
Transcriptome Meta-Analysis Confirms the Hidradenitis Suppurativa Pathogenic Triad: Upregulated Inflammation, Altered Epithelial Organization, and Dysregulated Metabolic Signaling


 , ,
, ,  , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
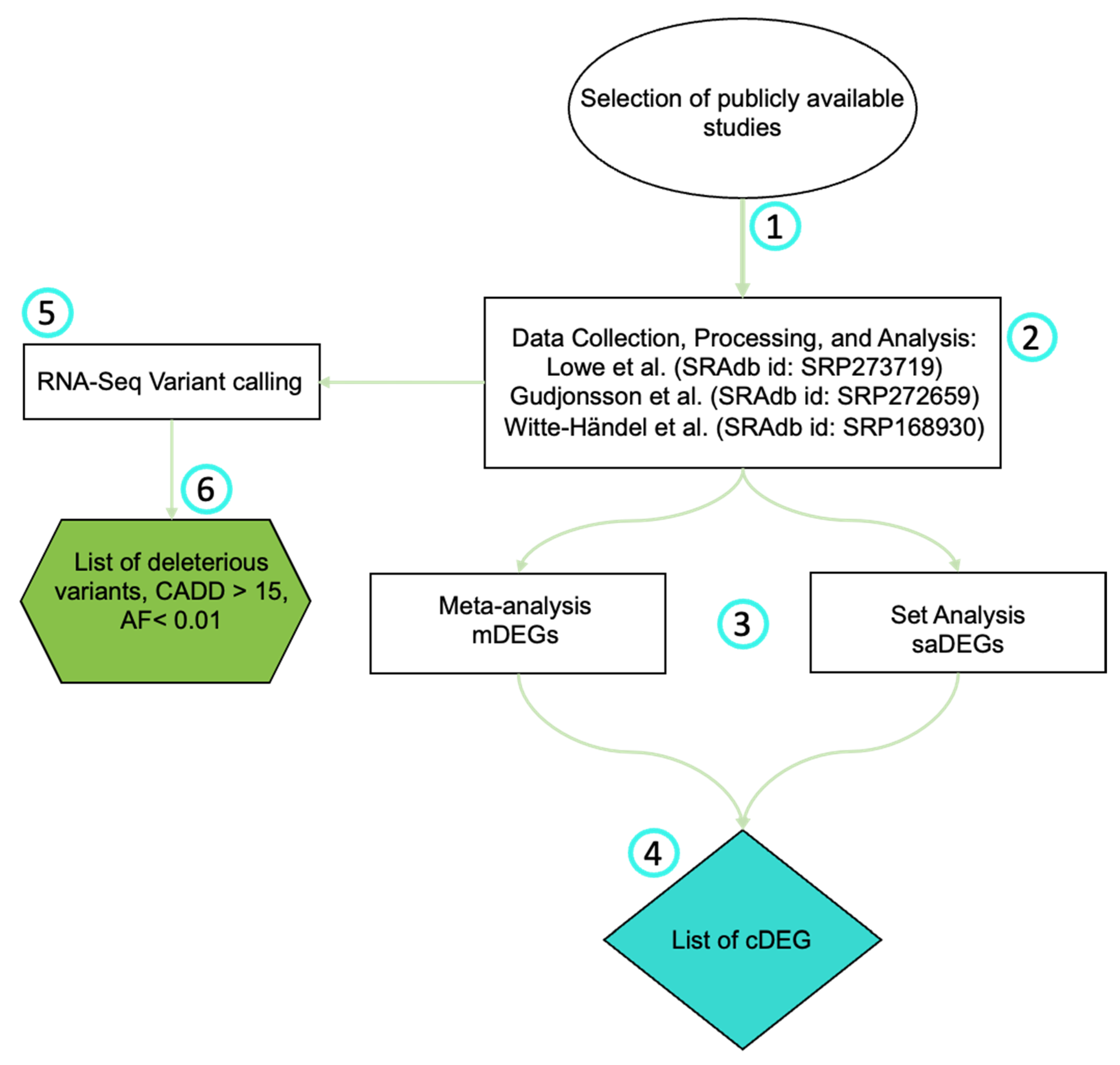
2. Materials and Methods
2.1. Selection of Publicly Available Studies
2.2. RNA-Seq Data Collection, Processing, and Analysis
2.3. Meta-Analysis
2.4. Pathway Analysis
2.5. RNA-Seq Variant Calling

2.6. Statistics
3. Results
3.1. Dataset Selection
3.2. Meta-Analisis
3.3. RNA-Seq Variant Calling
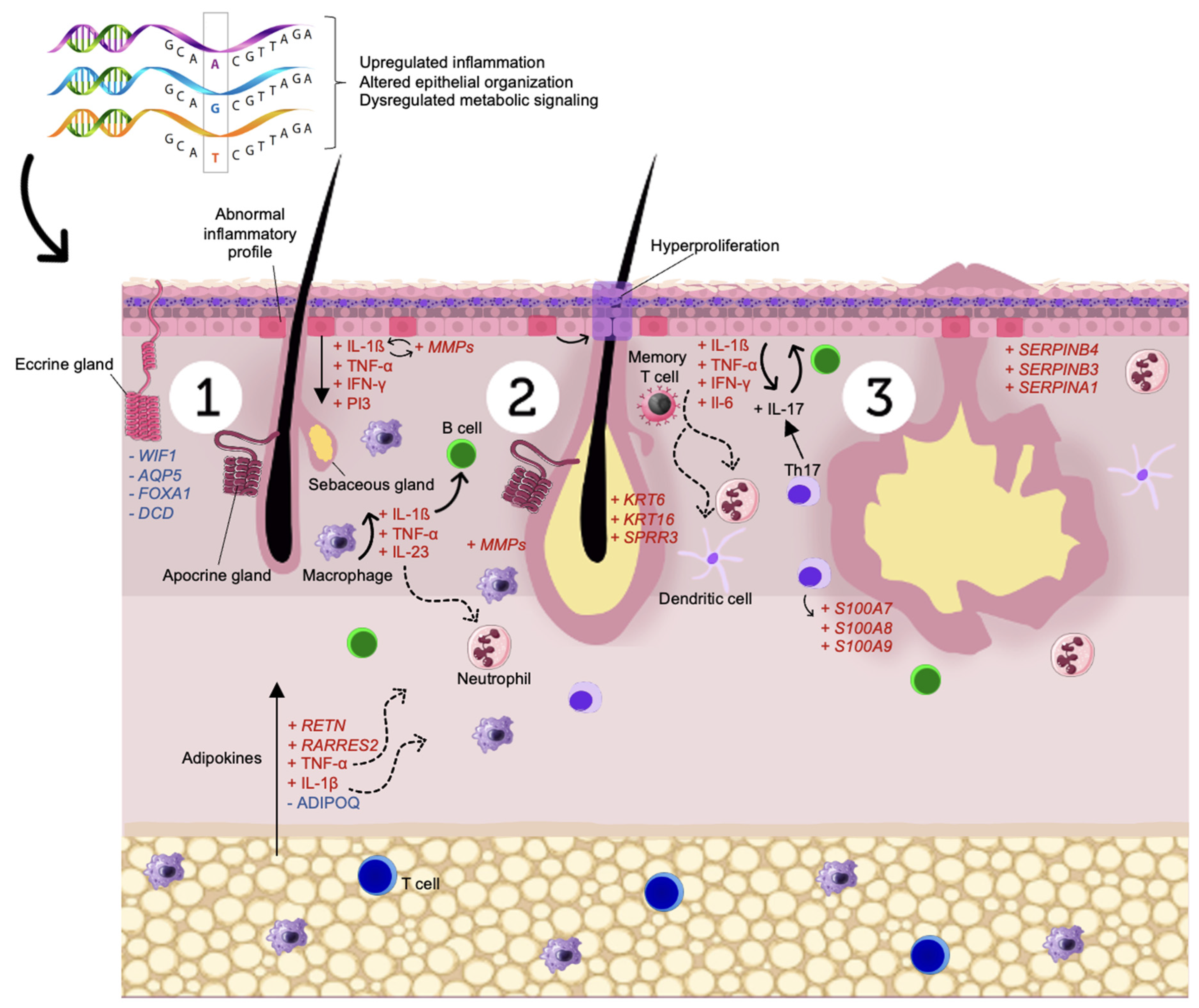
4. Discussion
4.1. Immune Dysregulation
4.2. Skin Homeostasis
4.3. Energy Metabolism
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Katoulis, A.C.; Liakou, A.I.; Rotsiamis, N.; Bonovas, S.; Bozi, E.; Rallis, E.; Christodoulou, C.; Rigopoulos, D. Descriptive Epidemiology of Hidradenitis Suppurativa in Greece: A Study of 152 Cases. Ski. Appendage Disord. 2017, 3, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Ralf Paus, L.; Kurzen, H.; Kurokawa, I.; Jemec, G.B.E.; Emtestam, L.; Sellheyer, K.; Giamarellos-Bourboulis, E.J.; Nagy, I.; Bechara, F.G.; Sartorius, K.; et al. What Causes Hidradenitis Suppurativa? Exp. Dermatol. 2008, 17, 455–456. [Google Scholar] [CrossRef]
- Dunstan, R.W.; Salte, K.M.; Todorović, V.; Lowe, M.; Wetter, J.B.; Harms, P.W.; Burney, R.E.; Scott, V.E.; Smith, K.M.; Rosenblum, M.D.; et al. Histologic Progression of Acne Inversa/Hidradenitis Suppurativa: Implications for Future Investigations and Therapeutic Intervention. Exp. Dermatol. 2021, 30, 820–830. [Google Scholar] [CrossRef]
- Jemec, G.B.E. Hidradenitis Suppurativa. N. Engl. J. Med. 2012, 366, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, Ł. Profound Consequences of Hidradenitis Suppurativa: A Review. Br. J. Dermatol. 2020, 183, e171–e177. [Google Scholar] [CrossRef] [PubMed]
- Dufour, D.N.; Emtestam, L.; Jemec, G.B. Hidradenitis Suppurativa: A Common and Burdensome, yet under-Recognised, Inflammatory Skin Disease. Postgrad. Med. J. 2014, 90, 216–221. [Google Scholar] [CrossRef] [PubMed]
- von Laffert, M.; Helmbold, P.; Wohlrab, J.; Fiedler, E.; Stadie, V.; Marsch, W.C. Hidradenitis Suppurativa (Acne Inversa): Early Inflammatory Events at Terminal Follicles and at Interfollicular Epidermis*. Exp. Dermatol. 2009, 19, 533–537. [Google Scholar] [CrossRef]
- von Laffert, M.; Stadie, V.; Wohlrab, J.; Marsch, W.C. Hidradenitis Suppurativa/Acne Inversa: Bilocated Epithelial Hyperplasia with Very Different Sequelae. Br. J. Dermatol. 2011, 164, 367–371. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Nogueira da Costa, A.; Makrantonaki, E.; Hou, X.X.; Almansouri, D.; Dudley, J.T.; Edwards, H.; Readhead, B.; Balthasar, O.; Jemec, G.B.E.; et al. Alterations in Innate Immunity and Epithelial Cell Differentiation Are the Molecular Pillars of Hidradenitis Suppurativa. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 846–861. [Google Scholar] [CrossRef]
- Duchatelet, S.; Miskinyte, S.; Delage, M.; Ungeheuer, M.-N.; Lam, T.; Benhadou, F.; del Marmol, V.; Vossen, A.R.J.V.; Prens, E.P.; Cogrel, O.; et al. Low Prevalence of GSC Gene Mutations in a Large Cohort of Predominantly Caucasian Patients with Hidradenitis Suppurativa. J. Investig. Dermatol. 2020, 140, 2085–2088.e14. [Google Scholar] [CrossRef]
- Pink, A.E.; Simpson, M.A.; Desai, N.; Dafou, D.; Hills, A.; Mortimer, P.; Smith, C.H.; Trembath, R.C.; Barker, J.N.W. Mutations in the γ-Secretase Genes NCSTN, PSENEN, and PSEN1 Underlie Rare Forms of Hidradenitis Suppurativa (Acne Inversa). J. Investig. Dermatol. 2012, 132, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Genovese, G.; Moltrasio, C.; Tricarico, P.M.; Gratton, R.; Piaserico, S.; Garcovich, S.; Boniotto, M.; Brandão, L.; Moura, R.; et al. Whole-Exome Sequencing in 10 Unrelated Patients with Syndromic Hidradenitis Suppurativa: A Preliminary Step for a Genotype-Phenotype Correlation. Dermatology 2022, 238, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Benhadou, F.; Byrd, A.S.; Chandran, N.S.; Giamarellos-Bourboulis, E.J.; Fabbrocini, G.; Frew, J.W.; Fujita, H.; González-López, M.A.; Guillem, P.; et al. What Causes Hidradenitis Suppurativa?—15 Years After. Exp. Dermatol. 2020, 29, 1154–1170. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Lin, M.-H.; Tian, X.; Cheng, H.-T.; Gridley, T.; Shen, J.; Kopan, R. γ-Secretase Functions through Notch Signaling to Maintain Skin Appendages but Is Not Required for Their Patterning or Initial Morphogenesis. Dev. Cell 2004, 7, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Plewig, G. Impaired Notch-MKP-1 Signalling in Hidradenitis Suppurativa: An Approach to Pathogenesis by Evidence from Translational Biology. Exp. Dermatol. 2013, 22, 172–177. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-Genome Sequencing Identifies Recurrent Mutations in Chronic Lymphocytic Leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key Issues in Conducting a Meta-Analysis of Gene Expression Microarray Datasets. PLoS Med. 2008, 5, e184. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef]
- Brandão, L.A.C.; Tricarico, P.M.; Gratton, R.; Agrelli, A.; Zupin, L.; Abou-Saleh, H.; Moura, R.; Crovella, S. Multiomics Integration in Skin Diseases with Alterations in Notch Signaling Pathway: PlatOMICs Phase 1 Deployment. Int. J. Mol. Sci. 2021, 22, 1523. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, Y.K.; Yoon, C.-H.; Kim, K.; Kim, K.-C. Meta-Analysis of Gene Expression Profiles in Long-Term Non-Progressors Infected with HIV-1. BMC Med. Genom. 2019, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.V.C.; Gratton, R.; de Melo, J.P.B.; Andrade-Santos, J.L.; Guimarães, R.L.; Crovella, S.; Tricarico, P.M.; Brandão, L.A.C. HIV-1 Infection Transcriptomics: Meta-Analysis of CD4+ T Cells Gene Expression Profiles. Viruses 2021, 13, 244. [Google Scholar] [CrossRef]
- Zhu, Y.; Stephens, R.M.; Meltzer, P.S.; Davis, S.R. SRAdb: Query and Use Public next-Generation Sequencing Data from within R. BMC Bioinform. 2013, 14, 19. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 9 June 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R Package Rsubread Is Easier, Faster, Cheaper and Better for Alignment and Quantification of RNA Sequencing Reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for Large-Scale Genome and Gene Function Analysis with the PANTHER Classification System (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Panther Classification System. Available online: http://www.pantherdb.org/ (accessed on 19 July 2022).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Wertenteil, S.; Baltz, R.; Strunk, A.; Finelt, N. Prevalence Estimates for Hidradenitis Suppurativa among Children and Adolescents in the United States: A Gender- and Age-Adjusted Population Analysis. J. Investig. Dermatol. 2018, 138, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
- Jemec, G.B.E.; Heidenheim, M.; Nielsen, N.H. The Prevalence of Hidradenitis Suppurativa and Its Potential Precursor Lesions. J. Am. Acad. Dermatol. 1996, 35, 191–194. [Google Scholar] [CrossRef]
- Kirby, J.S.; Zaenglein, A.L. Recognizing the Effects and Disparities of Pediatric Hidradenitis Suppurativa. JAMA Dermatol. 2021, 157, 379. [Google Scholar] [CrossRef]
- Lowe, M.M.; Naik, H.B.; Clancy, S.; Pauli, M.; Smith, K.M.; Bi, Y.; Dunstan, R.; Gudjonsson, J.E.; Paul, M.; Harris, H.; et al. Immunopathogenesis of Hidradenitis Suppurativa and Response to Anti–TNF-α Therapy. JCI Insight 2020, 5, e139932. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Tsoi, L.C.; Ma, F.; Billi, A.C.; van Straalen, K.R.; Vossen, A.R.J.V.; van der Zee, H.H.; Harms, P.W.; Wasikowski, R.; Yee, C.M.; et al. Contribution of Plasma Cells and B Cells to Hidradenitis Suppurativa Pathogenesis. JCI Insight 2020, 5, e139930. [Google Scholar] [CrossRef]
- Witte-Händel, E.; Wolk, K.; Tsaousi, A.; Irmer, M.L.; Mößner, R.; Shomroni, O.; Lingner, T.; Witte, K.; Kunkel, D.; Salinas, G.; et al. The IL-1 Pathway Is Hyperactive in Hidradenitis Suppurativa and Contributes to Skin Infiltration and Destruction. J. Investig. Dermatol. 2019, 139, 1294–1305. [Google Scholar] [CrossRef]
- Kolbinger, F.; Loesche, C.; Valentin, M.-A.; Jiang, X.; Cheng, Y.; Jarvis, P.; Peters, T.; Calonder, C.; Bruin, G.; Polus, F.; et al. β-Defensin 2 Is a Responsive Biomarker of IL-17A–Driven Skin Pathology in Patients with Psoriasis. J. Allergy Clin. Immunol. 2017, 139, 923–932. [Google Scholar] [CrossRef]
- Clausen, M.-L.; Jungersted, J.M.; Andersen, P.S.; Slotved, H.-C.; Krogfelt, K.A.; Agner, T. Human β-Defensin-2 as a Marker for Disease Severity and Skin Barrier Properties in Atopic Dermatitis. Br. J. Dermatol. 2013, 169, 587–593. [Google Scholar] [CrossRef]
- Cao, Y.; Harvey, B.P.; Hong, F.; Ruzek, M.; Wang, J.; Murphy, E.R.; Kaymakcalan, Z. Adalimumab Induces a Wound Healing Profile in Patients with Hidradenitis Suppurativa by Regulating Macrophage Differentiation and Matrix Metalloproteinase Expression. J. Investig. Dermatol. 2021, 141, 2730–2740.e9. [Google Scholar] [CrossRef]
- Alowami, S.; Qing, G.; Emberley, E.; Snell, L.; Watson, P.H. Psoriasin (S100A7) Expression Is Altered during Skin Tumorigenesis. BMC Dermatol. 2003, 3, 1. [Google Scholar] [CrossRef]
- Mirastschijski, U.; Lupše, B.; Maedler, K.; Sarma, B.; Radtke, A.; Belge, G.; Dorsch, M.; Wedekind, D.; McCawley, L.J.; Boehm, G.; et al. Matrix Metalloproteinase-3 Is Key Effector of TNF-α-Induced Collagen Degradation in Skin. Int. J. Mol. Sci. 2019, 20, 5234. [Google Scholar] [CrossRef] [PubMed]
- Pine, G.M.; Batugedara, H.M.; Nair, M.G. Here, There and Everywhere: Resistin-like Molecules in Infection, Inflammation, and Metabolic Disorders. Cytokine 2018, 110, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Er, L.-K.; Wu, S.; Hsu, L.-A.; Teng, M.-S.; Sun, Y.-C.; Ko, Y.-L. Pleiotropic Associations of RARRES2 Gene Variants and Circulating Chemerin Levels: Potential Roles of Chemerin Involved in the Metabolic and Inflammation-Related Diseases. Mediat. Inflamm. 2018, 2018, 4670521. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Pfundt, R.; Zeeuwen, P.; Molhuizen, H.; Schalkwijk, J. Transcriptional Regulation of the Elafin Gene in Human Keratinocytes. J. Investig. Dermatol. 2003, 120, 301–307. [Google Scholar] [CrossRef]
- Sivaprasad, U.; Kinker, K.G.; Ericksen, M.B.; Lindsey, M.; Gibson, A.M.; Bass, S.A.; Hershey, N.S.; Deng, J.; Medvedovic, M.; Khurana Hershey, G.K. SERPINB3/B4 Contributes to Early Inflammation and Barrier Dysfunction in an Experimental Murine Model of Atopic Dermatitis. J. Investig. Dermatol. 2015, 135, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.; Chaowattanapanit, S.; Kiratikanon, S.; Chaiwarith, R.; Choonhakarn, C.; Intachai, W.; Quarto, N.; Tongsima, S.; Ketudat Cairns, J.R.; Ngamphiw, C.; et al. SERPINA1, Generalized Pustular Psoriasis, and Adult-onset Immunodeficiency. J. Dermatol. 2021, 48, 1597–1601. [Google Scholar] [CrossRef]
- Lessard, J.C.; Piña-Paz, S.; Rotty, J.D.; Hickerson, R.P.; Kaspar, R.L.; Balmain, A.; Coulombe, P.A. Keratin 16 Regulates Innate Immunity in Response to Epidermal Barrier Breach. Proc. Natl. Acad. Sci. USA 2013, 110, 19537–19542. [Google Scholar] [CrossRef]
- Kainu, K.; Kivinen, K.; Zucchelli, M.; Suomela, S.; Kere, J.; Inerot, A.; Baker, B.S.; Powles, A.V.; Fry, L.; Samuelsson, L.; et al. Association of Psoriasis to PGLYRP and SPRR Genes at PSORS4 Locus on 1q Shows Heterogeneity between Finnish, Swedish and Irish Families. Exp. Dermatol. 2009, 18, 109–115. [Google Scholar] [CrossRef]
- Keil, K.P.; Mehta, V.; Branam, A.M.; Abler, L.L.; Buresh-Stiemke, R.A.; Joshi, P.S.; Schmitz, C.T.; Marker, P.C.; Vezina, C.M. Wnt Inhibitory Factor 1 (Wif1) Is Regulated by Androgens and Enhances Androgen-Dependent Prostate Development. Endocrinology 2012, 153, 6091–6103. [Google Scholar] [CrossRef]
- Yen, C.-L.E.; Brown, C.H.; Monetti, M.; Farese, R.V. A Human Skin Multifunctional O-Acyltransferase That Catalyzes the Synthesis of Acylglycerols, Waxes, and Retinyl Esters. J. Lipid Res. 2005, 46, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-A.; Ka, S.-O.; Moon, W.S.; Yi, H.-K.; Lee, Y.-H.; Kwon, K.-B.; Park, J.-W.; Park, B.-H. Effect of Dermcidin, an Antimicrobial Peptide, on Body Fat Mobilization in Normal Mice. J. Endocrinol. 2008, 198, 111–118. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahonen, M.A.; Höring, M.; Nguyen, V.D.; Qadri, S.; Taskinen, J.H.; Nagaraj, M.; Wabitsch, M.; Fischer-Posovszky, P.; Zhou, Y.; Liebisch, G.; et al. Insulin-Inducible THRSP Maintains Mitochondrial Function and Regulates Sphingolipid Metabolism in Human Adipocytes. Mol. Med. 2022, 28, 68. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, P.I.; Rogers, A.; Elliot, D.J.; Chau, N.; Hulin, J.-A.; Miners, J.O.; Meech, R. The Novel UDP Glycosyltransferase 3A2: Cloning, Catalytic Properties, and Tissue Distribution. Mol. Pharmacol. 2011, 79, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Massi, D.; Panelos, J. Notch Signaling and the Developing Skin Epidermis. In Notch Signaling in Embryology and Cancer; Springer: Berlin/Heidelberg, Germany, 2012; pp. 131–141. [Google Scholar]
- Radtke, F.; Fasnacht, N.; MacDonald, H.R. Notch Signaling in the Immune System. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Pavlovsky, M.; Sarig, O.; Eskin-Schwartz, M.; Malchin, N.; Bochner, R.; Mohamad, J.; Gat, A.; Peled, A.; Hafner, A.; Sprecher, E. A Phenotype Combining Hidradenitis Suppurativa with Dowling-Degos Disease Caused by a Founder Mutation in PSENEN. Br. J. Dermatol. 2018, 178, 502–508. [Google Scholar] [CrossRef]
- Jfri, A.; Litvinov, I.V.; Netchiporouk, E.; O’Brien, E. Novel Variants of MEFV and NOD2 Genes in Familial Hidradenitis Suppurativa: A Case Report. SAGE Open Med. Case Rep. 2020, 8, 2050313X2095311. [Google Scholar] [CrossRef]
- Higgins, R.; Pink, A.; Hunger, R.; Yawalkar, N.; Navarini, A.A. Generalized Comedones, Acne, and Hidradenitis Suppurativa in a Patient with an FGFR2 Missense Mutation. Front. Med. 2017, 4, 16. [Google Scholar] [CrossRef]
- Saito, N.; Minami-Hori, M.; Nagahata, H.; Nozaki, H.; Iinuma, S.; Igawa, S.; Kanno, K.; Kishibe, M.; Kanazawa, N.; Ishida-Yamamoto, A. Novel PSTPIP1 Gene Mutation in Pyoderma Gangrenosum, Acne and Suppurative Hidradenitis Syndrome. J. Dermatol. 2018, 45, e213–e214. [Google Scholar] [CrossRef]
- Witkowski, A.; Thweatt, J.; Smith, S. Mammalian ACSF3 Protein Is a Malonyl-CoA Synthetase That Supplies the Chain Extender Units for Mitochondrial Fatty Acid Synthesis. J. Biol. Chem. 2011, 286, 33729–33736. [Google Scholar] [CrossRef]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Arndt, B.; Krieger, T.; Kalinski, T.; Thielitz, A.; Reinhold, D.; Roessner, A.; Schraven, B.; Simeoni, L. The Transmembrane Adaptor Protein SIT Inhibits TCR-Mediated Signaling. PLoS ONE 2011, 6, e23761. [Google Scholar] [CrossRef]
- Saeki, M.; Irie, Y.; Ni, L.; Yoshida, M.; Itsuki, Y.; Kamisaki, Y. Monad, a WD40 Repeat Protein, Promotes Apoptosis Induced by TNF-α. Biochem. Biophys. Res. Commun. 2006, 342, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Brash, D.E.; Grossman, D. Keratinocyte Apoptosis in Epidermal Development and Disease. J. Investig. Dermatol. 2006, 126, 243–257. [Google Scholar] [CrossRef]
- Dong, Y.; Kang, H.; Liu, H.; Wang, J.; Guo, Q.; Song, C.; Sun, Y.; Zhang, Y.; Zhang, H.; Zhang, Z.; et al. Myoferlin, a Membrane Protein with Emerging Oncogenic Roles. BioMed Res. Int. 2019, 2019, 7365913. [Google Scholar] [CrossRef] [PubMed]
- González-López, M.A.; Ocejo-Viñals, J.G.; López-Sundh, A.E.; Guiral, S.; Ruiz-Solana, M.; Mata, C.; Portilla, V.; Corrales, A.; Blanco, R.; Hernández, J.L. Biomarkers of Endothelial Dysfunction and Atherosclerosis in Hidradenitis Suppurativa. J. Dermatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, Ł.; Batycka-Baran, A.; Bieniek, A.; Szepietowski, J.C. Decreased Number of Circulating Endothelial Progenitor Cells in Hidradenitis Suppurativa Patients. Dermatology 2015, 230, 228–233. [Google Scholar] [CrossRef]
- Shah, A.; Alhusayen, R.; Amini-Nik, S. The Critical Role of Macrophages in the Pathogenesis of Hidradenitis Suppurativa. Inflamm. Res. 2017, 66, 931–945. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-Wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Banerjee, A.; McNish, S.; Shanmugam, V.K. Interferon-Gamma (IFN-γ) Is Elevated in Wound Exudate from Hidradenitis Suppurativa. Immunol. Investig. 2017, 46, 149–158. [Google Scholar] [CrossRef]
- Hessam, S.; Sand, M.; Gambichler, T.; Skrygan, M.; Rüddel, I.; Bechara, F.G. Interleukin-36 in Hidradenitis Suppurativa: Evidence for a Distinctive Proinflammatory Role and a Key Factor in the Development of an Inflammatory Loop. Br. J. Dermatol. 2018, 178, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schäfer, M.; Coyle, A.J.; Renauld, J.-C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform Dermatitis Is Driven by IL-36–Mediated DC-Keratinocyte Crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef]
- Vossen, A.R.J.V.; van der Zee, H.H.; Prens, E.P. Hidradenitis Suppurativa: A Systematic Review Integrating Inflammatory Pathways Into a Cohesive Pathogenic Model. Front. Immunol. 2018, 9, 2965. [Google Scholar] [CrossRef] [PubMed]
- Keijsers, R.R.M.C.; Joosten, I.; van Erp, P.E.J.; Koenen, H.J.P.M.; van de Kerkhof, P.C.M. Cellular Sources of IL-17 in Psoriasis: A Paradigm Shift? Exp. Dermatol. 2014, 23, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Hotz, C.; Boniotto, M.; Guguin, A.; Surenaud, M.; Jean-Louis, F.; Tisserand, P.; Ortonne, N.; Hersant, B.; Bosc, R.; Poli, F.; et al. Intrinsic Defect in Keratinocyte Function Leads to Inflammation in Hidradenitis Suppurativa. J. Investig. Dermatol. 2016, 136, 1768–1780. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Cepok, S.; Sellner, J.; Grummel, V.; Weber, M.S.; Korn, T.; Berthele, A.; Hemmer, B. CXCL13 Is the Major Determinant for B Cell Recruitment to the CSF during Neuroinflammation. J. Neuroinflammation 2012, 9, 93. [Google Scholar] [CrossRef]
- Frew, J.W.; Grand, D.; Navrazhina, K.; Krueger, J.G. Beyond Antibodies: B Cells in Hidradenitis Suppurativa: Bystanders, Contributors or Therapeutic Targets? Exp. Dermatol. 2020, 29, 509–515. [Google Scholar] [CrossRef]
- Takahashi, K.; Yanagi, T.; Kitamura, S.; Hata, H.; Imafuku, K.; Iwami, D.; Hotta, K.; Morita, K.; Shinohara, N.; Shimizu, H. Successful Treatment of Hidradenitis Suppurativa with Rituximab for a Patient with Idiopathic Carpotarsal Osteolysis and Chronic Active Antibody-Mediated Rejection. J. Dermatol. 2018, 45, e116–e117. [Google Scholar] [CrossRef]
- Faivre, C.; Villani, A.P.; Aubin, F.; Lipsker, D.; Bottaro, M.; Cohen, J.-D.; Durupt, F.; Jeudy, G.; Sbidian, E.; Toussirot, E.; et al. Hidradenitis Suppurativa (HS): An Unrecognized Paradoxical Effect of Biologic Agents (BA) Used in Chronic Inflammatory Diseases. J. Am. Acad. Dermatol. 2016, 74, 1153–1159. [Google Scholar] [CrossRef]
- Verrier, T.; Solhonne, B.; Sallenave, J.-M.; Garcia-Verdugo, I. The WAP Protein Trappin-2/Elafin: A Handyman in the Regulation of Inflammatory and Immune Responses. Int. J. Biochem. Cell Biol. 2012, 44, 1377–1380. [Google Scholar] [CrossRef]
- Wiedow, O.; Schröder, J.M.; Gregory, H.; Young, J.A.; Christophers, E. Elafin: An Elastase-Specific Inhibitor of Human Skin. Purification, Characterization, and Complete Amino Acid Sequence. J. Biol. Chem. 1990, 265, 14791–14795. [Google Scholar] [CrossRef]
- Jensen, P.J.; Yang, T.; Yu, D.-W.; Baker, M.S.; Risse, B.; Sun, T.-T.; Lavker, R.M. Serpins in the Human Hair Follicle. J. Investig. Dermatol. 2000, 114, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Krueger, J.G.; Li, K.; Jabbari, A.; Brodmerkel, C.; Lowes, M.A.; Suárez-Fariñas, M. Meta-Analysis Derived (MAD) Transcriptome of Psoriasis Defines the “Core” Pathogenesis of Disease. PLoS ONE 2012, 7, e44274. [Google Scholar] [CrossRef]
- Kaakati, R.N.; Tanaka, J.; Liu, B.; Ward, R.; Macleod, A.S.; Green, C.L.; Jaleel, T. Atopic Dermatitis Is Associated with Hidradenitis Suppurativa Diagnosis: A Single Institution Retrospective Cohort Study. JAAD Int. 2021, 4, 18–24. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Moran, B.; Petrasca, A.; Smith, C.M. IL-17 in Inflammatory Skin Diseases Psoriasis and Hidradenitis Suppurativa. Clin. Exp. Immunol. 2020, 201, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, M.; Zhang, L. Keratin 6, 16 and 17—Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Hinchliffe, T.E.; Wu, T. Biomarkers of An Autoimmune Skin Disease—Psoriasis. Genom. Proteom. Bioinform. 2015, 13, 224–233. [Google Scholar] [CrossRef]
- Mommers, J.M.; van Rossum, M.M.; van Erp, P.E.J.; van de Kerkhof, P.C.M. Changes in Keratin 6 and Keratin 10 (Co-)Expression in Lesional and Symptomless Skin of Spreading Psoriasis. Dermatology 2000, 201, 15–20. [Google Scholar] [CrossRef]
- Tesfaigzi, J.; Carlson, D.M. Expression, Regulation, and Function of the SPR Family of Proteins. Cell Biochem. Biophys. 1999, 30, 243–265.e1. [Google Scholar] [CrossRef]
- Elias, P.M.; Wakefield, J.S. Mechanisms of Abnormal Lamellar Body Secretion and the Dysfunctional Skin Barrier in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2014, 134, 781–791. [Google Scholar] [CrossRef]
- Carregaro, F.; Stefanini, A.C.B.; Henrique, T.; Tajara, E.H. Study of Small Proline-Rich Proteins (SPRRs) in Health and Disease: A Review of the Literature. Arch. Dermatol. Res. 2013, 305, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Suárez-Fariñas, M.; Chiricozzi, A.; Nograles, K.E.; Shemer, A.; Fuentes-Duculan, J.; Cardinale, I.; Lin, P.; Bergman, R.; Bowcock, A.M.; et al. Broad Defects in Epidermal Cornification in Atopic Dermatitis Identified through Genomic Analysis. J. Allergy Clin. Immunol. 2009, 124, 1235–1244.e58. [Google Scholar] [CrossRef] [PubMed]
- Lukes, A.; Mun-Bryce, S.; Lukes, M.; Rosenberg, G.A. Extracellular Matrix Degradation by Metalloproteinases and Central Nervous System Diseases. Mol. Neurobiol. 1999, 19, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Join-Lambert, O.; Sabat, R. Aetiology and Pathogenesis of Hidradenitis Suppurativa. Br. J. Dermatol. 2020, 183, 999–1010. [Google Scholar] [CrossRef]
- Opdenakker, G.; van den Steen, P.E.; Dubois, B.; Nelissen, I.; van Coillie, E.; Masure, S.; Proost, P.; van Damme, J. Gelatinase B Functions as Regulator and Effector in Leukocyte Biology. J. Leukoc. Biol. 2001, 69, 851–859. [Google Scholar] [PubMed]
- Sanchez, J.; le Jan, S.; Muller, C.; François, C.; Renard, Y.; Durlach, A.; Bernard, P.; Reguiai, Z.; Antonicelli, F. Matrix Remodelling and MMP Expression/Activation Are Associated with Hidradenitis Suppurativa Skin Inflammation. Exp. Dermatol. 2019, 28, 593–600. [Google Scholar] [CrossRef]
- Rittié, L.; Sachs, D.L.; Orringer, J.S.; Voorhees, J.J.; Fisher, G.J. Eccrine Sweat Glands Are Major Contributors to Reepithelialization of Human Wounds. Am. J. Pathol. 2013, 182, 163–171. [Google Scholar] [CrossRef]
- Lu, C.P.; Polak, L.; Rocha, A.S.; Pasolli, H.A.; Chen, S.-C.; Sharma, N.; Blanpain, C.; Fuchs, E. Identification of Stem Cell Populations in Sweat Glands and Ducts Reveals Roles in Homeostasis and Wound Repair. Cell 2012, 150, 136–150. [Google Scholar] [CrossRef]
- Coates, M.; Mariottoni, P.; Corcoran, D.L.; Kirshner, H.F.; Jaleel, T.; Brown, D.A.; Brooks, S.R.; Murray, J.; Morasso, M.I.; MacLeod, A.S. The Skin Transcriptome in Hidradenitis Suppurativa Uncovers an Antimicrobial and Sweat Gland Gene Signature Which Has Distinct Overlap with Wounded Skin. PLoS ONE 2019, 14, e0216249. [Google Scholar] [CrossRef]
- Guo, L.; Chen, H.; Li, Y.; Zhou, Q.; Sui, Y. An Aquaporin 3-Notch1 Axis in Keratinocyte Differentiation and Inflammation. PLoS ONE 2013, 8, e80179. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Mentino, D.; de Marco, A.; del Vecchio, C.; Garra, S.; Cazzato, G.; Foti, C.; Crovella, S.; Calamita, G. Aquaporins Are One of the Critical Factors in the Disruption of the Skin Barrier in Inflammatory Skin Diseases. Int. J. Mol. Sci. 2022, 23, 4020. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Chanwangpong, A.; Schneider-Burrus, S.; Metternich, D.; Kokolakis, G.; Kurek, A.; Philipp, S.; Uribe, D.; Wolk, K.; Sterry, W. Increased Prevalence of Metabolic Syndrome in Patients with Acne Inversa. PLoS ONE 2012, 7, e31810. [Google Scholar] [CrossRef] [PubMed]
- Mintoff, D.; Benhadou, F.; Pace, N.P.; Frew, J.W. Metabolic Syndrome and Hidradenitis Suppurativa: Epidemiological, Molecular, and Therapeutic Aspects. Int. J. Dermatol. 2021, 61, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Shalom, G.; Freud, T.; Harman-Boehm, I.; Polishchuk, I.; Cohen, A.D. Hidradenitis Suppurativa and Metabolic Syndrome: A Comparative Cross-sectional Study of 3207 Patients. Br. J. Dermatol. 2015, 173, 464–470. [Google Scholar] [CrossRef]
- Alberti, K.G.M.; Zimmet, P.; Shaw, J. The Metabolic Syndrome—A New Worldwide Definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Cameron, A.J.; Boyko, E.J.; Sicree, R.A.; Zimmet, P.Z.; Söderberg, S.; Alberti, K.G.M.M.; Tuomilehto, J.; Chitson, P.; Shaw, J.E. Central Obesity as a Precursor to the Metabolic Syndrome in the AusDiab Study and Mauritius. Obesity 2008, 16, 2707–2716. [Google Scholar] [CrossRef]
- Kromann, C.; Ibler, K.; Kristiansen, V.; Jemec, G. The Influence of Body Weight on the Prevalence and Severity of Hidradenitis Suppurativa. Acta Derm. Venereol. 2014, 94, 553–557. [Google Scholar] [CrossRef]
- Balgobind, A.; Finelt, N.; Strunk, A.; Garg, A. Association between Obesity and Hidradenitis Suppurativa among Children and Adolescents: A Population-Based Analysis in the United States. J. Am. Acad. Dermatol. 2020, 82, 502–504. [Google Scholar] [CrossRef]
- Kaleta, K.P.; Nikolakis, G.; Hossini, A.M.; Balthasar, O.; Almansouri, D.; Vaiopoulos, A.; Knolle, J.; Boguslawska, A.; Wojas-Pelc, A.; Zouboulis, C.C. Metabolic Disorders/Obesity Is a Primary Risk Factor in Hidradenitis Suppurativa: An Immunohistochemical Real-World Approach. Dermatology 2022, 238, 251–259. [Google Scholar] [CrossRef]
- Zouboulis, V.A.; Zouboulis, K.C.; Zouboulis, C.C. Hidradenitis Suppurativa and Comorbid Disorder Biomarkers, Druggable Genes, New Drugs and Drug Repurposing—A Molecular Meta-Analysis. Pharmaceutics 2021, 14, 44. [Google Scholar] [CrossRef]
- Curat, C.A.; Wegner, V.; Sengenès, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumié, A. Macrophages in Human Visceral Adipose Tissue: Increased Accumulation in Obesity and a Source of Resistin and Visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef]
- Wolk, K.; Sabat, R. Adipokines in Psoriasis: An Important Link between Skin Inflammation and Metabolic Alterations. Rev. Endocr. Metab. Disord. 2016, 17, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Kovács, D.; Fazekas, F.; Oláh, A.; Törőcsik, D. Adipokines in the Skin and in Dermatological Diseases. Int. J. Mol. Sci. 2020, 21, 9048. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The Fat-Derived Hormone Adiponectin Reverses Insulin Resistance Associated with Both Lipoatrophy and Obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Smith, U. Cytokines Promote Wnt Signaling and Inflammation and Impair the Normal Differentiation and Lipid Accumulation in 3T3-L1 Preadipocytes. J. Biol. Chem. 2006, 281, 9507–9516. [Google Scholar] [CrossRef]
- Gustafson, B.; Hammarstedt, A.; Andersson, C.X.; Smith, U. Inflamed Adipose Tissue. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2276–2283. [Google Scholar] [CrossRef]
- Andersson, C.X.; Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. Inflamed Adipose Tissue, Insulin Resistance and Vascular Injury. Diabetes. Metab. Res. Rev. 2008, 24, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kihara, S.; Arita, Y.; Nishida, M.; Matsuyama, A.; Okamoto, Y.; Ishigami, M.; Kuriyama, H.; Kishida, K.; Nishizawa, H.; et al. Adipocyte-Derived Plasma Protein, Adiponectin, Suppresses Lipid Accumulation and Class A Scavenger Receptor Expression in Human Monocyte-Derived Macrophages. Circulation 2001, 103, 1057–1063. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Argueta, J.G.M.; Masuhiro, Y.; Kagishita, M.; Nonaka, K.; Saito, T.; Hanazawa, S.; Yamashita, Y. Adiponectin Inhibits Toll-like Receptor Family-Induced Signaling. FEBS Lett. 2005, 579, 6821–6826. [Google Scholar] [CrossRef]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Enrich, B.; Tilg, H. Adiponectin Induces the Anti-Inflammatory Cytokines IL-10 and IL-1RA in Human Leukocytes. Biochem. Biophys. Res. Commun. 2004, 323, 630–635. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| SRA | Title | Samples Included in Our Study | Main Findings |
|---|---|---|---|
| SRP273719 | Immunopathogenesis of hidradenitis suppurativa and response to anti–TNF-α therapy | 42 samples (HS skin lesion pre-TNF = 19: HS skin lesion mild-moderate HS = 7: healthy skin control = 16) | Highly enriched pathways in HS lesioned skin are immune related. Signatures of complement activation, B cell signaling, and pathways involving phagocytosis were found to be unique to HS. TNF-α–regulated genes, IFN-γ, and IL-1β were selected as the major drivers of the inflammatory pathways in HS skin lesions. Nonetheless, IL-1 receptor antagonist, IL-1RN, and IL-10RA, 2 potent immunoregulatory molecules, were relatively reduced in HS skin. Alongside, α-catenin and sirtuin 1, both important for regulation of cell proliferation and survival, were reduced in HS skin. |
| SRP272659 | Contribution of plasma cells and B cells to hidradenitis suppurativa pathogenesis | 32 samples (HS skin lesion = 22: healthy skin control = 10) | Several upregulated genes in the skin were associated with B cell responses, including immunoglobulin genes such as IGLV3-27, CD19, and CD79a. Other important genes found were the antimicrobial gene DEFB4A; CXCL13, a B cell chemoattractant, and the neutrophil chemokine CXCL1. In summary, they found B cells, and in particular plasma cells, as a potential therapeutic target in HS. |
| SRP168930 | The IL-1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction | 7 samples (HS skin lesion = 3: healthy skin control = 4) | IL-1β is highly active in HS, contributing to local and systemic inflammation. IL-1β induces expression of many molecules involved in extracellular matrix destruction including MMPs, ADAM12, serpinA1, COL3A1, and COL10A1, and immune cell infiltration such as CXCL1, CXCL6, CCL7, CXCL10, CXCL16, CXCL13, CCL24, CCL2, CCL8, and CCL20. IL-1β and, therefore, MMP1, MMP3, MMP9, MMP10, CCL2, CXCL1, IL-6, and IL-32 were upregulated when compared with healthy control skin and psoriasis lesions. |
| Pathway Identifier | Pathway Name | Entities Found | FDR |
|---|---|---|---|
| R-HSA-198933 | Immunoregulatory interactions between a lymphoid and a non-lymphoid cell | 81 | 7.2839 × 10−31 |
| R-HSA-1474244 | Extracellular matrix organization | 109 | 2.4259 × 10−14 |
| R-HSA-380108 | Chemokine receptors bind chemokines | 35 | 1.4862 × 10−11 |
| R-HSA-373076 | Class A/1 (Rhodopsin-like receptors) | 106 | 2.2223 × 10−11 |
| R-HSA-202430 | Translocation of ZAP-70 to immunological synapse | 16 | 1.2195 × 10−9 |
| R-HSA-389948 | PD-1 signaling | 18 | 1.55 × 10−9 |
| R-HSA-6798695 | Neutrophil degranulation | 132 | 5.2463 × 10−9 |
| R-HSA-500792 | GPCR ligand binding | 126 | 5.25 × 10−9 |
| R-HSA-909733 | Interferon alpha/beta signaling | 33 | 5.2463 × 10−9 |
| R-HSA-202433 | Generation of second messenger molecules | 22 | 5.2463 × 10−9 |
| R-HSA-202427 | Phosphorylation of CD3 and TCR zeta chains | 17 | 5.2463 × 10−9 |
| R-HSA-1474228 | Degradation of the extracellular matrix | 55 | 1.2723 × 10−8 |
| R-HSA-877300 | Interferon gamma signaling | 40 | 3.5328 × 0−8 |
| R-HSA-1442490 | Collagen degradation | 32 | 6.6811 × 10−8 |
| R-HSA-375276 | Peptide ligand-binding receptors | 67 | 7.395 × 10−8 |
| R-HSA-6809371 | Formation of the cornified envelope | 50 | 8.368 × 10−8 |
| R-HSA-6785807 | Interleukin-4 and Interleukin-13 signaling | 44 | 1.1813 × 10−7 |
| R-HSA-6783783 | Interleukin-10 signaling | 25 | 2.22 × 10−7 |
| R-HSA-1474290 | Collagen formation | 37 | 7.0322 × 10−7 |
| R-HSA-418594 | G alpha (i) signaling events | 108 | 1.4245 × 10−6 |
| Gene | DEGs | SNP ID | Ref | Alt | Distribution of Genotypes among HS Patients | Wilcoxon Test | Exonic Function | HGVS | AF |
|---|---|---|---|---|---|---|---|---|---|
| NCSTN | - | rs35603924 | G | C | GG (50)/GC (1) | 0.1087 | nonsynonymous SNV | c.G231C:p.E77D | 0.00432 |
| APH1A | - | rs996158631 | A | T | AA (50)/AT (1) | 0.5630 | nonsynonymous SNV | c.T123A:p.D41E | 0.00010 |
| APH1B | - | rs142676640 | C | T | CC (50)/CT (1) | 0.2229 | nonsynonymous SNV | c.C640T:p.R214 | 0.000699 |
| PSEN2 | - | rs143912759 | C | A | CC (50)/CA (1) | 0.4288 | nonsynonymous SNV | c.C1139A:p.T380K | 0.00026 |
| PSEN1 | - | rs1174374799 | - | T | −(50)/−T (1) | 0.4542 | frameshift insertion | c.526dupT:p.S178Ffs * 10 | 0.000006573 |
| PSENEN | - | rs751542345 | T | G | TT (50)/TG (1) | 0.1519 | stopgain | c.T168G:p.Y56X | 0.000008 |
| FGFR2 | - | rs56226109 | G | A | GG (49)/GA (2) | 0.2413 | nonsynonymous SNV | c.C170T:p.S57L | 0.003722 |
| MEFV | up | rs28940579 | A | G | AA (50)/AG (1) | 0.0215 | nonsynonymous SNV | c.T2177C:p.V726A | 0.001440 |
| MEFV | up | rs104895094 | T | C | TT (50)/TC (1) | 0.0996 | nonsynonymous SNV | c.A2084G:p.K695R | 0.005245 |
| NOD2 | - | rs104895452 | C | A | CC (50)/CA (1) | 0.9148 | nonsynonymous SNV | c.C2672A:p.A891D | 0.000707 |
| NOD2 | - | rs5743279 | G | A | GG (50)/GA (1) | 0.1775 | nonsynonymous SNV | c.G2288A:p.R763Q | 0.001217 |
| NOD2 | - | rs5743272 | A | G | AA (49)/AG (2) | 0.0699 | nonsynonymous SNV | c.A974G:p.H325R | 0.000392 |
| NOD2 | - | rs35285618 | G | A | GG (50)/GA (1) | 0.2845 | nonsynonymous SNV | c.G2042A:p.R681H | 0.00198 |
| NOD2 | - | rs2066847 | - | C | −(50)/−C (1) | 0.6377 | frameshift insertion | c.2936dupC:p.L980Pfs * 2 | 0.015002 |
| NOD2 | - | rs34684955 | G | A | GG (50)/GA (1) | 0.2845 | nonsynonymous SNV | c.G337A:p.A113T | 0.00251 |
| NOD2 | - | rs5743278 | C | G | CC (50)/CG (1) | 0.1038 | nonsynonymous SNV | c.C2093G:p.A698G | 0.00371 |
| NOD2 | - | rs576658764 | C | T | CC (50)/CT (1) | 0.2845 | nonsynonymous SNV | c.C1540T:p.R514W | 0.00007 |
| PSTPIP1 | up | rs34240327 | G | C | GG (49)/GC (2) | 0.4935 | nonsynonymous SNV | c.G773C:p.G258A | 0.00461 |
| Genes | DEGs | SNP ID | Ref | Alt | Distribution of Genotypes among HS Patients | Wilcoxon Test | HGVS | AF | CADD Score |
|---|---|---|---|---|---|---|---|---|---|
| ACSF3 | - | rs144681140 | G | A | GG (49)/GA (2) | 0.0295 | c.G1406A:p.R469Q | 0.0032 | 22.9 |
| KLF4 | - | rs139237114 | G | A | GG (49)/GA (2) | 0.0385 | c.C859T:p.H287Y | 0.0016 | 23.8 |
| DUSP23 | - | rs11544443 | A | T | AA (49)/AT (2) | 0.0475 | c.A371T:p.E124V | 0.0022 | 25.1 |
| BTN2A1 | - | rs143104579 | G | A | GG (48)/GA (3) | 0.045 | c.G188A:p.R63H | 0.0096 | 18.01 |
| FLOT2 | - | rs3736238 | C | T | CC (49)/CT (2) | 0.0284 | c.G982A:p.A328T | 0.0119 | 18.44 |
| GPANK1 | - | rs35265780 | G | A | GG (49)/GA (2) | 0.04 | c.C335T:p.A112V | 0.0096 | 32 |
| FNIP2 | - | rs62001914 | C | A | CC (49)/CA (2) | 0.0476 | c.C1653A:p.S551R | 0.0092 | 24.8 |
| CORO1B | - | rs145707942 | C | G | CC (49)/CG (2) | 0.0462 | c.G367C:p.E123Q | 0.0002 | 24.2 |
| ADCY4 | - | rs61745073 | T | A | TT (49)/TA (2) | 0.0357 | c.A1358T:p.E453V | 0.0022 | 22.7 |
| AKR1C3 | down | rs34186955 | C | T | CC (49)/CT (2) | 0.0295 | c.C538T:p.P180S | 0.0086 | 23.3 |
| ALDH6A1 | down | rs139579994 | G | A | GG (49)/GA (2) | 0.0395 | c.C716T:p.P239L | 0.0018 | 27.9 |
| GSDMD | up | rs62000416 | C | A | CC (49)/CA (2) | 0.0315 | c.C556A:p.L186M | 0.0056 | 23.4 |
| YTHDF1 | - | rs141487890 | G | A | GG (49)/GA (2) | 0.0344 | c.C437T:p.A146V | 0.0008 | 24 |
| MYOF | - | rs61861290 | G | A | GG (48)/GA (3) | 0.0207 | c.C4576T:p.P1526S | 0.0062 | 26.2 |
| SIT1 | up | rs138786883 | C | A | CC (49)/CA (2) | 0.0496 | c.G520T:p.A174S | 0.0032 | 17.54 |
| RBMXL1 | - | rs139713926 | T | C | TT (49)/TC (2) | 0.0242 | c.A701G:p.Y234C | 0.0022 | 25.5 |
| GALNT7 | - | rs144873913 | C | A | CC (48)/CA (2) | 0.0496 | c.C1585A:p.P529T | 0.0014 | 30 |
| PHACTR4 | - | rs72661785 | G | C | GG (49)/GC (2) | 0.043 | c.G1609C:p.A537P | 0.0036 | 26.3 |
| WDR92 | - | rs138784630 | C | T | CC (49)CT (2) | 0.04 | c.G841A:p.A281T | 0.0072 | 23.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, A.S.L.E.; Bloise, G.; Moltrasio, C.; Coelho, A.; Agrelli, A.; Moura, R.; Tricarico, P.M.; Jamain, S.; Marzano, A.V.; Crovella, S.; et al. Transcriptome Meta-Analysis Confirms the Hidradenitis Suppurativa Pathogenic Triad: Upregulated Inflammation, Altered Epithelial Organization, and Dysregulated Metabolic Signaling. Biomolecules 2022, 12, 1371. https://doi.org/10.3390/biom12101371
de Oliveira ASLE, Bloise G, Moltrasio C, Coelho A, Agrelli A, Moura R, Tricarico PM, Jamain S, Marzano AV, Crovella S, et al. Transcriptome Meta-Analysis Confirms the Hidradenitis Suppurativa Pathogenic Triad: Upregulated Inflammation, Altered Epithelial Organization, and Dysregulated Metabolic Signaling. Biomolecules. 2022; 12(10):1371. https://doi.org/10.3390/biom12101371
Chicago/Turabian Stylede Oliveira, Ana Sofia Lima Estevao, Giovanna Bloise, Chiara Moltrasio, Antonio Coelho, Almerinda Agrelli, Ronald Moura, Paola Maura Tricarico, Stéphane Jamain, Angelo Valerio Marzano, Sergio Crovella, and et al. 2022. "Transcriptome Meta-Analysis Confirms the Hidradenitis Suppurativa Pathogenic Triad: Upregulated Inflammation, Altered Epithelial Organization, and Dysregulated Metabolic Signaling" Biomolecules 12, no. 10: 1371. https://doi.org/10.3390/biom12101371
APA Stylede Oliveira, A. S. L. E., Bloise, G., Moltrasio, C., Coelho, A., Agrelli, A., Moura, R., Tricarico, P. M., Jamain, S., Marzano, A. V., Crovella, S., & Cavalcanti Brandão, L. A. (2022). Transcriptome Meta-Analysis Confirms the Hidradenitis Suppurativa Pathogenic Triad: Upregulated Inflammation, Altered Epithelial Organization, and Dysregulated Metabolic Signaling. Biomolecules, 12(10), 1371. https://doi.org/10.3390/biom12101371