
Improving Precise Genome Editing Using Donor DNA/gRNA Hybrid Duplex Generated by Complementary Bases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. Construction of Plasmids
2.3. Yeast Transformation
2.4. In Vitro Synthesis of gRNA
2.5. Preparation of DGybrid
2.6. Native-PAGE Analysis of DGybrid
2.7. Introduction of DGybrid into Cells by Electroporation
2.8. Evaluation of Genome-Editing Efficiency
2.9. Evaluation of Introduction Efficiency of Nucleic Acids into Cells Using Flow Cytometry
3. Results
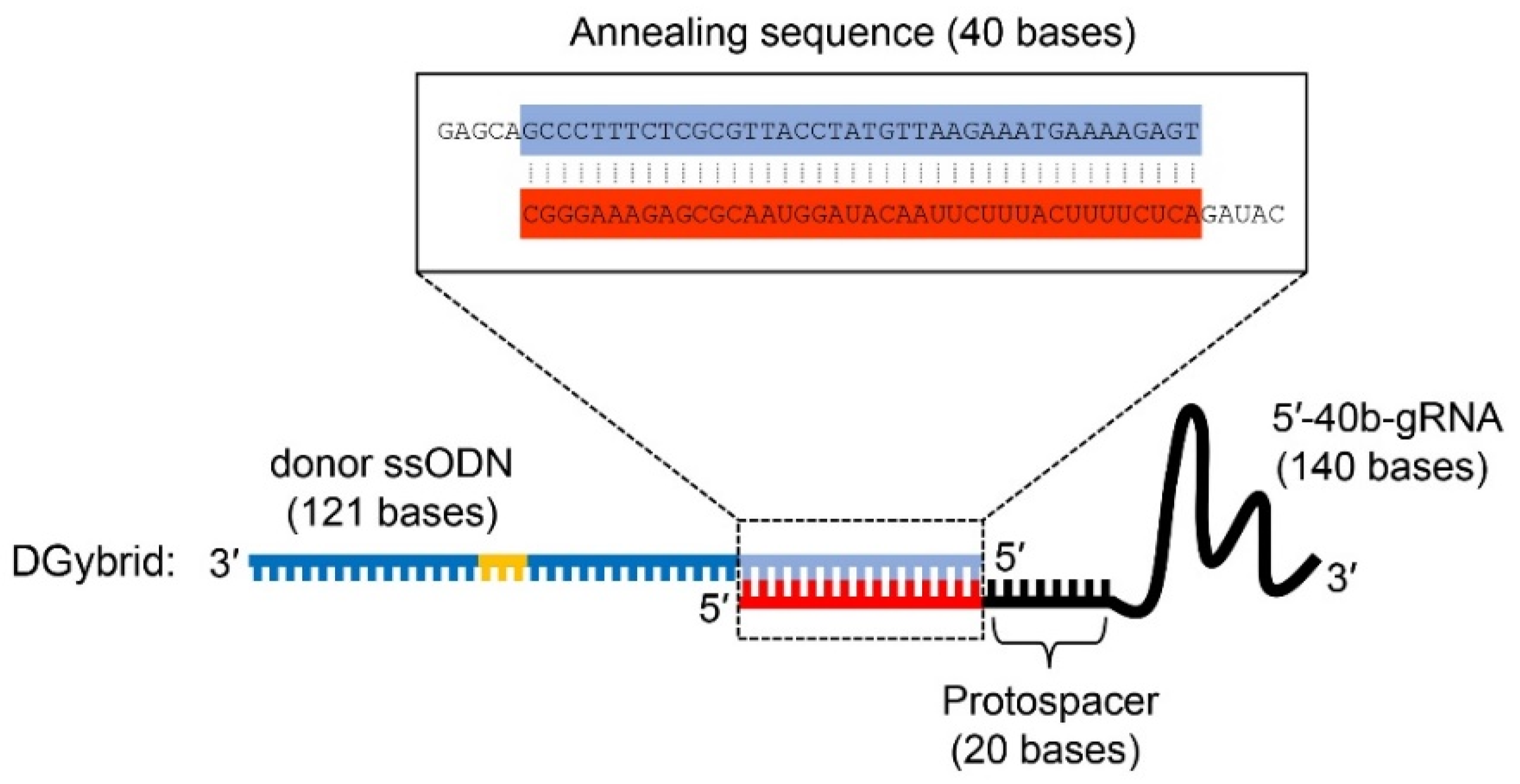
3.1. Preparation of DGybrid
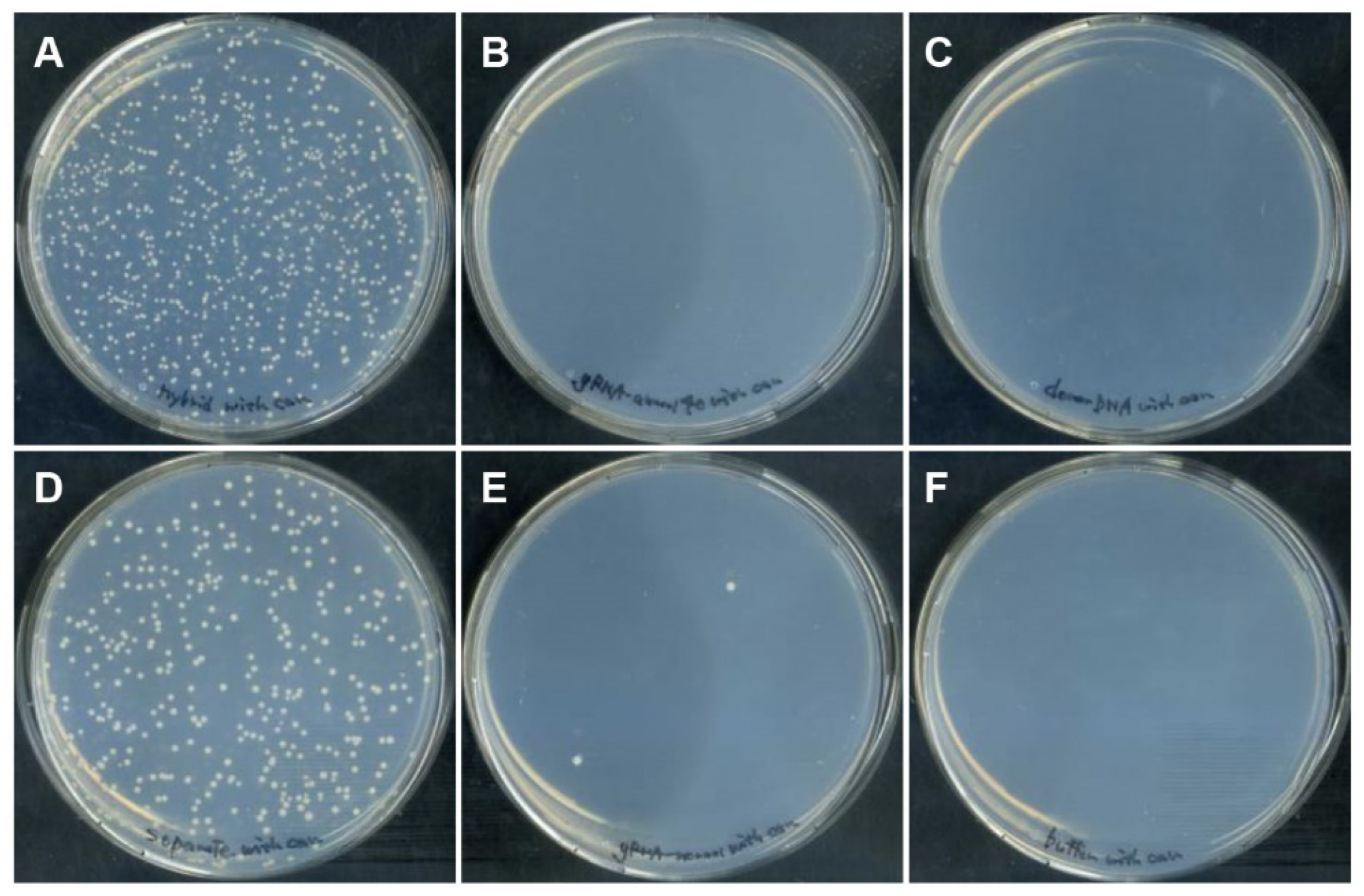
3.2. Genome Editing via HDR with DGybrid
3.3. Properties of DNA/RNA Hybrid Contributing to Improved Editing Efficiency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Adli, M. The CRISPR Tool Kit for Genome Editing and Beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Tsai, Y.T.; Justus, S.; Lee, T.T.; Zhang, L.; Lin, C.S.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 2016, 24, 1388–1394. [Google Scholar] [CrossRef]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.J.; Mitros, T.; Munoz, D.P.; Boffelli, D.; et al. Selection-free Genome Editing of the Sickle Mutation in Human Adult Hematopoietic Stem/progenitor Cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-mediated Genome Engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef]
- Sanchez-Leon, S.; Gil-Humanes, J.; Ozuna, C.V.; Gimenez, M.J.; Sousa, C.; Voytas, D.F.; Barro, F. Low-gluten, Nontransgenic Wheat Engineered with CRISPR/Cas9. Plant Biotechnol. J. 2018, 16, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Jakociunas, T.; Jensen, M.K.; Keasling, J.D. CRISPR/Cas9 Advances Engineering of Microbial Cell Factories. Metab. Eng. 2016, 34, 44–59. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-strand DNA Break Repair by The Nonhomologous DNA End-joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors Determining DNA Double-strand Break Repair Pathway Choice in G2 Phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome Engineering in Saccharomyces cerevisiae Using CRISPR-Cas Systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef]
- Yoshimi, K.; Kunihiro, Y.; Kaneko, T.; Nagahora, H.; Voigt, B.; Mashimo, T. ssODN-mediated Knock-in with CRISPR-Cas for Large Genomic Regions in Zygotes. Nat. Commun. 2016, 7, 10431. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.K.; Puchta, H. CRISPR/Cas-mediated Gene Targeting in Plants: Finally a Turn for the Better for Homologous Recombination. Plant Cell Rep. 2019, 38, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Carlson-Stevermer, J.; Abdeen, A.A.; Kohlenberg, L.; Goedland, M.; Molugu, K.; Lou, M.; Saha, K. Assembly of CRISPR Ribonucleoproteins with Biotinylated Oligonucleotides via an RNA Aptamer for Precise Gene Editing. Nat. Commun. 2017, 8, 1711. [Google Scholar] [CrossRef]
- Li, G.; Wang, H.; Zhang, X.; Wu, Z.; Yang, H. A Cas9-transcription Factor Fusion Protein Enhances Homology-directed Repair Efficiency. J. Biol. Chem. 2021, 296, 100525. [Google Scholar] [CrossRef]
- Aird, E.J.; Lovendahl, K.N.; St Martin, A.; Harris, R.S.; Gordon, W.R. Increasing Cas9-mediated Homology-directed Repair Efficiency Through Covalent Tethering of DNA Repair Template. Commun. Biol. 2018, 1, 54. [Google Scholar] [CrossRef]
- Savic, N.; Ringnalda, F.C.; Lindsay, H.; Berk, C.; Bargsten, K.; Li, Y.; Neri, D.; Robinson, M.D.; Ciaudo, C.; Hall, J.; et al. Covalent Linkage of the DNA RepairTemplate to the CRISPR-Cas9 Nuclease Enhances Homology-Directed Repair. eLife 2018, 7, e33761. [Google Scholar] [CrossRef]
- Gu, B.; Posfai, E.; Rossant, J. Efficient Generation of Targeted Large Insertions by Microinjection into Two-cell-stage Mouse Embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef]
- Ling, X.; Xie, B.; Gao, X.; Chang, L.; Zheng, W.; Chen, H.; Huang, Y.; Tan, L.; Li, M.; Liu, T. Improving the Efficiency of Precise Genome Editing with Site-specific Cas9-oligonucleotide Conjugates. Sci. Adv. 2020, 6, eaaz0051. [Google Scholar] [CrossRef]
- Lee, K.; Mackley, V.A.; Rao, A.; Chong, A.T.; Dewitt, M.A.; Corn, J.E.; Murthy, N. Synthetically Modified Guide RNA and Donor DNA are a Versatile Platform for CRISPR-Cas9 Engineering. eLife 2017, 6, e25312. [Google Scholar] [CrossRef]
- Yanagawa, Y.; Kawano, H.; Kobayashi, T.; Miyahara, H.; Okino, A.; Mitsuhara, I. Direct Protein Introduction into Plant Cells Using a Multi-gas Plasma Jet. PLoS ONE 2017, 12, e0171942. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef] [PubMed]
- Mumberg, D.; Muller, R.; Funk, M. Yeast Vectors for the Controlled Expression of Heterologous Proteins in Different Genetic Backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]
- Kweon, J.; Jang, A.H.; Kim, D.E.; Yang, J.W.; Yoon, M.; Rim Shin, H.; Kim, J.S.; Kim, Y. Fusion Guide RNAs for Orthogonal Gene Manipulation with Cas9 and Cpf1. Nat. Commun. 2017, 8, 1723. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Bussey, H. Yeast Arginine Permease: Nucleotide Sequence of the CAN1 Gene. Curr. Genet. 1986, 10, 587–592. [Google Scholar] [CrossRef]
- Satomura, A.; Nishioka, R.; Mori, H.; Sato, K.; Kuroda, K.; Ueda, M. Precise Genome-wide Base Editing by the CRISPR Nickase System in Yeast. Sci. Rep. 2017, 7, 2095. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Liu, J.H.; Xi, K.; Zhang, X.; Bao, L.; Zhang, X.; Tan, Z.J. Structural Flexibility of DNA-RNA Hybrid Duplex: Stretching and Twist-Stretch Coupling. Biophys. J. 2019, 117, 74–86. [Google Scholar] [CrossRef]
- Suresh, G.; Priyakumar, U.D. DNA-RNA Hybrid Duplexes with Decreasing Pyrimidine Content in the DNA Strand Provide Structural Snapshots for the A- to B-form Conformational Transition of Nucleic Acids. Phys. Chem. Chem. Phys. 2014, 16, 18148–18155. [Google Scholar] [CrossRef]
- DeWitt, M.A.; Corn, J.E.; Carroll, D. Genome Editing via delivery of Cas9 ribonucleoprotein. Methods 2017, 121–122, 9–15. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic Lipid-mediated Delivery of Proteins Enables Efficient Protein-based Genome Editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly Efficient RNA-guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Ueda, M. CRISPR Nickase-Mediated Base Editing in Yeast. Methods Mol. Biol. 2021, 2196, 27–37. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiba, W.; Amai, T.; Ueda, M.; Kuroda, K. Improving Precise Genome Editing Using Donor DNA/gRNA Hybrid Duplex Generated by Complementary Bases. Biomolecules 2022, 12, 1621. https://doi.org/10.3390/biom12111621
Aiba W, Amai T, Ueda M, Kuroda K. Improving Precise Genome Editing Using Donor DNA/gRNA Hybrid Duplex Generated by Complementary Bases. Biomolecules. 2022; 12(11):1621. https://doi.org/10.3390/biom12111621
Chicago/Turabian StyleAiba, Wataru, Takamitsu Amai, Mitsuyoshi Ueda, and Kouichi Kuroda. 2022. "Improving Precise Genome Editing Using Donor DNA/gRNA Hybrid Duplex Generated by Complementary Bases" Biomolecules 12, no. 11: 1621. https://doi.org/10.3390/biom12111621
APA StyleAiba, W., Amai, T., Ueda, M., & Kuroda, K. (2022). Improving Precise Genome Editing Using Donor DNA/gRNA Hybrid Duplex Generated by Complementary Bases. Biomolecules, 12(11), 1621. https://doi.org/10.3390/biom12111621