
Replicative Instability Drives Cancer Progression

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
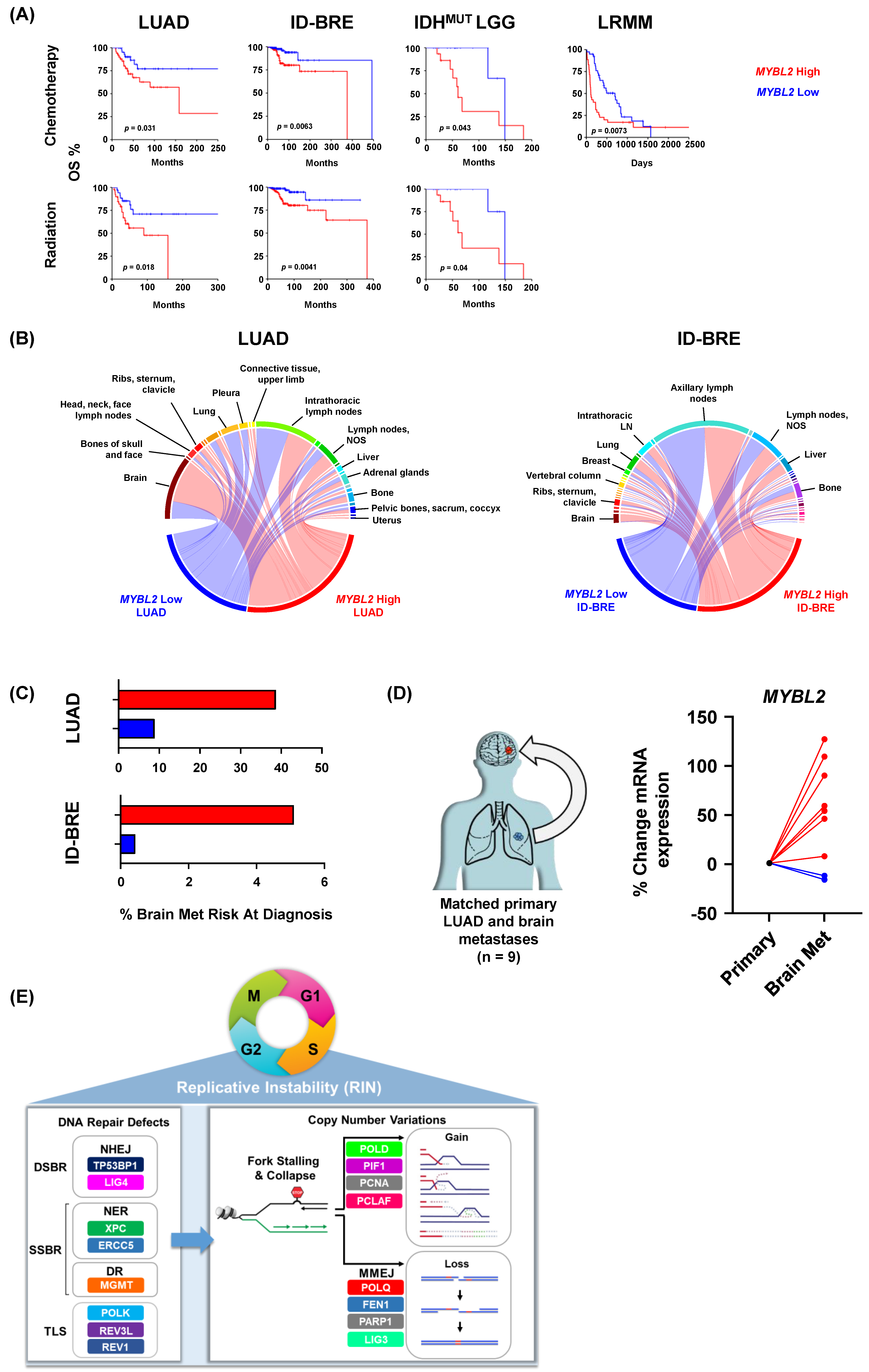{kind=link}
Abstract
1. Background
2. Methods
2.1. TCGA Pan-Cancer Analysis
2.2. Survival Analyses
2.3. DNA Repair Pathway WE Score
2.4. Catalogue of Somatic Mutations in Cancer (COSMIC) v3.2 Single Base Substitution (SBS) Analysis
2.5. RS Score
2.6. MPS
2.7. RSS Site Alteration Analysis
2.8. RSS Site Functional Analysis
2.9. Tumor Microenvironment Analysis
2.10. ORIEN Therapy Response
2.11. FUSED
2.12. ORIEN Metastatic Dissemination
2.13. Statistical Analyses
3. Results
3.1. Pan-Cancer Analysis Identifies MYBL2 Expression as a Robust Marker of Poor Patient Outcomes
3.2. MYBL2 High Tumors Are Characterized by Genomic Instability despite Containing Wildtype BRCA
3.3. Heterozygous Loss of Repair Effectors Underly Defective DNA Repair in MYBL2 High Tumors
3.4. Defective SSBR and TLS Are Linked to Increased Replication Stress and Distinct Genomic Footprints
3.5. Recurrent Copy Number Alterations at RSS Sites Rewire Transcriptional Programs and Impact Hallmark of Cancer Master Regulators
3.6. MYBL2 High Tumors Exhibit Immunosuppressive Microenvironments
3.7. Elevated MYBL2 Identifies Patients at Risk for Therapy Failure and Distant Metastases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RIN | replicative instability |
| MYBL2 | Myb proto-oncogene like 2 |
| TCGA | The Cancer Genome Atlas |
| OS | overall survival |
| PFS | progression free survival |
| DSS | disease-specific survival |
| LUAD | lung adenocarcinoma |
| IDHMUT LGG | isocitrate dehydrogenase-mutant lower grade glioma |
| PAAD | pancreatic adenocarcinoma |
| UCEC | uterine corpus endometrial carcinoma |
| SARC | sarcoma |
| FGA | fraction of the genome altered |
| MSI | microsatellite instability |
| HR | homologous recombination |
| RPS | repair proficiency score |
| Combined-HRD score | combined homologous recombination deficiency score |
| WE score | weighted expression score |
| SSBR | single-strand break repair |
| DSBR | double-strand break repair |
| TLS | translesion synthesis |
| NER | nucleotide excision repair |
| NHEJ | non-homologous end-joining |
| DR | direct reversal repair |
| COSMIC | Catalog of Somatic Mutations in Cancer |
| SBS | single-base substitution |
| RS score | replication stress score |
| MPS | mutational position score |
| ERFS | early replicating fragile sites |
| MiDAS | mitotic DNA synthesis sites |
| RSS sites | replication stress sensitive sites |
| MDSC | myeloid derived suppressor cell |
| ORIEN | Oncology Research Information Exchange Network |
| ID-BRE | invasive ductal breast cancer |
| LRMM | late-relapse multiple myeloma |
| FUSED | FUSion Error-prone repair Detection |
| CIN | chromosomal instability |
| MMEJ | microhomology mediated end-joining |
| FA | Fanconi Anemia |
| GO | Gene Ontology |
References
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Levine, D.A. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Lazar, A.J.; McLellan, M.D.; Bailey, M.H.; Miller, C.A.; Appelbaum, E.L.; Cordes, M.G.; Fronick, C.C.; Fulton, L.A.; Fulton, R.S.; Mardis, E.R.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Cunningham, C.E.; MacAuley, M.J.; Yadav, G.; Vizeacoumar, F.S.; Freywald, A.; Vizeacoumar, F.J. Targeting the CINful genome: Strategies to overcome tumor heterogeneity. Prog. Biophys. Mol. Biol. 2019, 147, 77–91. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA -Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.B.; Wages, N.A.; Grant, P.A.; Stukenberg, P.T.; Gentzler, R.D.; Hall, R.D.; Akerley, W.L.; Varghese, T.K.; Arnold, S.M.; Williams, T.M.; et al. MYBL2-Driven Transcriptional Programs Link Replication Stress and Error-prone DNA Repair with Genomic Instability in Lung Adenocarcinoma. Front. Oncol. 2021, 10, 585551. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using ‘ggplot2.’ R Package Version 0.4.5. 2019. Available online: https://CRAN.R-project.org/package=survminer (accessed on 15 August 2022).
- Scharer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Yi, C.; He, C. DNA Repair by Reversal of DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012575. [Google Scholar] [CrossRef]
- Stinson, B.M.; Loparo, J.J. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu. Rev. Biochem. 2021, 90, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Simko, V. R Package ‘Corrplot’: Visualization of a Correlation Matrix (Version 0.92). 2021. Available online: https://github.com/taiyun/corrplot (accessed on 15 August 2022).
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Kassambara, A. Ggpubr: ‘ggplot2’ Based Publication Ready Plots. R Package Version 0.4.0. 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 15 August 2022).
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef]
- Macheret, M.; Bhowmick, R.; Sobkowiak, K.; Padayachy, L.; Mailler, J.; Hickson, I.D.; Halazonetis, T.D. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. 2020, 30, 997–1008. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling Intercellular Communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. Available online: https://igraph.org (accessed on 15 August 2022).
- Pedersen, T.L. Ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. R Package Version 2.0.5. 2021. Available online: https://CRAN.R-project.org/package=ggraph (accessed on 15 August 2022).
- Jiménez-Sánchez, A.; Cast, O.; Miller, M.L. Comprehensive Benchmarking and Integration of Tumor Microenvironment Cell Estimation Methods. Cancer Res. 2019, 79, 6238–6246. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, D.; LeCain, M.; Sheer, D. The role of microhomology in genomic structural variation. Trends Genet. 2014, 30, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Weiss, J.; Xu, W. Swimplot: Tools for Creating Swimmers Plots Using ‘ggplot2’. R Package Version 1.2.0. 2021. Available online: https://CRAN.R-project.org/package=swimplot (accessed on 15 August 2022).
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across the Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSIsensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014, 30, 1015–1016. [Google Scholar] [CrossRef]
- Pitroda, S.P.; Pashtan, I.M.; Logan, H.L.; Budke, B.; Darga, T.E.; Weichselbaum, R.R.; Connell, P.P. DNA Repair Pathway Gene Expression Score Correlates with Repair Proficiency and Tumor Sensitivity to Chemotherapy. Sci. Transl. Med. 2014, 6, ra42–ra229. [Google Scholar] [CrossRef]
- Leu, S.; von Felten, S.; Frank, S.; Vassella, E.; Vajtai, I.; Taylor, E.; Schulz, M.; Hutter, G.; Hench, J.; Schucht, P.; et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro-Oncology 2013, 15, 469–479. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The genome as a record of environmental exposure. Mutagenesis 2015, 30, 763–770. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. Causes of Genome Instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Mrasek, K.; Schoder, C.; Teichmann, A.C.; Behr, K.; Franze, B.; Wilhelm, K.; Blaurock, N.; Claussen, U.; Liehr, T.; Weise, A. Global Screening and Extended Nomenclature for 230 Aphidicolin-Inducible Fragile Sites, including 61 Yet Unreported Ones. Int. J. Oncol. 2010, 36, 929–940. Available online: http://www.spandidos-publications.com/ijo/36/4/929 (accessed on 31 March 2022).
- Briu, L.-M.; Maric, C.; Cadoret, J.-C. Replication Stress, Genomic Instability, and Replication Timing: A Complex Relationship. IJMS 2021, 22, 4764. [Google Scholar] [CrossRef] [PubMed]
- Stok, C.; Kok, Y.P.; van den Tempel, N.; van Vugt, M.A.T.M. Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res. 2021, 49, 4239–4257. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Kotecha, R.; Chao, S.T.; Ahluwalia, M.S.; Sahgal, A.; Chang, E.L. Current approaches to the management of brain metastases. Nat. Rev. Clin. Oncol. 2020, 17, 279–299. [Google Scholar] [CrossRef]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.-K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Sig. Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA polymerase θ (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, B.B.; Smith, J.P.; Zhang, Q.; Jiang, Z.; Hampton, O.A.; Churchman, M.L.; Arnold, S.M.; Owen, D.H.; Gray, J.E.; Dillon, P.M.; et al. Replicative Instability Drives Cancer Progression. Biomolecules 2022, 12, 1570. https://doi.org/10.3390/biom12111570
Morris BB, Smith JP, Zhang Q, Jiang Z, Hampton OA, Churchman ML, Arnold SM, Owen DH, Gray JE, Dillon PM, et al. Replicative Instability Drives Cancer Progression. Biomolecules. 2022; 12(11):1570. https://doi.org/10.3390/biom12111570
Chicago/Turabian StyleMorris, Benjamin B., Jason P. Smith, Qi Zhang, Zhijie Jiang, Oliver A. Hampton, Michelle L. Churchman, Susanne M. Arnold, Dwight H. Owen, Jhanelle E. Gray, Patrick M. Dillon, and et al. 2022. "Replicative Instability Drives Cancer Progression" Biomolecules 12, no. 11: 1570. https://doi.org/10.3390/biom12111570
APA StyleMorris, B. B., Smith, J. P., Zhang, Q., Jiang, Z., Hampton, O. A., Churchman, M. L., Arnold, S. M., Owen, D. H., Gray, J. E., Dillon, P. M., Soliman, H. H., Stover, D. G., Colman, H., Chakravarti, A., Shain, K. H., Silva, A. S., Villano, J. L., Vogelbaum, M. A., Borges, V. F., ... Mayo, M. W. (2022). Replicative Instability Drives Cancer Progression. Biomolecules, 12(11), 1570. https://doi.org/10.3390/biom12111570