
Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action




Abstract
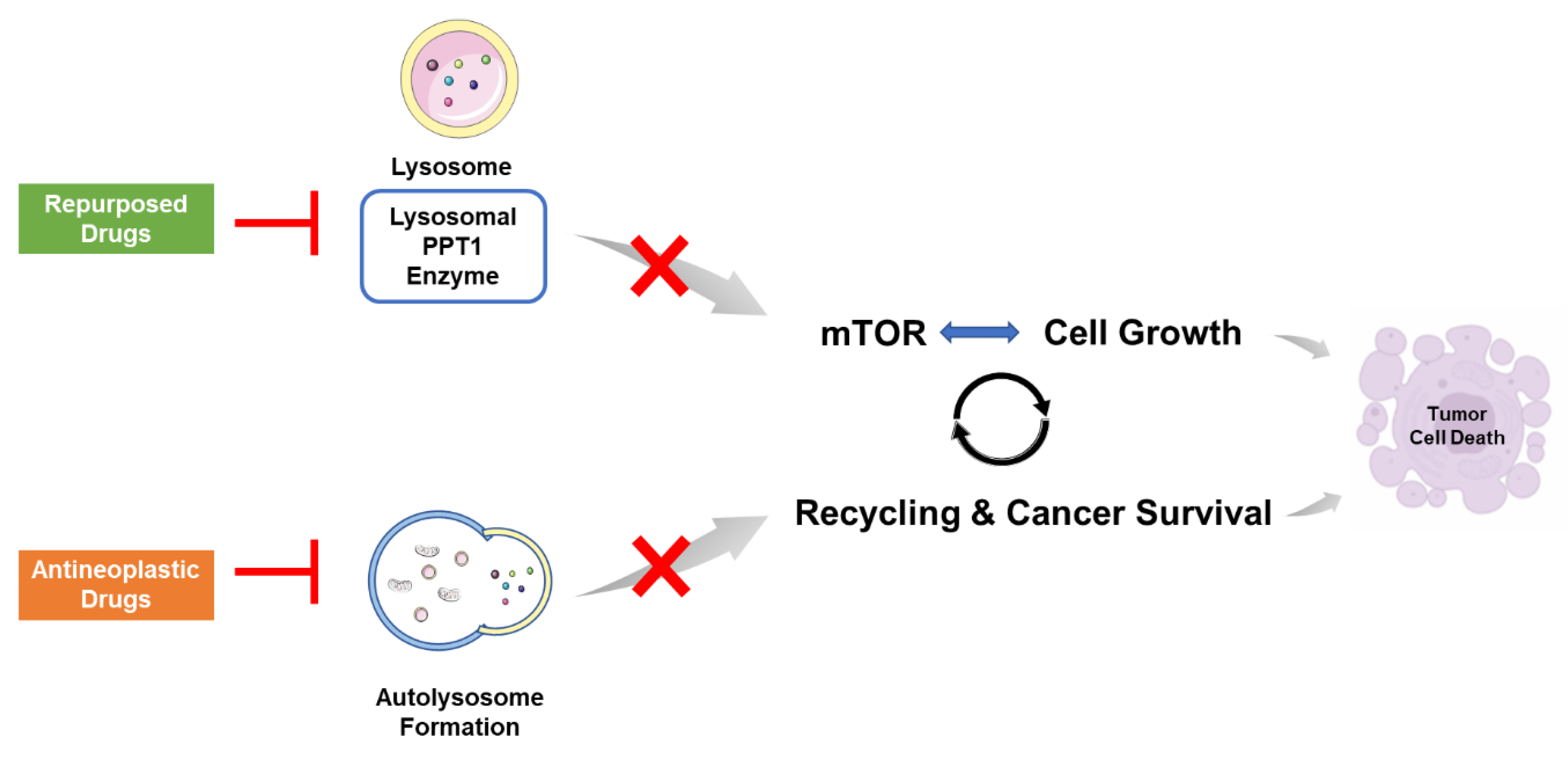
:1. Introduction
2. Materials and Methods
2.1. Biosafety Studies in MRC-5 Fibroblast Cells
2.1.1. Cell Culture
2.1.2. Drug Treatment
2.1.3. Morphological Analysis
2.1.4. MTT Assay
2.1.5. Selectivity Index (SI)
2.1.6. Statistical Analysis
2.2. Immunohistochemistry Studies in MCF-7 and HT-29 Cancer Cells
2.2.1. Cell culture and Drug Treatment
2.2.2. Collection, Fixation, and HistogelTM Processing of Cultured Cells
2.2.3. CMA Construction and Immunocytochemistry Expression Analysis
3. Results and Discussion
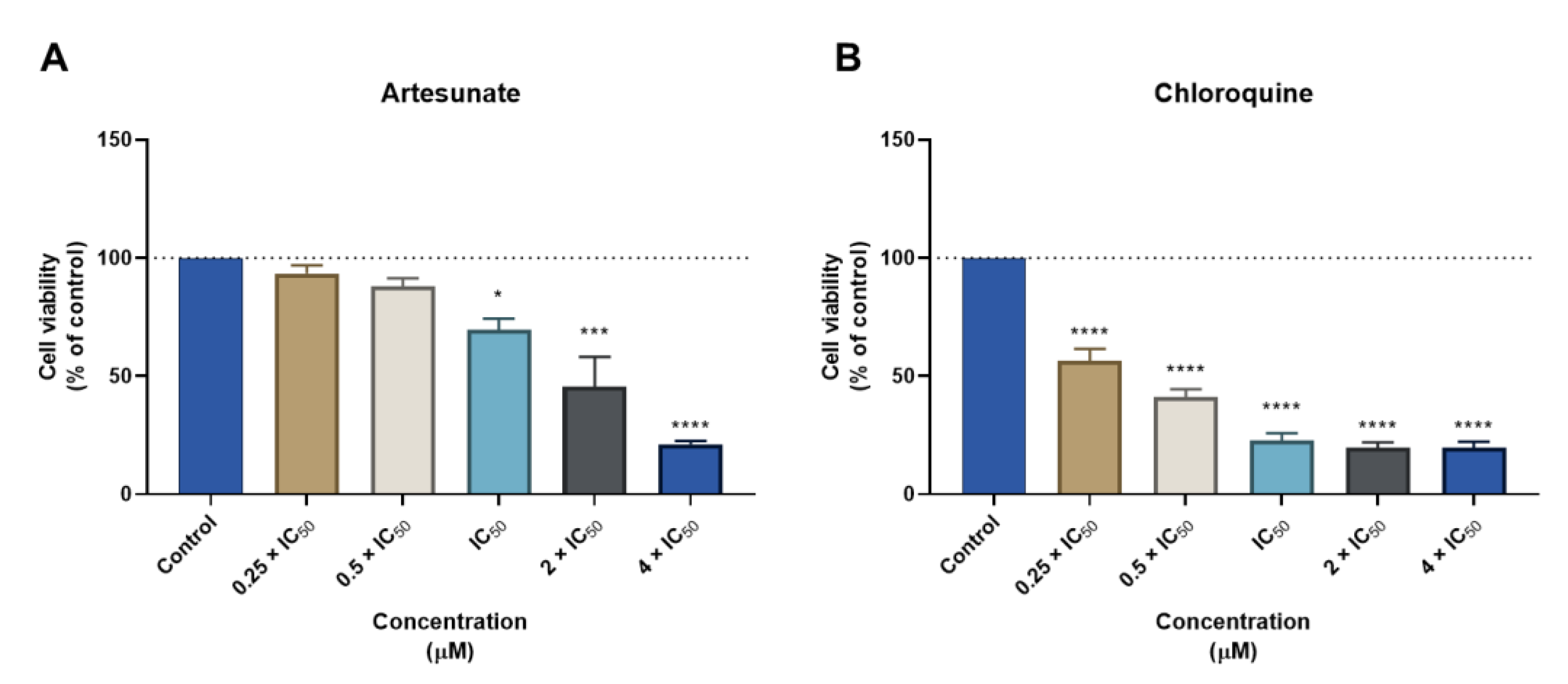
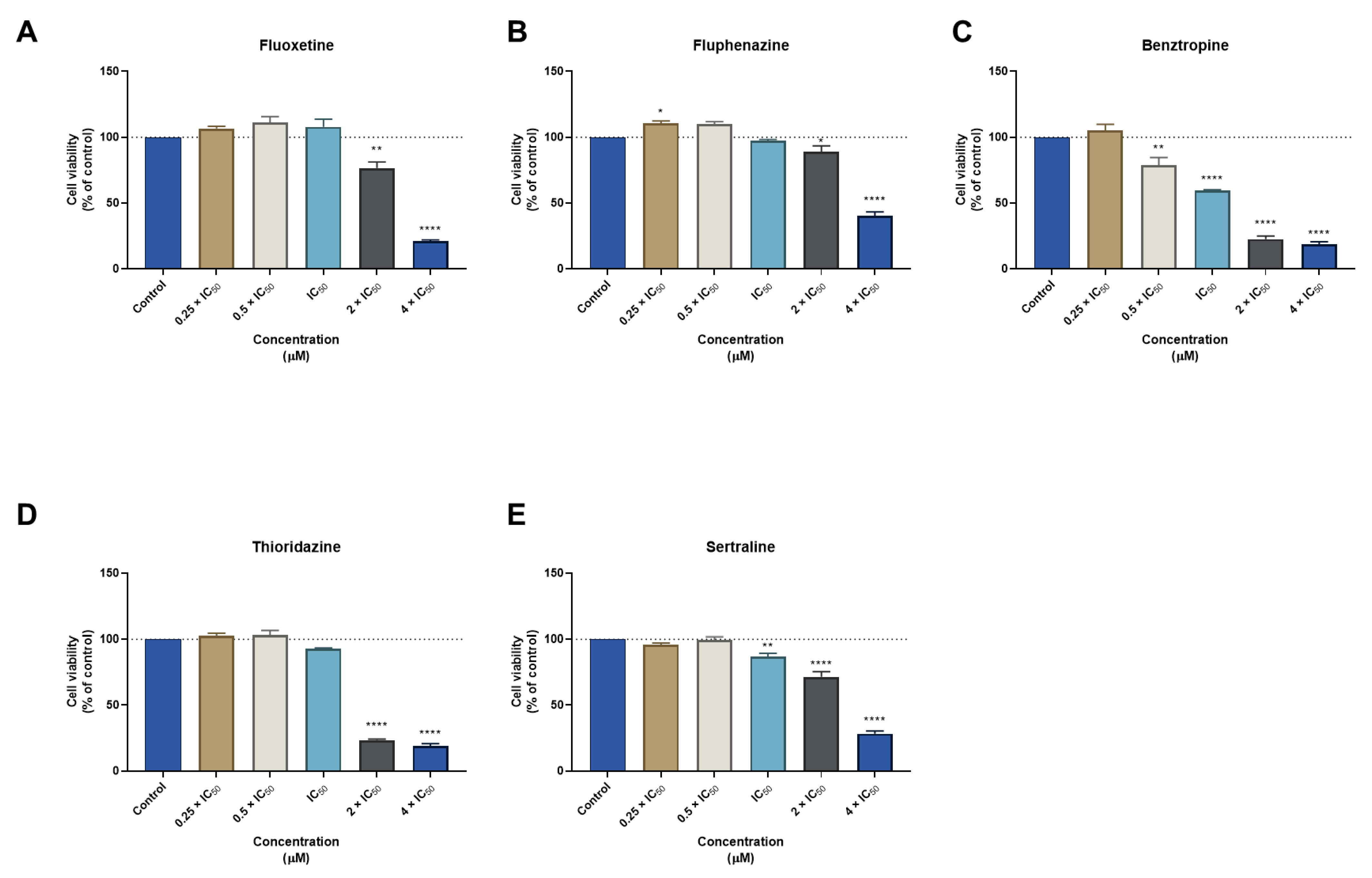
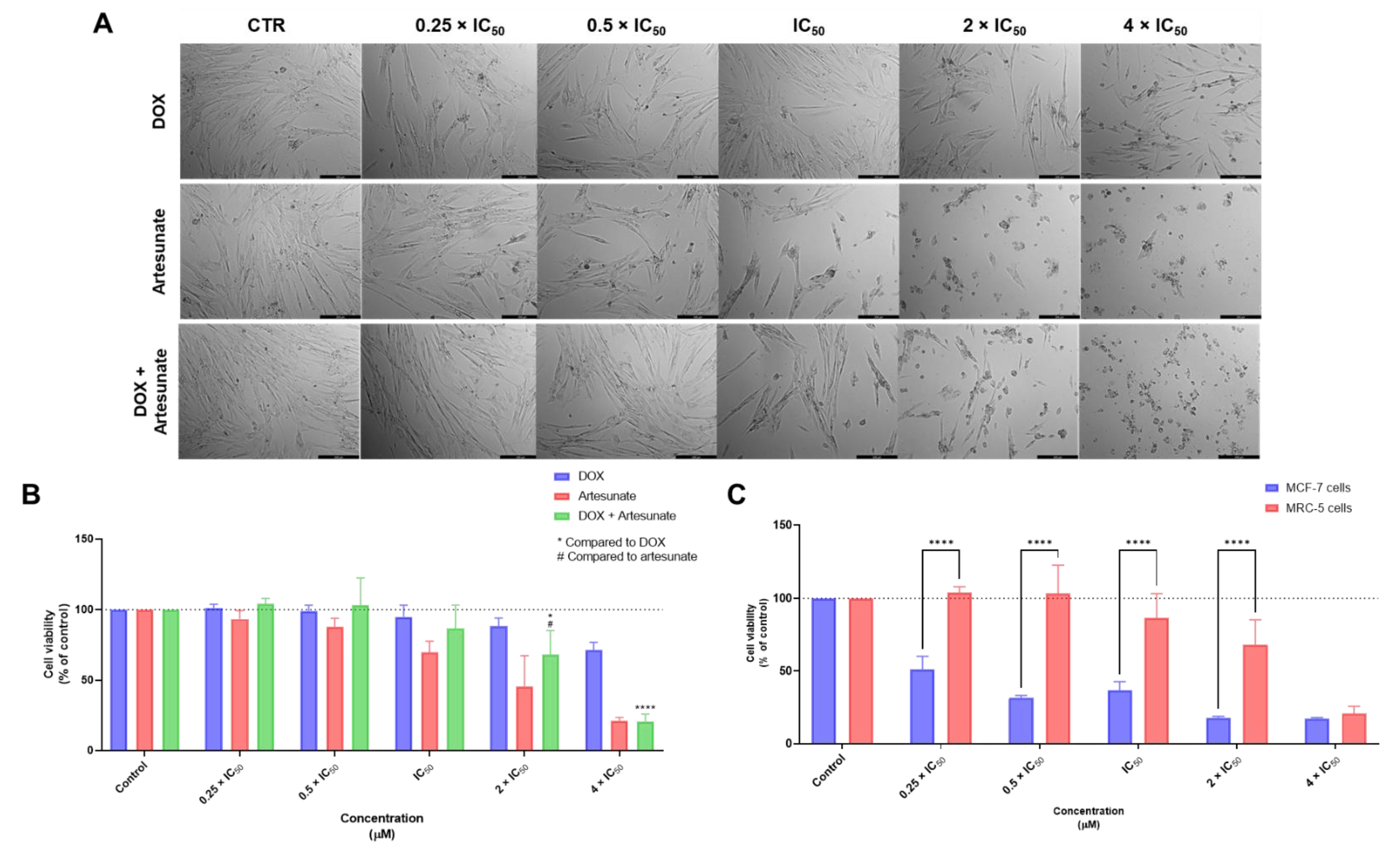
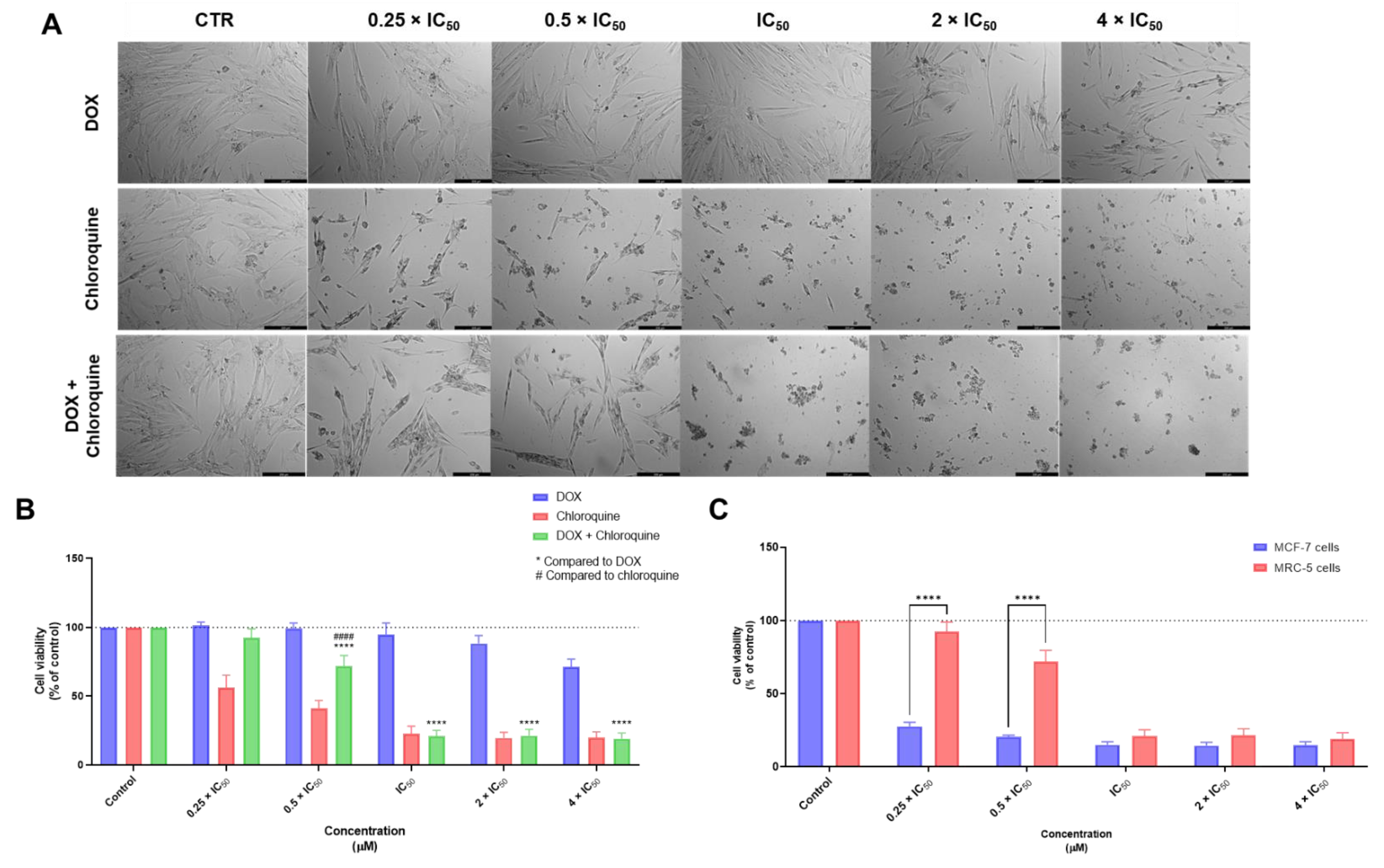
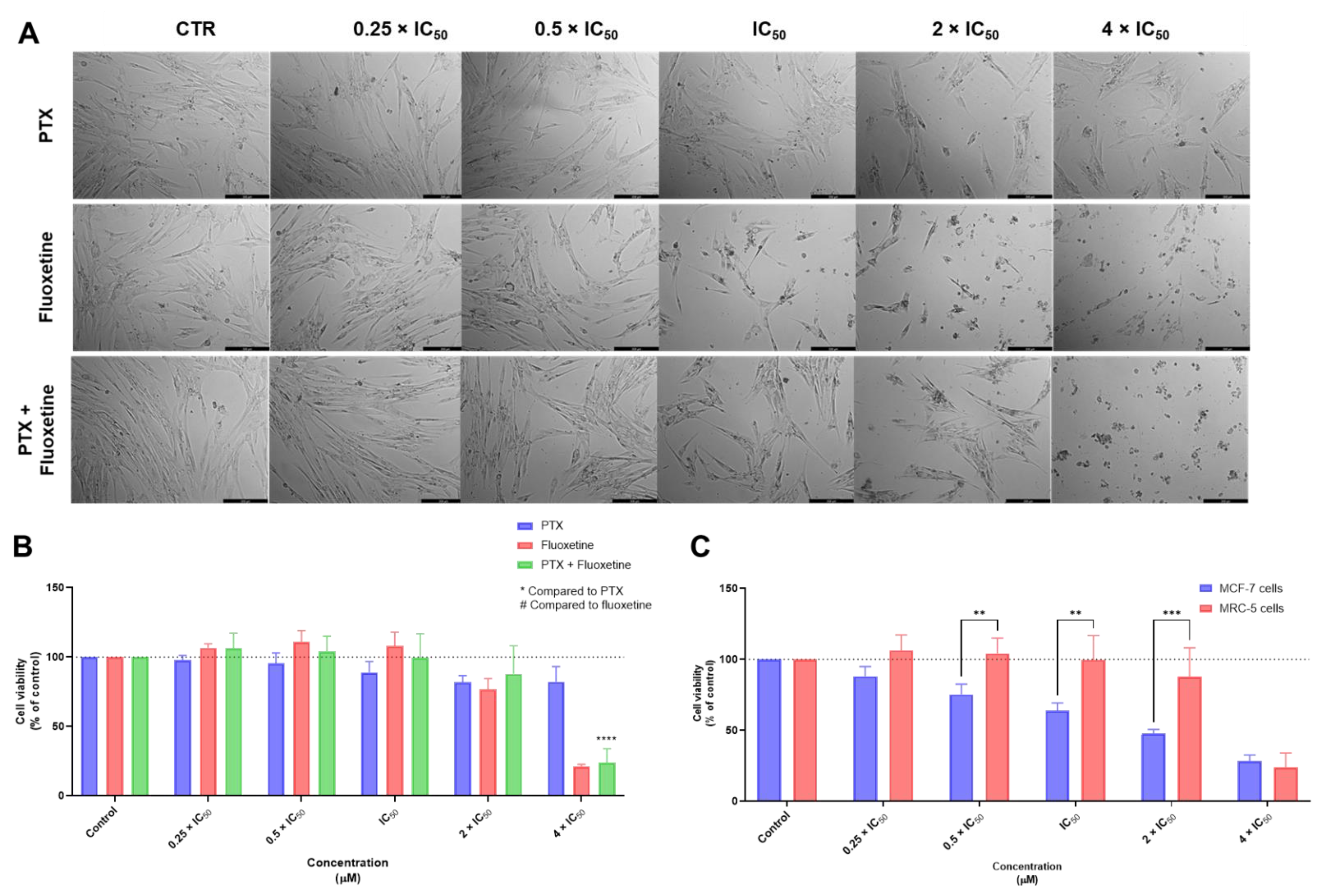
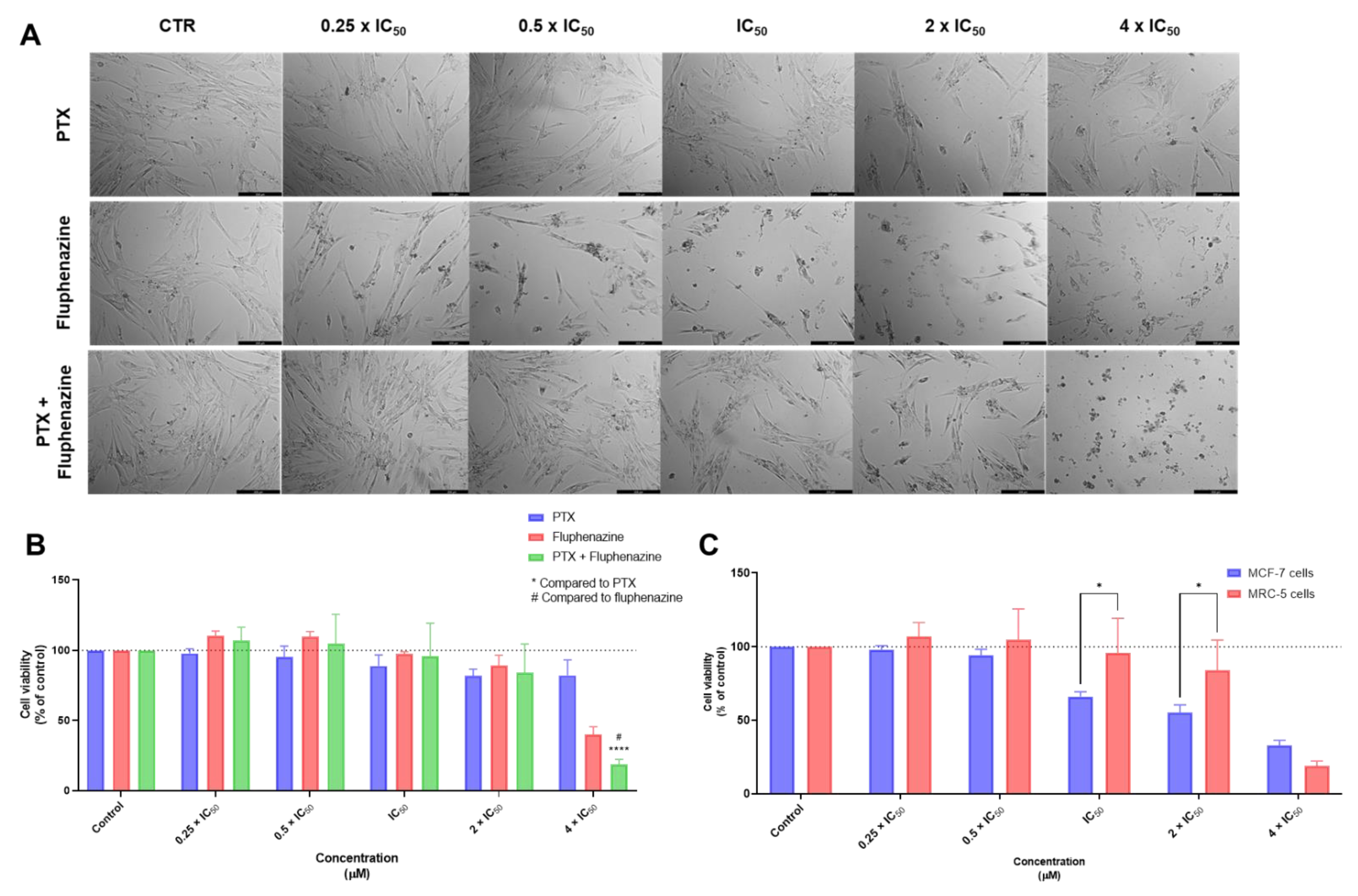
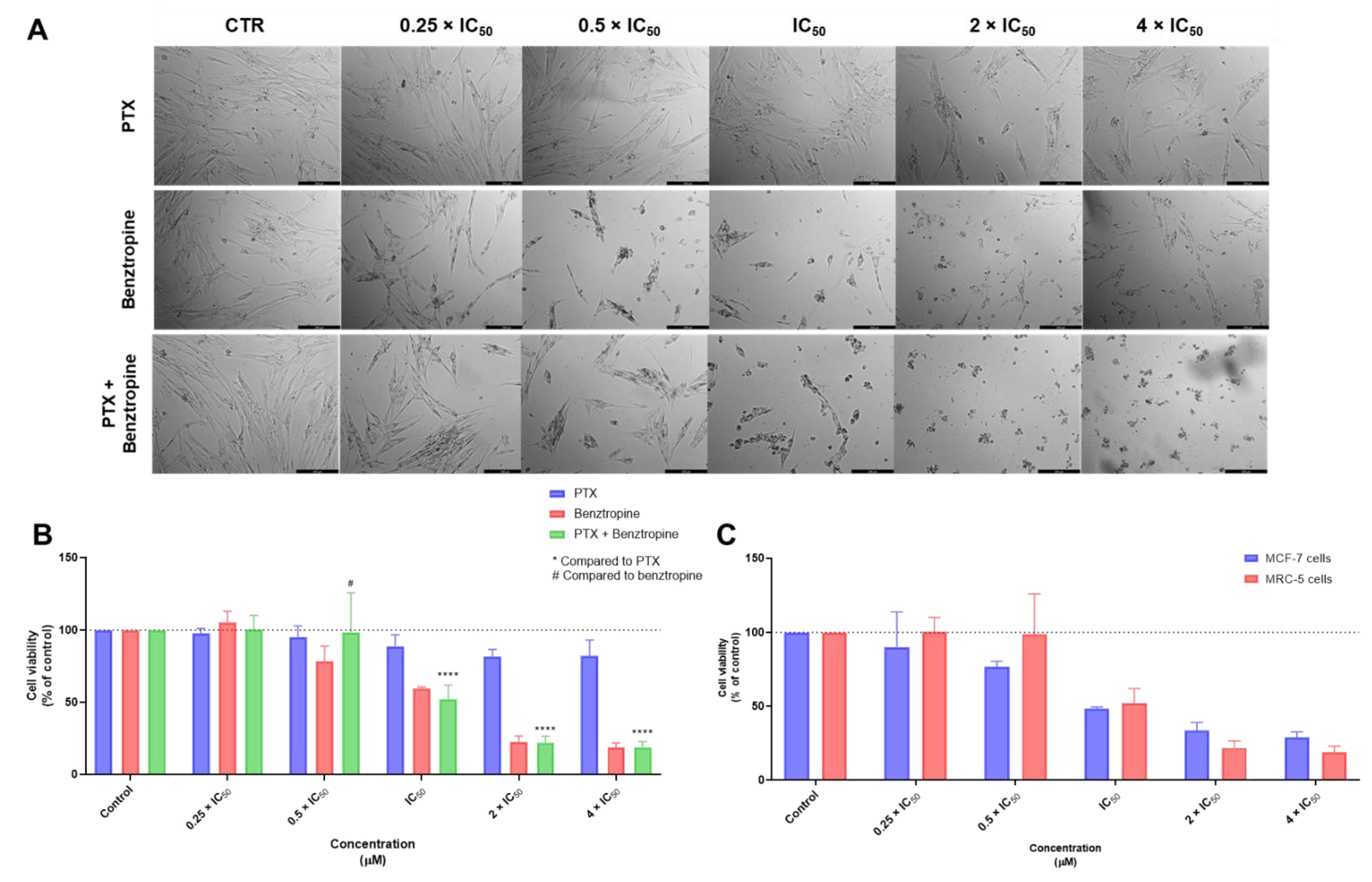
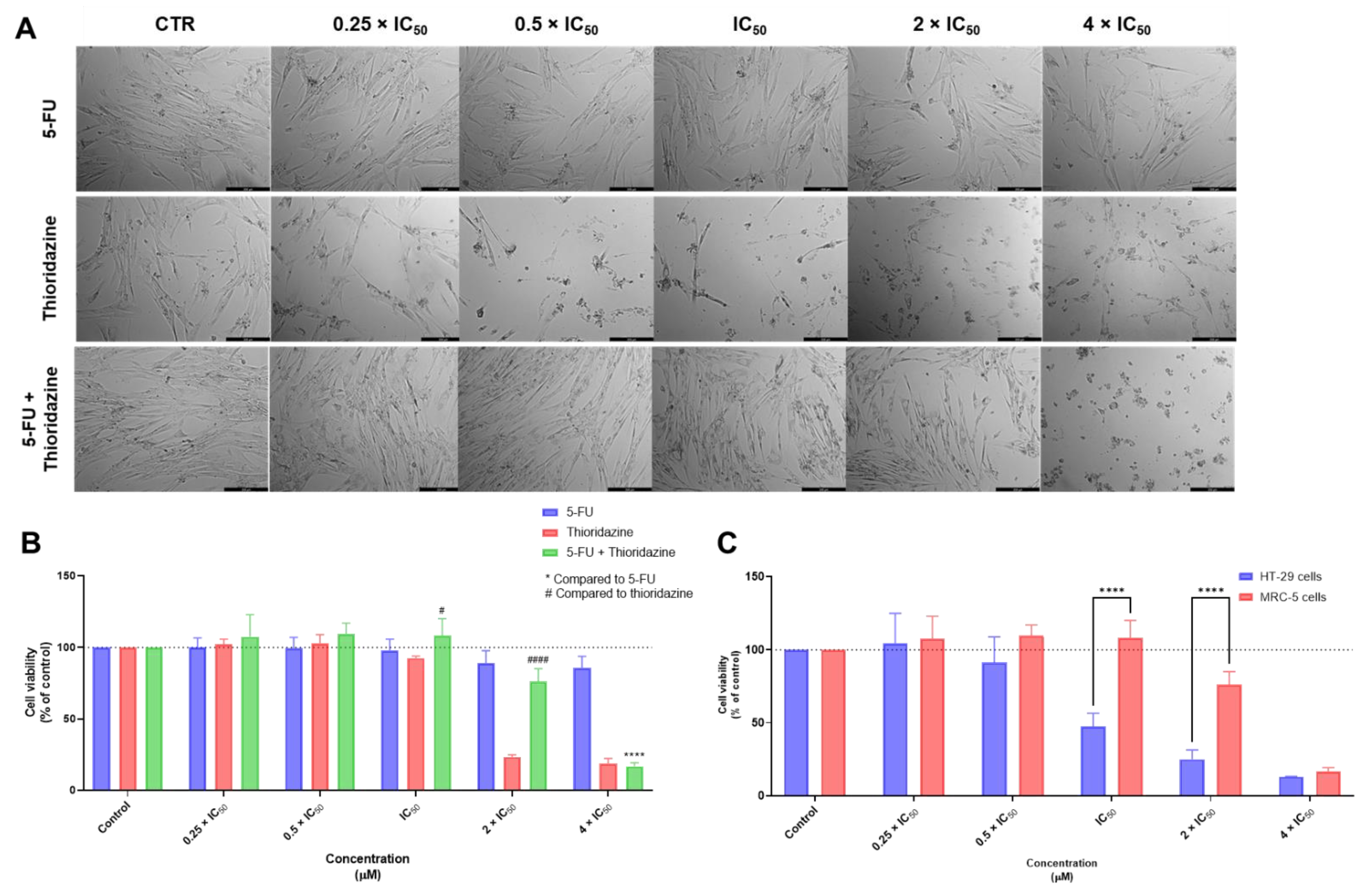
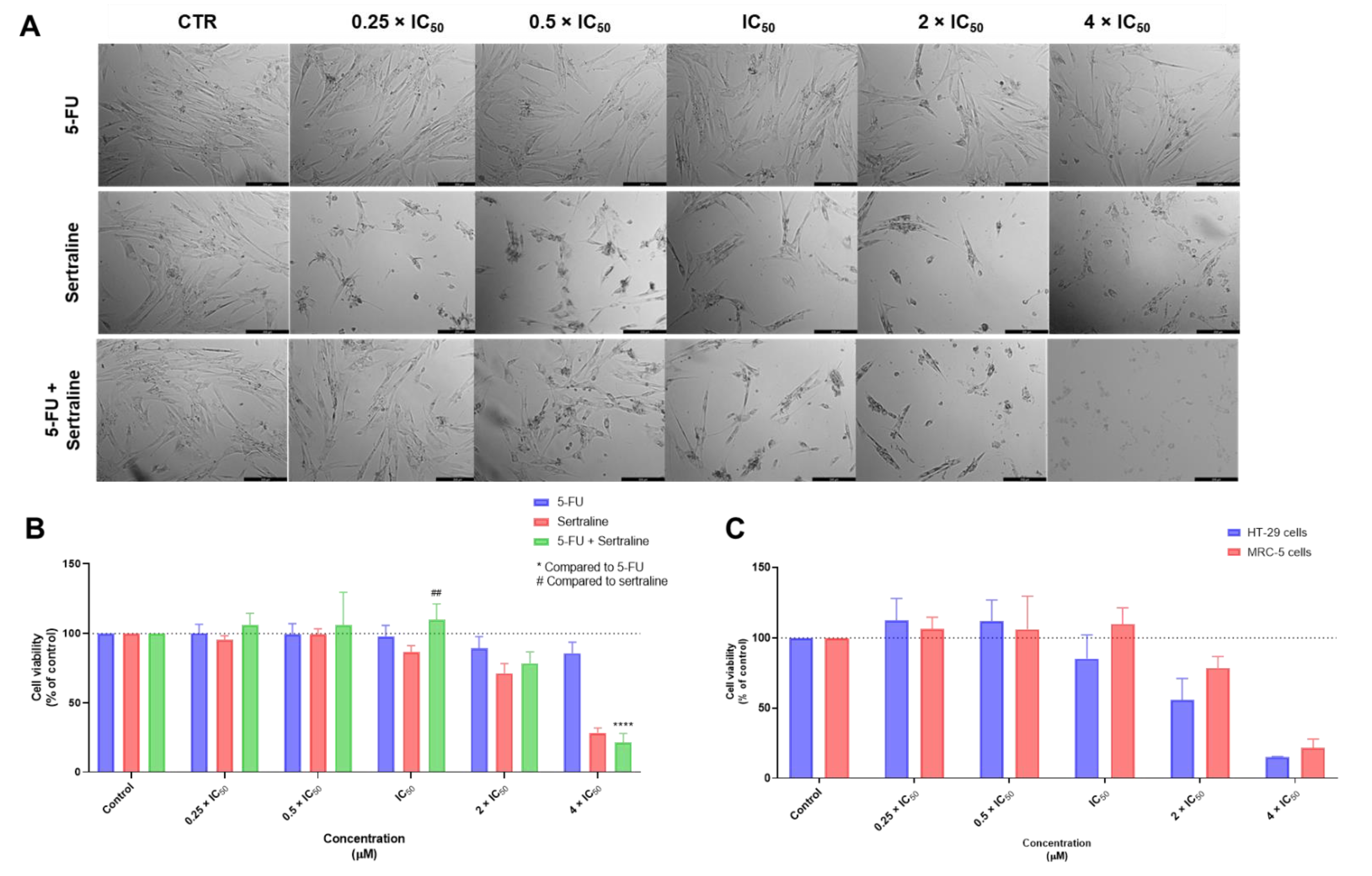
3.1. Biosafety Studies in MRC-5 Fibroblast Cells
3.1.1. Single Drug Treatments
3.1.2. Combination Treatments
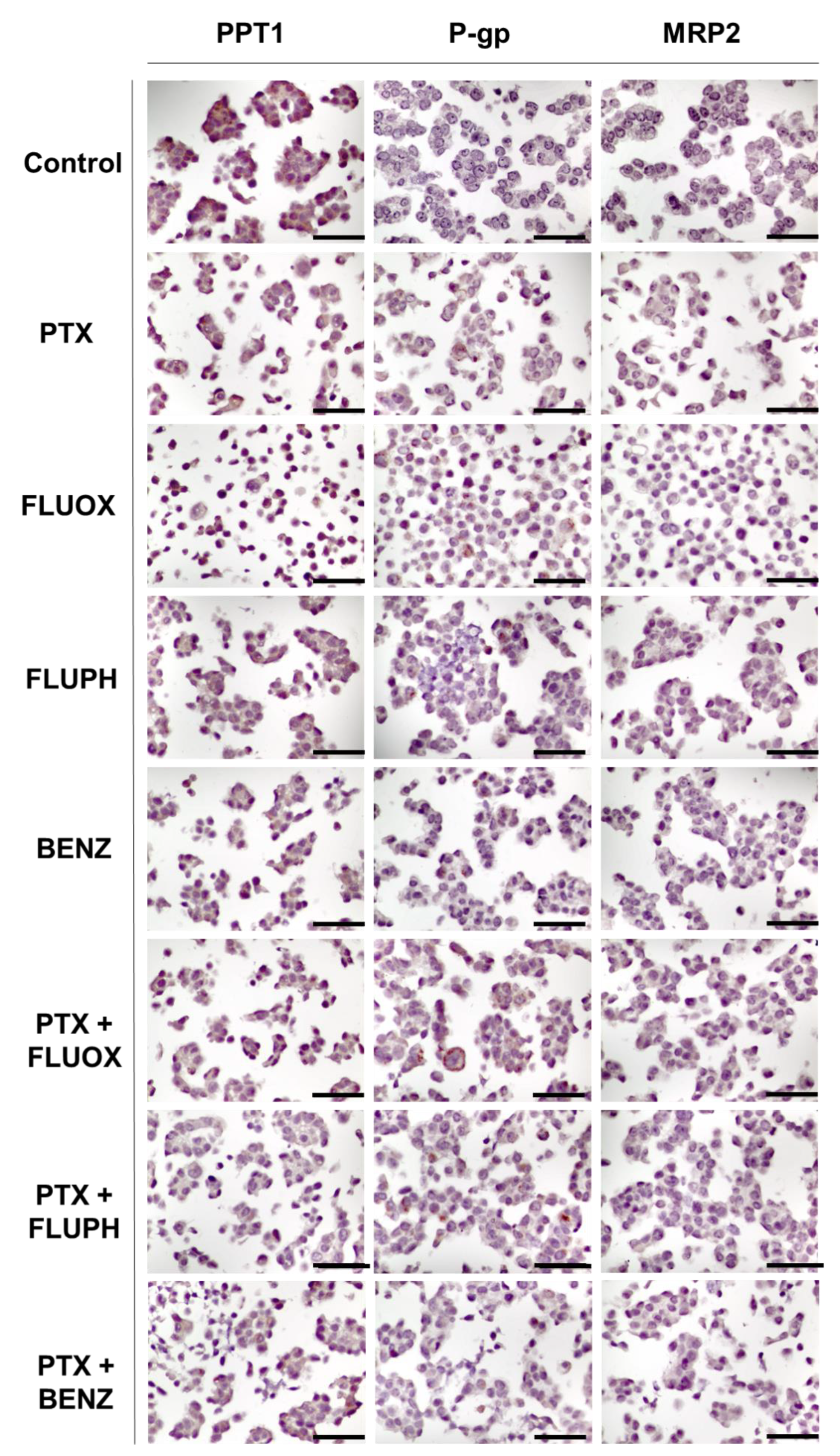
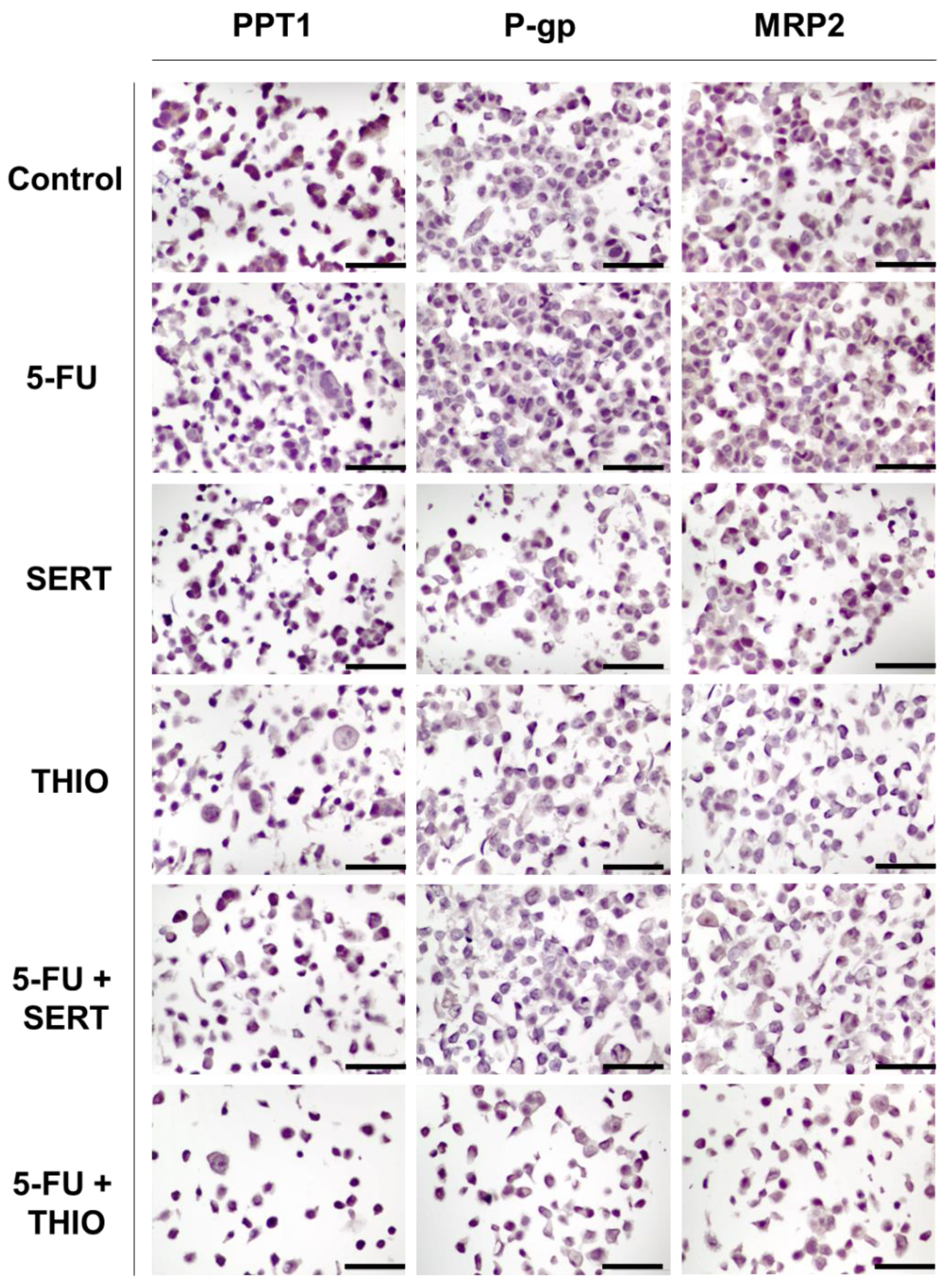
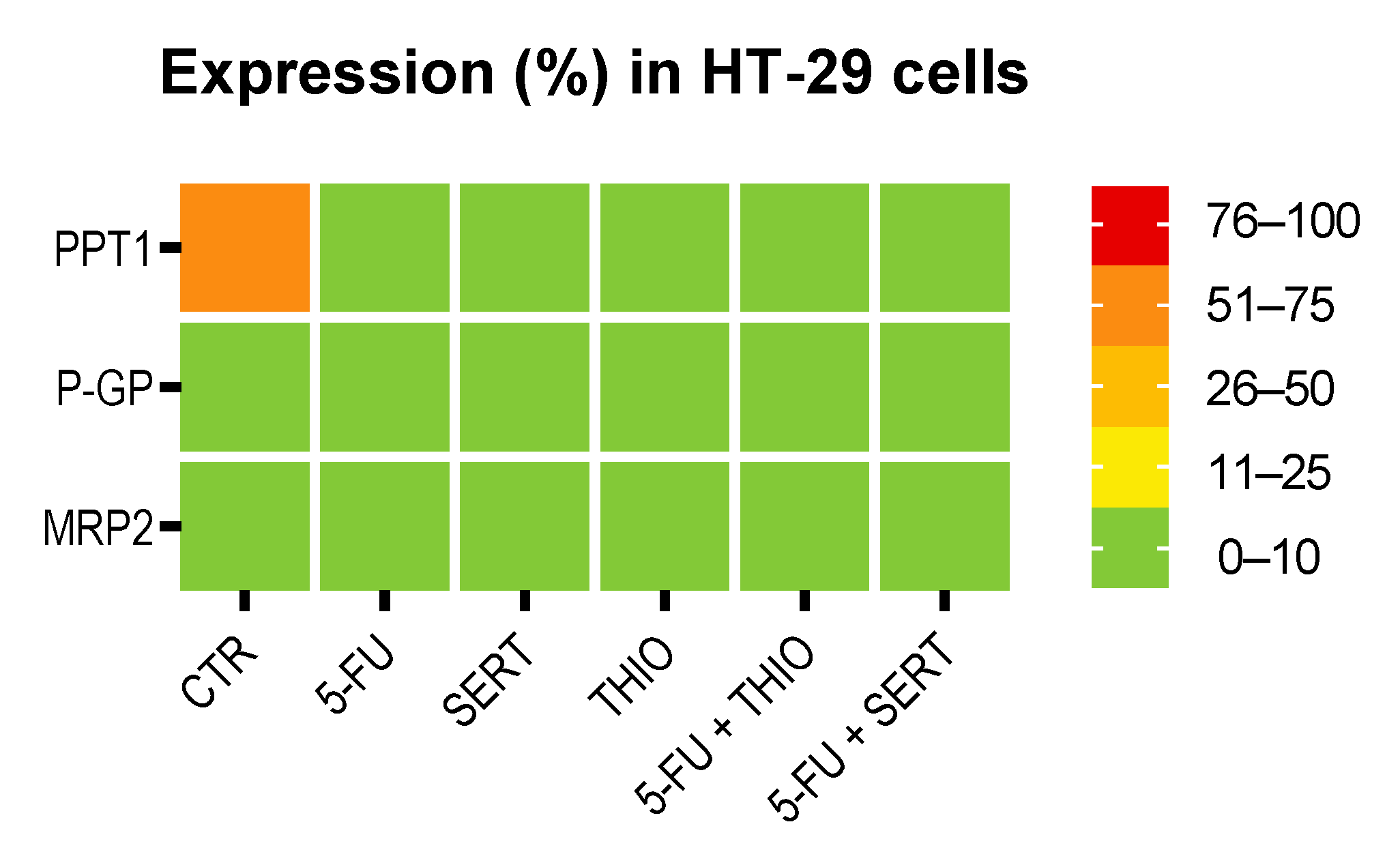
3.2. Immunohistochemistry Studies in MCF-7 and HT-29 Cancer Cells
3.2.1. MCF-7 Cells Treated with DOX + Antimalarial Drugs
3.2.2. MCF-7 Cells Treated with PTX + CNS Drugs
3.2.3. HT-29 Cells Treated with 5-FU + CNS Drugs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA. Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Entezar-Almahdi, E.; Mohammadi-Samani, S.; Tayebi, L.; Farjadian, F. Recent Advances in Designing 5-Fluorouracil Delivery Systems: A Stepping Stone in the Safe Treatment of Colorectal Cancer. Int. J. Nanomed. 2020, 15, 5445–5458. [Google Scholar] [CrossRef]
- Ben-Hamo, R.; Zilberberg, A.; Cohen, H.; Bahar-Shany, K.; Wachtel, C.; Korach, J.; Aviel-Ronen, S.; Barshack, I.; Barash, D.; Levanon, K.; et al. Resistance to Paclitaxel Is Associated with a Variant of the Gene BCL2 in Multiple Tumor Types. Npj Precis. Oncol. 2019, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin Pathways. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Antoszczak, M.; Markowska, A.; Markowska, J.; Huczyński, A. Old Wine in New Bottles: Drug Repurposing in Oncology. Eur. J. Pharmacol. 2020, 866, 172784. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent Advances of Cocktail Chemotherapy by Combination Drug Delivery Systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, D.; Wu, B.; Quan, Y.; Liu, D.; Li, Y.; Zhang, X. Synergistic Activity of an Antimetabolite Drug and Tyrosine Kinase Inhibitors against Breast Cancer Cells. Chem. Pharm. Bull. 2017, 65, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.-S.; Choi, J.Y. Synergistic Anti-Cancer Effect of Phenformin and Oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Chen, Y.; Wang, Z.; Wang, J.; Wan, J.; Yu, C.; Liu, X.; Li, W. Synergistic Anti-Tumour Effects of Tetrandrine and Chloroquine Combination Therapy in Human Cancer: A Potential Antagonistic Role for P21. Br. J. Pharmacol. 2015, 172, 2232–2245. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Xiao, X.; Zhang, C.; Yu, W.; Guo, W.; Zhang, Z.; Li, Z.; Feng, X.; Hao, J.; Zhang, K.; et al. Melatonin Synergizes the Chemotherapeutic Effect of 5-Fluorouracil in Colon Cancer by Suppressing PI3K/AKT and NF-ΚB/INOS Signaling Pathways. J. Pineal Res. 2017, 62, e12380. [Google Scholar] [CrossRef]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A Curative Combination Cancer Therapy Achieves High Fractional Cell Killing through Low Cross-Resistance and Drug Additivity. Elife 2019, 8, e50036. [Google Scholar] [CrossRef]
- Duarte, D.; Cardoso, A.; Vale, N. Synergistic Growth Inhibition of HT-29 Colon and MCF-7 Breast Cancer Cells with Simultaneous and Sequential Combinations of Antineoplastics and CNS Drugs. Int. J. Mol. Sci. 2021, 22, 7408. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells. Biomolecules 2020, 10, 1623. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Duarte, D.; Rêma, A.; Amorim, I.; Vale, N. Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells. Biomolecules 2022, 12, 190. [Google Scholar] [CrossRef]
- Magalhães, A.C.; Ricardo, S.; Moreira, A.C.; Nunes, M.; Tavares, M.; Pinto, R.J.; Gomes, M.S.; Pereira, L. InfectionCMA: A Cell MicroArray Approach for Efficient Biomarker Screening in In Vitro Infection Assays. Pathogens 2022, 11, 313. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Vale, N. How Antimalarials and Antineoplastic Drugs Can Interact in Combination Therapies: A Perspective on the Role of PPT1 Enzyme. Curr. Drug Metab. 2021, 22, 1009–1016. [Google Scholar] [CrossRef]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-KB P65 and Strategies for Therapeutic Manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef]
- Finch, A.; Pillans, P. P-Glycoprotein and Its Role in Drug-Drug Interactions. Aust. Prescr. 2014, 37, 137–139. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Hoffmann, U.; Kroemer, H.K. Structure and Function of the MRP2 (ABCC2) Protein and Its Role in Drug Disposition. Expert Opin. Drug Metab. Toxicol. 2006, 2, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 Is a Promising Molecular Target in the Diagnosis of Cancer (Review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, P.F.; Alves, J.M.; Damasceno, J.L.; Oliveira, R.A.M.; Júnior Dias, H.; Crotti, A.E.M.; Tavares, D.C. Cytotoxicity Screening of Essential Oils in Cancer Cell Lines. Rev. Bras. Farmacogn. 2015, 25, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Mellado, M.; Soto, M.; Madrid, A.; Montenegro, I.; Jara-Gutiérrez, C.; Villena, J.; Werner, E.; Godoy, P.; Aguilar, L.F. In Vitro Antioxidant and Antiproliferative Effect of the Extracts of Ephedra Chilensis K Presl Aerial Parts. BMC Complement. Altern. Med. 2019, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.; Silva, P.M.A.; Coelho, R.; Pinto, C.; Resende, A.; Bousbaa, H.; Almeida, G.M.; Ricardo, S. Generation of Two Paclitaxel-Resistant High-Grade Serous Carcinoma Cell Lines With Increased Expression of P-Glycoprotein. Front. Oncol. 2021, 11, 4326. [Google Scholar] [CrossRef]
- Jiang, P.D.; Zhao, Y.L.; Shi, W.; Deng, X.Q.; Xie, G.; Mao, Y.Q.; Li, Z.G.; Zheng, Y.Z.; Yang, S.Y.; Wei, Y.Q. Cell Growth Inhibition, G2/M Cell Cycle Arrest, and Apoptosis Induced by Chloroquine in Human Breast Cancer Cell Line Bcap-37. Cell. Physiol. Biochem. 2008, 22, 431–440. [Google Scholar] [CrossRef]
- Samutprasert, P.; Chiablaem, K.; Teeraseranee, C.; Phaiyarin, P.; Pukfukdee, P.; Pienpinijtham, P.; Svasti, J.; Palaga, T.; Lirdprapamongkol, K.; Wanichwecharungruang, S. Epigallocatechin Gallate-Zinc Oxide Co-Crystalline Nanoparticles as an Anticancer Drug That Is Non-Toxic to Normal Cells. RSC Adv. 2018, 8, 7369–7376. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Wang, X.; Fan, C.; Mao, X. The Role of Ki67 in Evaluating Neoadjuvant Endocrine Therapy of Hormone Receptor-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 1426. [Google Scholar] [CrossRef]
- Schluter, C.; Duchrow, M.; Wohlenberg, C.; Becker, M.H.G.; Key, G.; Flad, H.D.; Gerdes, J. The Cell Proliferation-Associated Antigen of Antibody Ki-67: A Very Large, Ubiquitous Nuclear Protein with Numerous Repeated Elements, Representing a New Kind of Cell Cycle-Maintaining Proteins. J. Cell Biol. 1993, 123, 513–522. [Google Scholar] [CrossRef]
- Chen, A. PARP Inhibitors: Its Role in Treatment of Cancer. Chin. J. Cancer 2011, 30, 463. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-Signaling Pathway in Cancer. OncoTargets Ther. 2018, 11, 2063. [Google Scholar] [CrossRef] [Green Version]
- Yap, S.Q.; Mathavarajah, S.; Huber, R.J. The Converging Roles of Batten Disease Proteins in Neurodegeneration and Cancer. Iscience 2021, 24, 102337. [Google Scholar] [CrossRef] [PubMed]
- Famta, P.; Shah, S.; Chatterjee, E.; Singh, H.; Dey, B.; Guru, S.K.; Singh, S.B.; Srivastava, S. Exploring New Horizons in Overcoming P-Glycoprotein-Mediated Multidrug-Resistant Breast Cancer via Nanoscale Drug Delivery Platforms. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100054. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; O’Brien, J.P.; Boccia, J.; Casals, D.; Bertino, J.R.; Melamed, M.R. Expression of the Multidrug Resistance Gene Product (P-Glycoprotein) in Human Normal and Tumor Tissues. J. Histochem. Cytochem. 1990, 38, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.; Sureshkumar, R.; Zehravi, M.; Akter, R.; Ali, F.; Ramproshad, S.; Mondal, B.; Tagde, P.; Ahmed, Z.; Khan, F.S.; et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life 2022, 12, 897. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, E.; Banik, K.; Harsha, C.; Sailo, B.L.; Thakur, K.K.; Khwairakpam, A.D.; Vikkurthi, R.; Devi, T.B.; Gupta, S.C.; Kunnumakkara, A.B. Phytochemicals in Cancer Cell Chemosensitization: Current Knowledge and Future Perspectives. Semin. Cancer Biol. 2022, 80, 306–339. [Google Scholar] [CrossRef]
- Turner, A.P.; Alam, C.; Bendayan, R. Efflux Transporters in Cancer Resistance: Molecular and Functional Characterization of P-Glycoprotein. In Drug Efflux Pumps in Cancer Resistance Pathways: From Molecular Recognition and Characterization to Possible Inhibition Strategies in Chemotherapy; Academic Press: Cambridge, MA, USA, 2020; pp. 1–30. [Google Scholar] [CrossRef]
- Guerra, A.R.; Duarte, M.F.; Duarte, I.F. Targeting Tumor Metabolism with Plant-Derived Natural Products: Emerging Trends in Cancer Therapy. J. Agric. Food Chem. 2018, 66, 10663–10685. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor Microenvironment and Epithelial Mesenchymal Transition as Targets to Overcome Tumor Multidrug Resistance. Drug Resist. Updates 2020, 53, 100715. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Chen, Z.S. Multidrug Resistance Proteins (MRPs) and Cancer Therapy. AAPS J. 2015, 17, 802. [Google Scholar] [CrossRef] [Green Version]
- Kranz, J.; Hessel, S.; Aretz, J.; Seidel, A.; Petzinger, E.; Geyer, J.; Lampen, A. The Role of the Efflux Carriers Abcg2 and Abcc2 for the Hepatobiliary Elimination of Benzo[a]Pyrene and Its Metabolites in Mice. Chem. Biol. Interact. 2014, 224, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.J.; Bahoush, G.; Zaker, F.; Ansari, S.; Rafsanjani, K.A.; Sharafi, H. Association of -24CT, 1249GA, and 3972CT ABCC2 Gene Polymorphisms with Methotrexate Serum Levels and Toxic Side Effects in Children with Acute Lymphoblastic Leukemia. Pediatr. Hematol. Oncol. 2014, 31, 169–177. [Google Scholar] [CrossRef]
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M.; et al. Role of Multidrug Resistance Protein 2 (MRP2) in Chemoresistance and Clinical Outcome in Oesophageal Squamous Cell Carcinoma. Br. J. Cancer 2011, 104, 707. [Google Scholar] [CrossRef] [PubMed]
- Huisman, M.T.; Chhatta, A.A.; Van Tellingen, O.; Beijnen, J.H.; Schinkel, A.H. MRP2 (ABCC2) Transports Taxanes and Confers Paclitaxel Resistance and Both Processes Are Stimulated by Probenecid. Int. J. Cancer 2005, 116, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Tam, A.; Santi, S.A.; Parissenti, A.M. Role of Autophagy and Lysosomal Drug Sequestration in Acquired Resistance to Doxorubicin in MCF-7 Cells. BMC Cancer 2016, 16, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, Y.; Jing, L.; Wang, J.; Yan, Y.; Feng, Y.; Zhang, Y.; Liu, Z.; Ma, L.; Diao, A. Lysosome Inhibitors Enhance the Chemotherapeutic Activity of Doxorubicin in HepG2 Cells. Chemotherapy 2017, 62, 85–93. [Google Scholar] [CrossRef]
- Sonee, M.; Barrón, E.; Yarber, F.A.; Hamm-Alvarez, S.F. Taxol Inhibits Endosomal-Lysosomal Membrane Trafficking at Two Distinct Steps in CV-1 Cells. Am. J. Physiol. 1998, 275, C1630–C1639. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, Y.-T.; Xiang, M.; Qiu, J.-L.; Luo, S.-Q.; Lin, F. Enhanced Lysosomal Function Is Critical for Paclitaxel Resistance in Cancer Cells: Reversed by Artesunate. Acta Pharmacol. Sin. 2020, 42, 624. [Google Scholar] [CrossRef]
- Lund, K.; Olsen, C.E.; Wong, J.J.W.; Olsen, P.A.; Solberg, N.T.; Høgset, A.; Krauss, S.; Selbo, P.K. 5-FU Resistant EMT-like Pancreatic Cancer Cells Are Hypersensitive to Photochemical Internalization of the Novel Endoglin-Targeting Immunotoxin CD105-Saporin. J. Exp. Clin. Cancer Res. 2017, 36, 187. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | HT-29 | MCF-7 |
|---|---|---|
| IC50 (µM) | IC50 (µM) | |
| DOX | N.D. | 0.17 |
| PTX | N.D | 2.78 (nM) |
| 5-FU | 3 | N.D. |
| FLUOX | 6.12 | 7.78 |
| BENZ | 18.23 | 21.71 |
| THIO | 4.26 | 5.72 |
| SERT | 2.45 | 2.22 |
| FLUPH | 1.86 | 2.68 |
| ART | 17.88 | 11.60 |
| CQ | 32.13 | 63.98 * |
| Drug | IC50 (Non-Tumoral Cells) | IC50 (Tumoral Cells) | SI |
|---|---|---|---|
| DOX | 0.43 µM | 0.17 µM (MCF-7) | 2.53 |
| PTX | 2.61 nM | 2.78 nM (MCF-7) | 0.94 |
| 5-FU | 5.78 µM | 3.00 µM (HT-29) | 1.93 |
| Drug | IC50 (Non-Tumoral Cells) | IC50 (Tumoral Cells) | SI |
|---|---|---|---|
| ART | 14.70 µM | 11.60 µM | 1.27 |
| CQ | 14.85 µM | 63.98 µM | 0.23 |
| Drug | IC50 (Non-Tumoral Cells) | IC50 (Tumoral Cells) | SI |
|---|---|---|---|
| FLUOX | 16.59 µM | 7.78 µM (MCF-7) | 2.13 |
| FLUPH | 6.87 µM | 2.68 µM (MCF-7) | 2.56 |
| BENZ | 19.91 µM | 21.71 µM (MCF-7) | 0.92 |
| THIO | 5.46 µM | 4.26 µM (HT-29) | 1.28 |
| SERT | 5.20 µM | 2.45 µM (HT-29) | 2.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, D.; Nunes, M.; Ricardo, S.; Vale, N. Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action. Biomolecules 2022, 12, 1490. https://doi.org/10.3390/biom12101490
Duarte D, Nunes M, Ricardo S, Vale N. Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action. Biomolecules. 2022; 12(10):1490. https://doi.org/10.3390/biom12101490
Chicago/Turabian StyleDuarte, Diana, Mariana Nunes, Sara Ricardo, and Nuno Vale. 2022. "Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action" Biomolecules 12, no. 10: 1490. https://doi.org/10.3390/biom12101490
APA StyleDuarte, D., Nunes, M., Ricardo, S., & Vale, N. (2022). Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action. Biomolecules, 12(10), 1490. https://doi.org/10.3390/biom12101490