed]
- Somborac-Bacura, A.; van der Toorn, M.; Franciosi, L.; Slebos, D.J.; Zanic-Grubisic, T.; Bischoff, R.; van Oosterhout, A.J. Cigarette smoke induces endoplasmic reticulum stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp. Physiol. 2013, 98, 316–325. [Google Scholar] [CrossRef]
- Yamada, Y.; Tomaru, U.; Ishizu, A.; Ito, T.; Kiuchi, T.; Ono, A.; Miyajima, S.; Nagai, K.; Higashi, T.; Matsuno, Y.; et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice. Lab. Investig. 2015, 95, 625–634. [Google Scholar] [CrossRef]
- Mahalanobish, S.; Dutta, S.; Saha, S.; Sil, P.C. Melatonin induced suppression of ER stress and mitochondrial dysfunction inhibited NLRP3 inflammasome activation in COPD mice. Food Chem. Toxicol. 2020, 144, 111588. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.Z.; Wang, W. Inhibition of endoplasmic reticulum stress alleviates cigarette smoke-induced airway inflammation and emphysema. Oncotarget 2017, 8, 77685–77695. [Google Scholar] [CrossRef]
- Barnes, P.J. Senescence in COPD and its comorbidities. Annu. Rev. Physiol. 2017, 79, 517–539. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef]
- Koloko, N.M.; Brandsma, C.A.; Gosens, R.; Prakash, Y.S.; Burgess, J.K. The stress of lung aging: Endoplasmic reticulum and senescence Tete-a-Tete. Physiology (Bethesda) 2021, 36, 150–159. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; de Vries, M.; Timens, W.; van den Berge, M.; Demaria, M.; Oliver, B.; Heijink, I.H.; Brandsma, C.A. Link between increased cellular senescence and extracellular matrix changes in COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L48–L60. [Google Scholar] [CrossRef]
- Monkley, S.; Overed-Sayer, C.; Parfrey, H.; Rassl, D.; Crowther, D.; Escudero-Ibarz, L.; Davis, N.; Carruthers, A.; Berks, R.; Coetzee, M.; et al. Sensitization of the UPR by loss of PPP1R15A promotes fibrosis and senescence in IPF. Sci. Rep. 2021, 11, 21584. [Google Scholar] [CrossRef]
- Lin, L.; Hou, G.; Han, D.; Kang, J.; Wang, Q. Ursolic acid protected lung of rats from damage induced by cigarette smoke extract. Front. Pharmacol. 2019, 10, 700. [Google Scholar] [CrossRef]
- Murata, K.; Fujita, N.; Takahashi, R. Ninjinyoeito ameliorated cigarette smoke extract-induced apoptosis and inflammation through JNK signaling inhibition in human lung fibroblasts. BMC Complement. Med. Ther. 2022, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, Y.; Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med. 2008, 45, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, Y.; Hiramatsu, N.; Kato, H.; Sakoh, T.; Nakajima, S.; Hayakawa, K.; Saito, Y.; Johno, H.; Takahashi, S.; Gu, L.; et al. Induction of CCAAT/enhancer-binding protein-homologous protein by cigarette smoke through the superoxide anion-triggered PERK-eIF2alpha pathway. Toxicology 2011, 287, 105–112. [Google Scholar] [CrossRef]
- Lee, J.; Jang, J.; Park, S.M.; Yang, S.R. An update on the role of nrf2 in respiratory disease: Molecular mechanisms and therapeutic approaches. Int. J. Mol. Sci. 2021, 22, 8406. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P.A.; Ferrari, M.; Stranieri, C.; Vallerio, P.; Mozzini, C.; Garbin, U.; Zambon, G.; Cominacini, L. Nrf2 expression is increased in peripheral blood mononuclear cells derived from mild-moderate ex-smoker COPD patients with persistent oxidative stress. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yin, Y.; Hou, G.; Han, D.; Kang, J.; Wang, Q. Ursolic acid attenuates cigarette smoke-induced emphysema in rats by regulating PERK and Nrf2 pathways. Pulm. Pharmacol. Ther. 2017, 44, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, M.; Li, D.; Zhao, Y.; Yu, D.; Zhang, R.; Chen, W.; Pi, J.; Chen, R.; Cui, L.; et al. Effects of Real-Ambient PM2.5 Exposure on Lung Damage Modulated by Nrf2−/−. Front. Pharmacol. 2021, 12, 662664. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef]
- Kono, Y.; Colley, T.; To, M.; Papaioannou, A.I.; Mercado, N.; Baker, J.R.; To, Y.; Abe, S.; Haruki, K.; Ito, K.; et al. Cigarette smoke-induced impairment of autophagy in macrophages increases galectin-8 and inflammation. Sci. Rep. 2021, 11, 335. [Google Scholar] [CrossRef]
- Li, Y.; Yu, G.; Yuan, S.; Tan, C.; Lian, P.; Fu, L.; Hou, Q.; Xu, B.; Wang, H. Cigarette Smoke-Induced pulmonary inflammation and autophagy are attenuated in Ephx2-Deficient mice. Inflammation 2017, 40, 497–510. [Google Scholar] [CrossRef]
- He, B.; Chen, Q.; Zhou, D.; Wang, L.; Liu, Z. Role of reciprocal interaction between autophagy and endoplasmic reticulum stress in apoptosis of human bronchial epithelial cells induced by cigarette smoke extract. IUBMB Life 2019, 71, 66–80. [Google Scholar] [CrossRef]
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kadota, T.; Tsubouchi, K.; Yoshida, M.; Minagawa, S.; Hara, H.; Kawamoto, H.; Watanabe, N.; et al. Chaperone-Mediated Autophagy Suppresses Apoptosis via Regulation of the Unfolded Protein Response during Chronic Obstructive Pulmonary Disease Pathogenesis. J. Immunol. 2020, 205, 1256–1267. [Google Scholar] [CrossRef]
- Lin, F.; Liao, C.; Zhang, J.; Sun, Y.; Lu, W.; Bai, Y.; Liao, Y.; Li, M.; Qi, Y.; Chen, Y. Hydrogen sulfide inhibits bronchial epithelial cell epithelial mesenchymal transition through regulating endoplasm reticulum stress. Front. Mol. Biosci. 2022, 9, 828766. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Rab, A.; Twitty, G.; Stevenson, L.; Fortenberry, J.; Piotrowski, A.; Dumanski, J.P.; Bebok, Z. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J. Biol. Chem. 2008, 283, 12154–12165. [Google Scholar] [CrossRef] [PubMed]
- Dransfield, M.; Rowe, S.; Vogelmeier, C.F.; Wedzicha, J.; Criner, G.J.; Han, M.K.; Martinez, F.J.; Calverley, P. Cystic fibrosis transmembrane conductance regulator: Roles in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2022, 205, 631–640. [Google Scholar] [CrossRef]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway epithelium dysfunction in cystic fibrosis and COPD. Mediat. Inflamm. 2018, 2018, 1309746. [Google Scholar] [CrossRef]
- Kaza, N.; Lin, V.Y.; Stanford, D.; Hussain, S.S.; Falk, L.E.; Kim, H.; Borgonovi, M.; Conrath, K.; Mutyam, V.; Byzek, S.A.; et al. Evaluation of a novel CFTR potentiator in COPD ferrets with acquired CFTR dysfunction. Eur. Respir. J. 2022, 60, 2101581. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ahmad, I.; Lam, A.; Carlisle, M.A.; Li, C.; Wells, J.M.; Raju, S.V.; Athar, M.; Rowe, S.M.; Dransfield, M.T.; et al. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight 2018, 3, e120694. [Google Scholar] [CrossRef]
- Lin, F.; Liao, C.; Sun, Y.; Zhang, J.; Lu, W.; Bai, Y.; Liao, Y.; Li, M.; Ni, X.; Hou, Y.; et al. Hydrogen sulfide inhibits cigarette Smoke-Induced endoplasmic reticulum stress and apoptosis in bronchial epithelial cells. Front. Pharmacol. 2017, 8, 675. [Google Scholar] [CrossRef]
- Ding, H.B.; Liu, K.X.; Huang, J.F.; Wu, D.W.; Chen, J.Y.; Chen, Q.S. Protective effect of exogenous hydrogen sulfide on pulmonary artery endothelial cells by suppressing endoplasmic reticulum stress in a rat model of chronic obstructive pulmonary disease. Biomed. Pharmacother. 2018, 105, 734–741. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhang, W.; Qiao, J.; Peng, Z.; Chai, X. Melatonin protects against COPD by attenuating apoptosis and endoplasmic reticulum stress via upregulating SIRT1 expression in rats. Can. J. Physiol. Pharmacol. 2019, 97, 386–391. [Google Scholar] [CrossRef]
- Yu, G.; Zeng, X.; Wang, H.; Hou, Q.; Tan, C.; Xu, Q.; Wang, H. 14,15-epoxyeicosatrienoic Acid suppresses cigarette smoke extract-induced apoptosis in lung epithelial cells by inhibiting endoplasmic reticulum stress. Cell. Physiol. Biochem. 2015, 36, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Li, F.; Liu, Y.W.; Shi, Y.J.; Li, Z.H.; Cao, G.K.; Zhu, W. Adiponectin attenuates endoplasmic reticulum stress and alveolar epithelial apoptosis in COPD rats. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4999–5007. [Google Scholar] [PubMed]
- Fan, L.; Li, L.; Yu, X.; Liang, Z.; Cai, T.; Chen, Y.; Xu, Y.; Hu, T.; Wu, L.; Lin, L. Jianpiyifei II granules suppress apoptosis of bronchial epithelial cells in chronic obstructive pulmonary disease via inhibition of the reactive oxygen Species-Endoplasmic reticulum Stress-Ca2+ signaling pathway. Front. Pharmacol. 2020, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Luo, B.; Ting, Y.; He, S.; Xie, L.; Sun, S. SIRT1 attenuates endoplasmic reticulum stress and apoptosis in rat models of COPD. Growth Factors 2020, 38, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zhang, L.; Yao, J.; Yao, S.; Yuan, T. AMPK alleviates endoplasmic reticulum stress by inducing the ER-chaperone ORP150 via FOXO1 to protect human bronchial cells from apoptosis. Biochem. Biophys. Res. Commun. 2018, 497, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Park, S.Y.; Park, S.; Hong, G.H.; Moon, K.A.; Kim, Y.S.; Oh, Y.M.; Kwon, H.S.; Kim, T.B.; Moon, H.B.; et al. Progranulin protects lung epithelial cells from cigarette smoking-induced apoptosis. Respirology 2017, 22, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Michaeloudes, C.; Liang, Y.; Bhavsar, P.K.; Chung, K.F.; Ip, M.; Mak, J. ORMDL3 regulates cigarette smoke-induced endoplasmic reticulum stress in airway smooth muscle cells. J. Allergy Clin. Immunol. 2022, 149, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ye, L.; Zhu, G.; Zeng, Y.; Yang, C.; Cai, H.; Mo, Y.; Song, X.; Gao, X.; Peng, W.; et al. ROS-Responsive miR-150-5p downregulation contributes to cigarette Smoke-Induced COPD via targeting IRE1alpha. Oxid. Med. Cell. Longev. 2022, 2022, 5695005. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model(s) | Reference(s) | |
|---|---|---|
| ER stress inhibitor | ||
| 4-PBA | In vivo (murine); in vitro (BEAS-2B) | [130] |
| Salubrinal | Human subjects; in vivo (murine); in vitro (HBECs) | [157] |
| H2S | Human subjects; in vivo (rat); in vitro (16HBE) | [152,158,159] |
| Melatonin | In vivo (murine, rat); in vitro (L-132) | [129,160] |
| Ursolic acid | In vivo (rat) | [137,143] |
| 14,15-EET | In vitro (BEAS-2B) | [161] |
| Adiponectin | In vivo (rat) | [162] |
| Herbal ingredients or mixtures | In vivo (murine, rat); in vitro (BEAS-2B, 16HBE, A549, HBECs, HFL1) | [12,117,118,138,163] |
| Key mediator | ||
| SIRT1 | In vivo (rat); in vitro (A549) | [117,118,160,164] |
| AMPK | In vitro (HBEpC) | [165] |
| PGRN | In vivo (murine); in vitro (A549) | [166] |
| ORMDL3 | In vitro (HASMC) | [167] |
| miR-150-5p | Human subjects; in vivo (murine); in vitro (HBECs) | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Yang, A.; Yu, G.; Wang, H. Endoplasmic Reticulum Stress in Chronic Obstructive Pulmonary Disease: Mechanisms and Future Perspectives. Biomolecules 2022, 12, 1637. https://doi.org/10.3390/biom12111637
Yu Y, Yang A, Yu G, Wang H. Endoplasmic Reticulum Stress in Chronic Obstructive Pulmonary Disease: Mechanisms and Future Perspectives. Biomolecules. 2022; 12(11):1637. https://doi.org/10.3390/biom12111637
Chicago/Turabian StyleYu, Yue, Ailin Yang, Ganggang Yu, and Haoyan Wang. 2022. "Endoplasmic Reticulum Stress in Chronic Obstructive Pulmonary Disease: Mechanisms and Future Perspectives" Biomolecules 12, no. 11: 1637. https://doi.org/10.3390/biom12111637
APA StyleYu, Y., Yang, A., Yu, G., & Wang, H. (2022). Endoplasmic Reticulum Stress in Chronic Obstructive Pulmonary Disease: Mechanisms and Future Perspectives. Biomolecules, 12(11), 1637. https://doi.org/10.3390/biom12111637