
Radical Mediated Rapid In Vitro Formation of c-Type Cytochrome

 , , ,
, , ,
Abstract
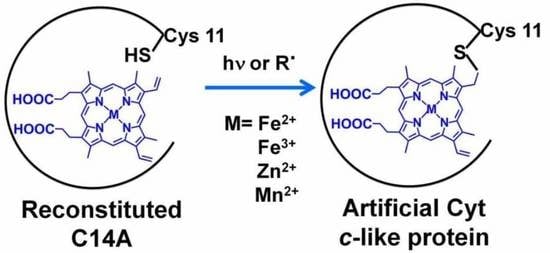
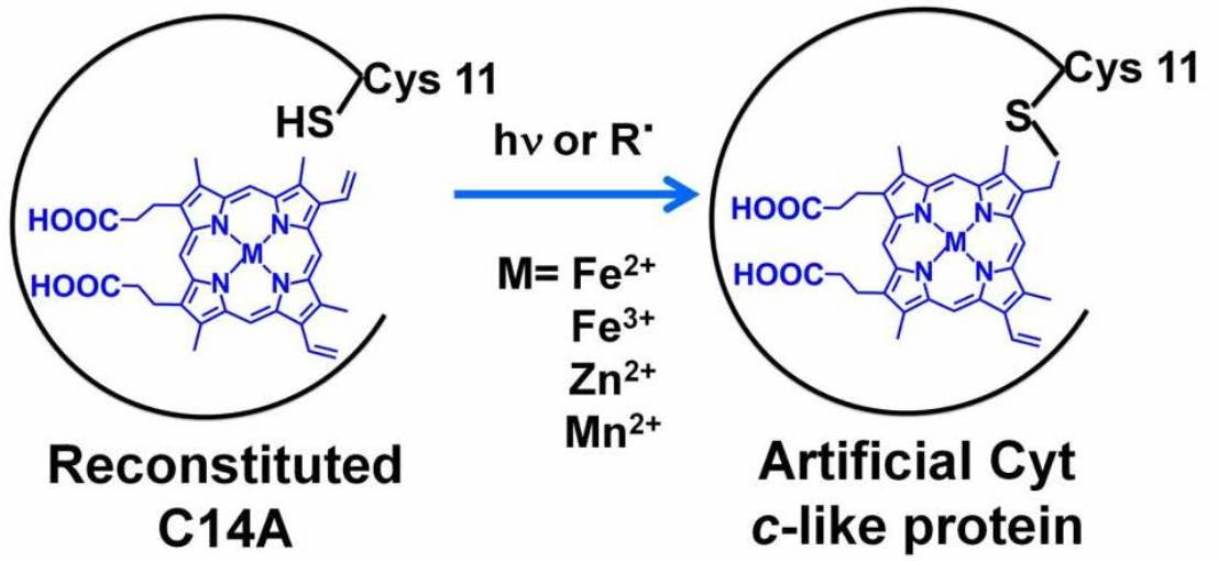
1. Introduction
2. Results
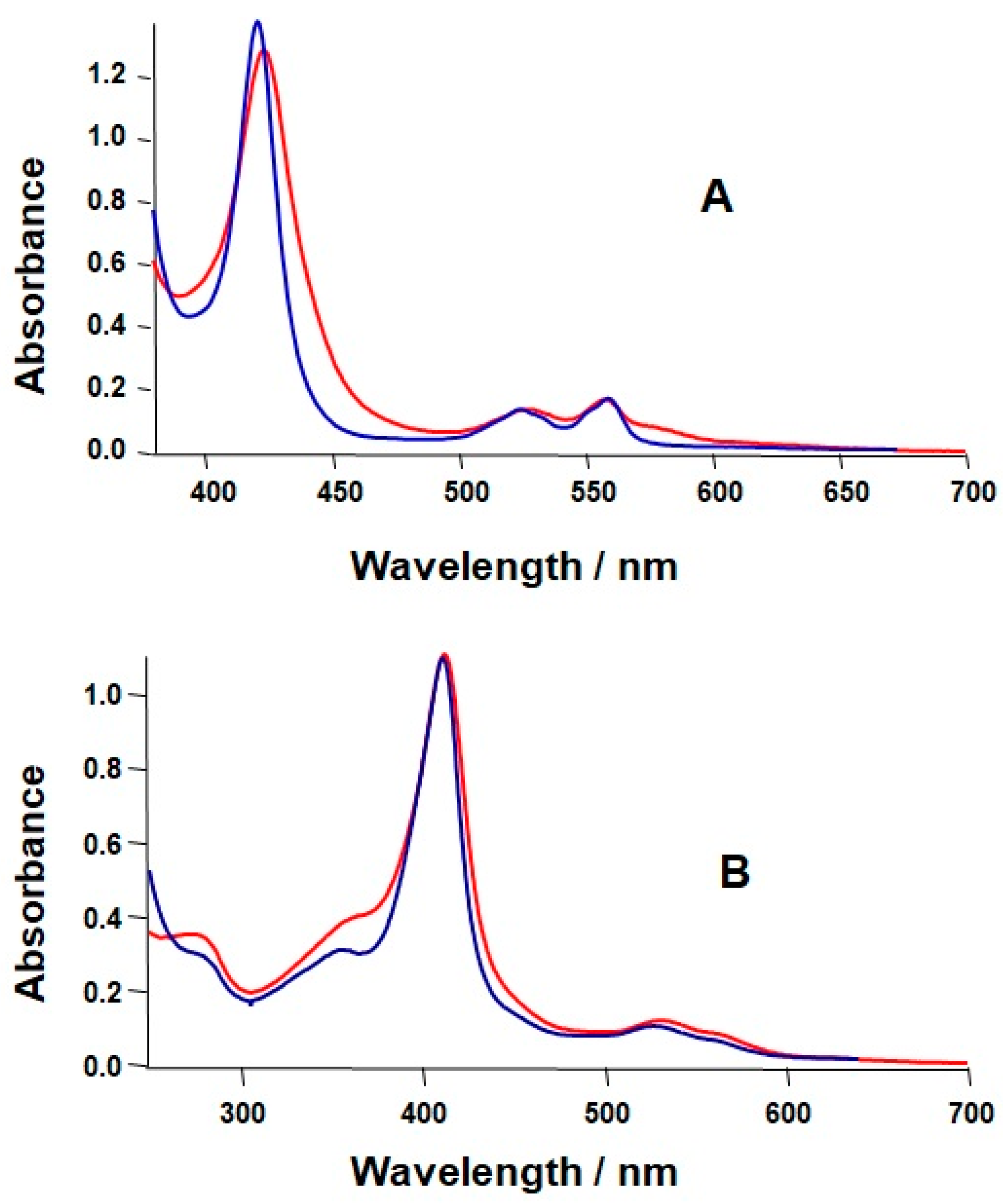
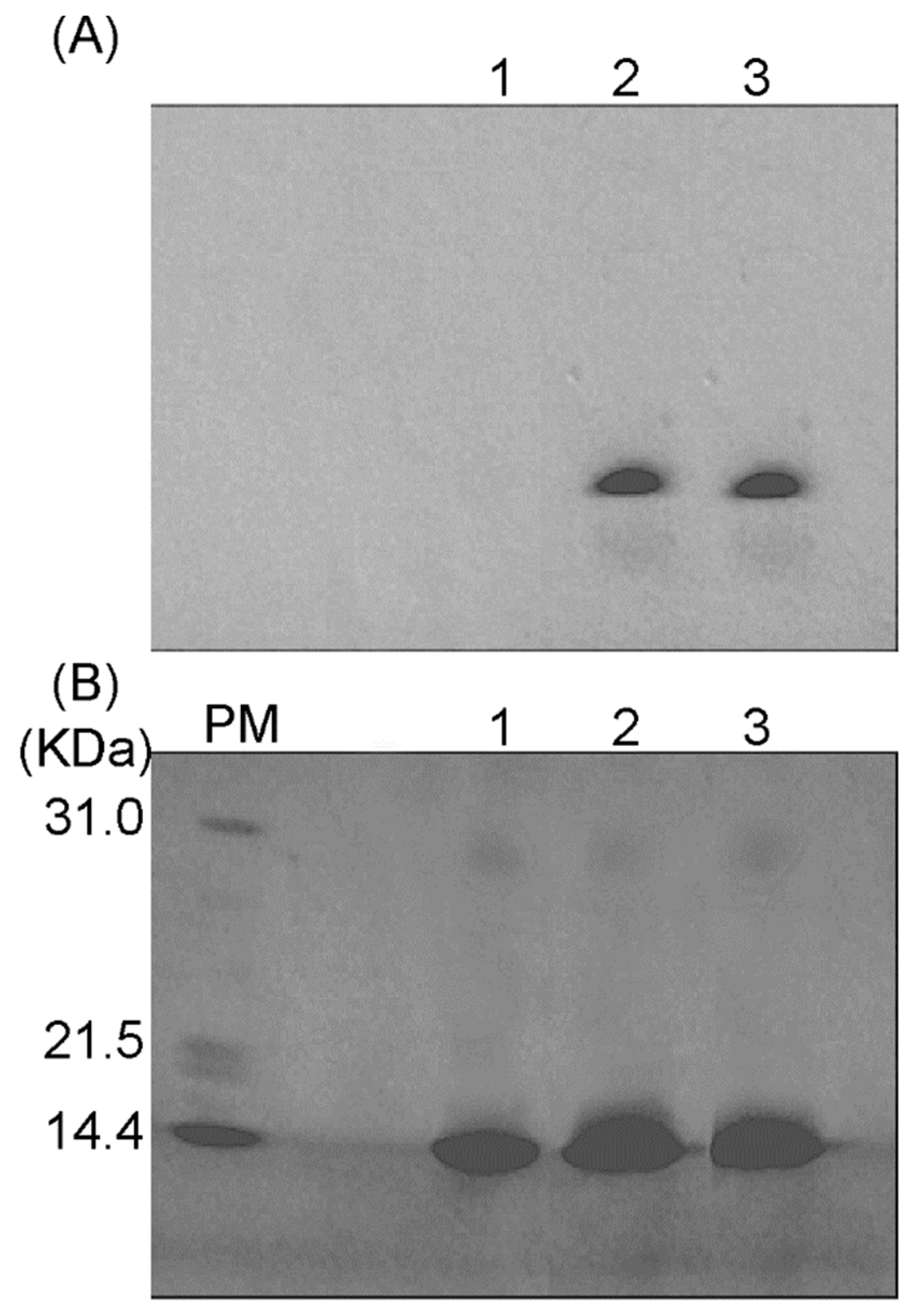
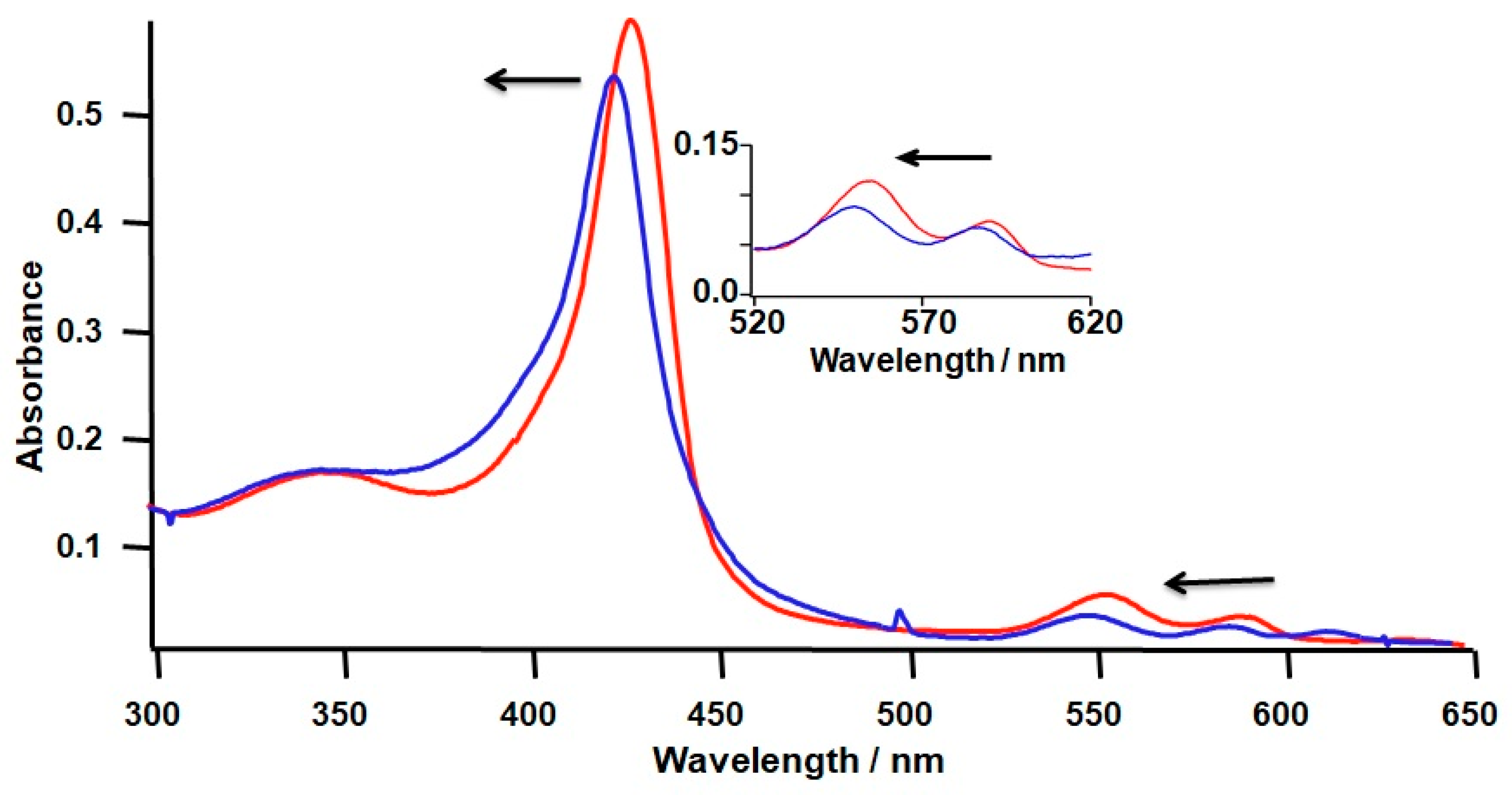
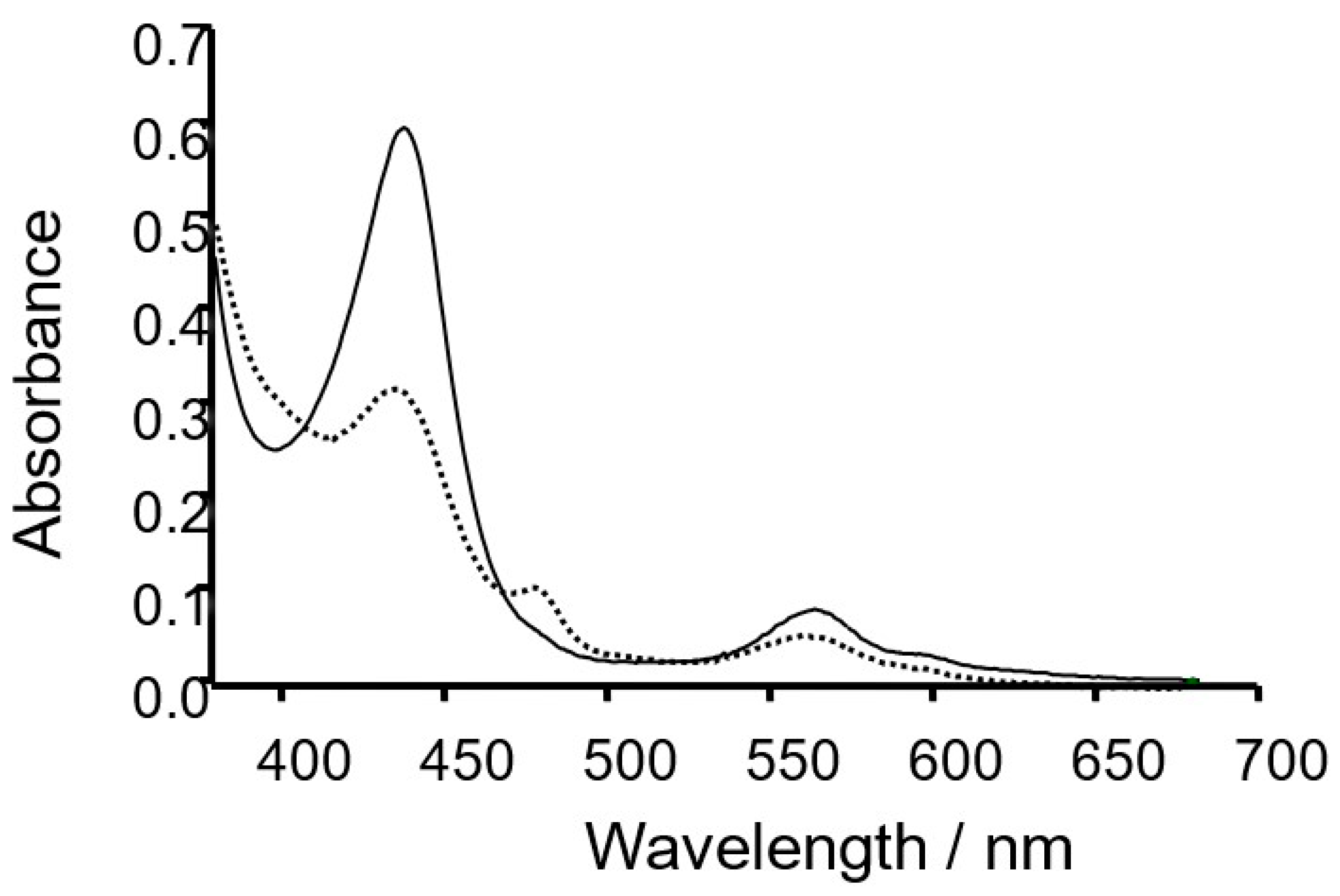
2.1. Formation of a Covalent Linkage in Ferric or Ferrous Heme Reconstituted Cytochrome c552 (C14A) in the Presence of Light
2.2. Formation of a Covalent Linkage in Zn-PPIX Reconstituted C14A in the Presence of Light
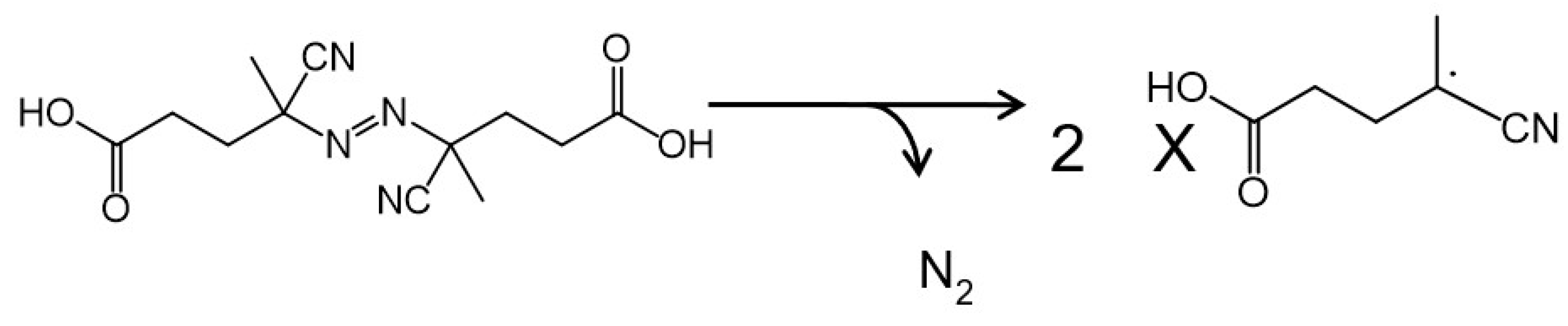
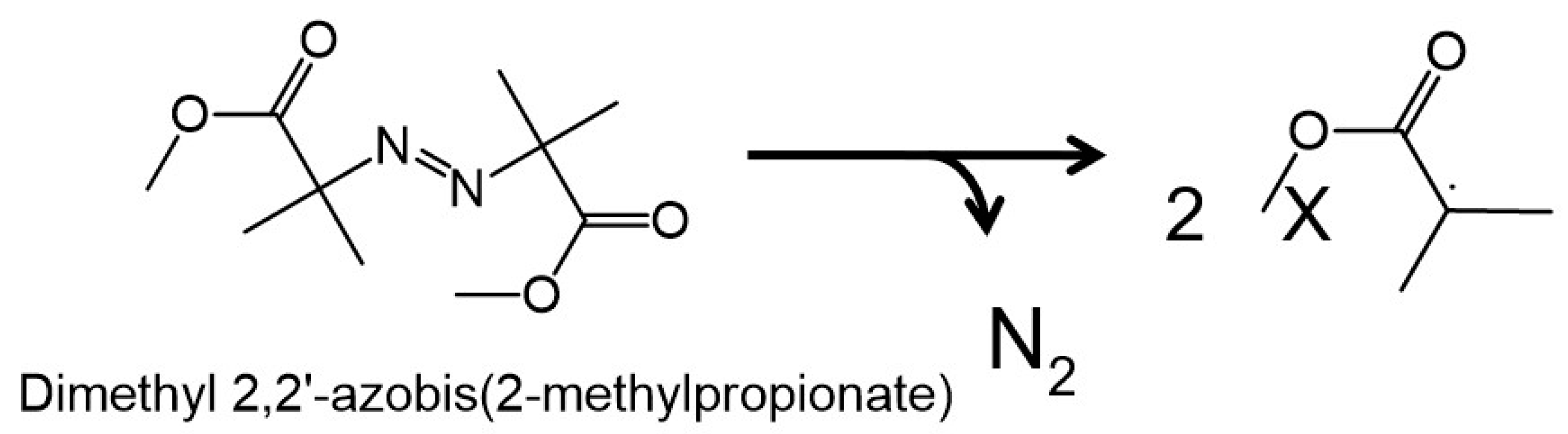
2.3. Introduction of Radical Initiators
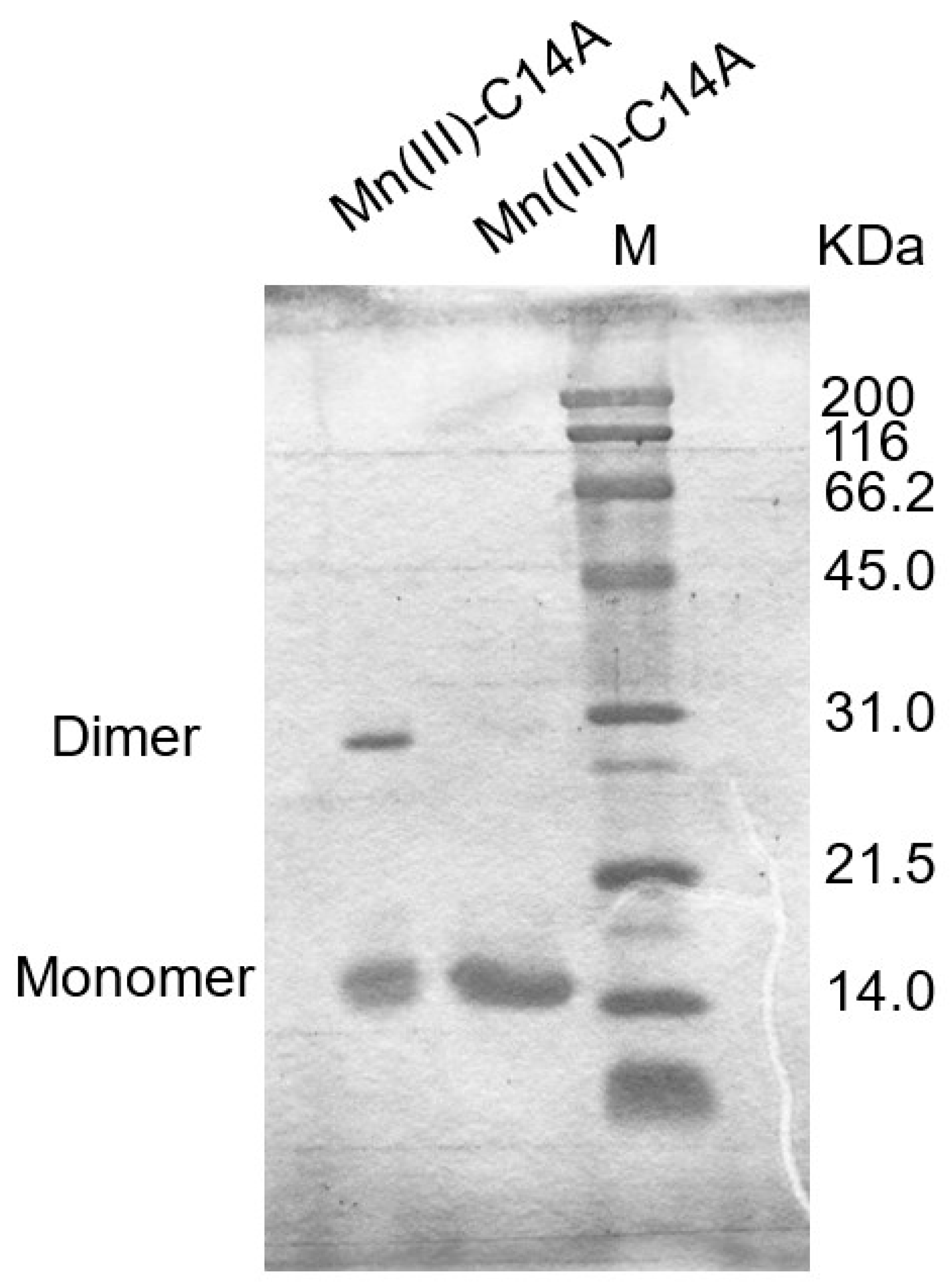
2.4. Covalent Linkage Formation in Other Reconstituted Proteins
3. Discussion
3.1. Light Promoted Covalent Linkage Formation
3.2. Effects of a Radical Initiator on the Covalent Linkage Formation
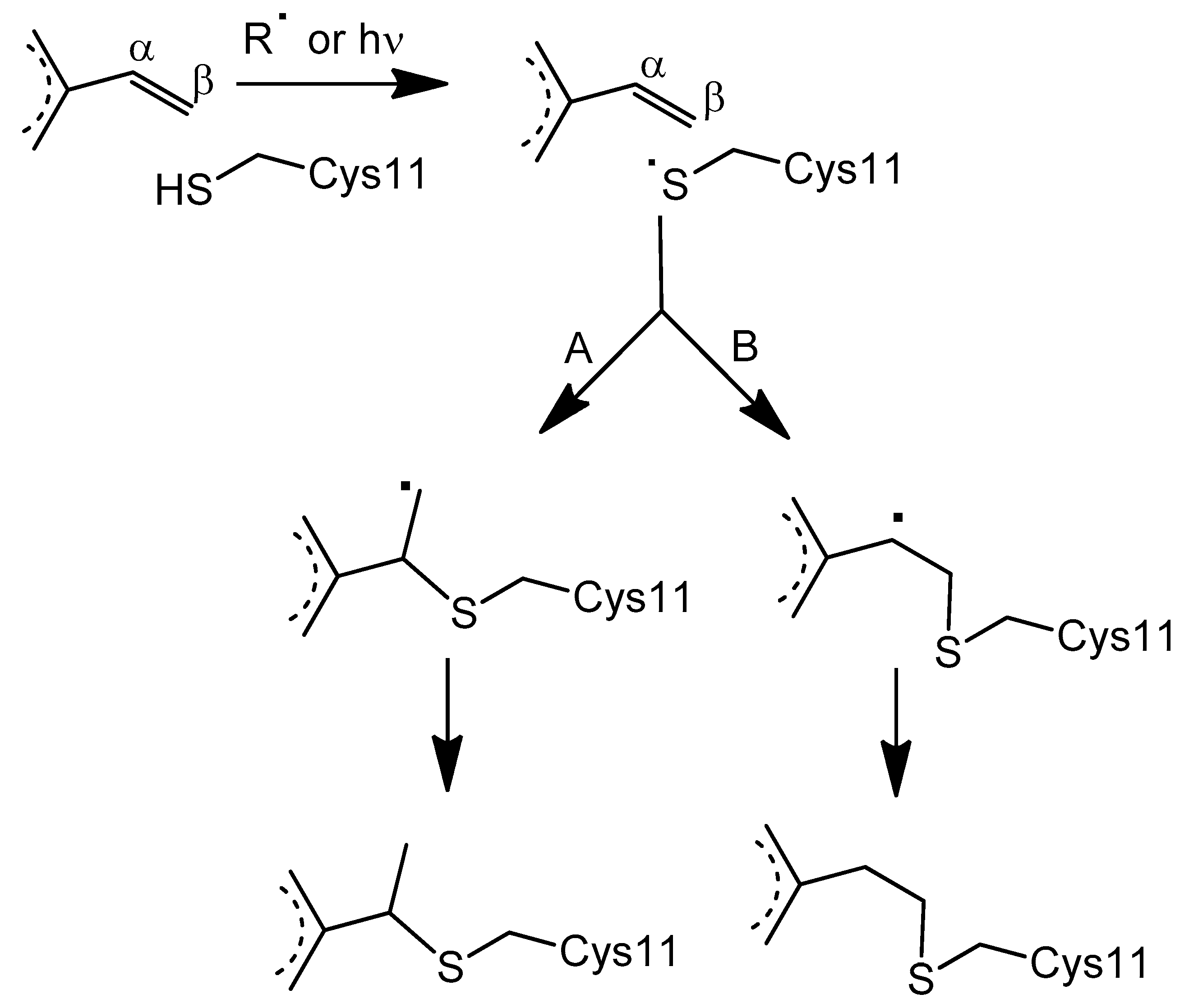
3.3. Proposed Mechanism for the Covalent Linkage Reaction
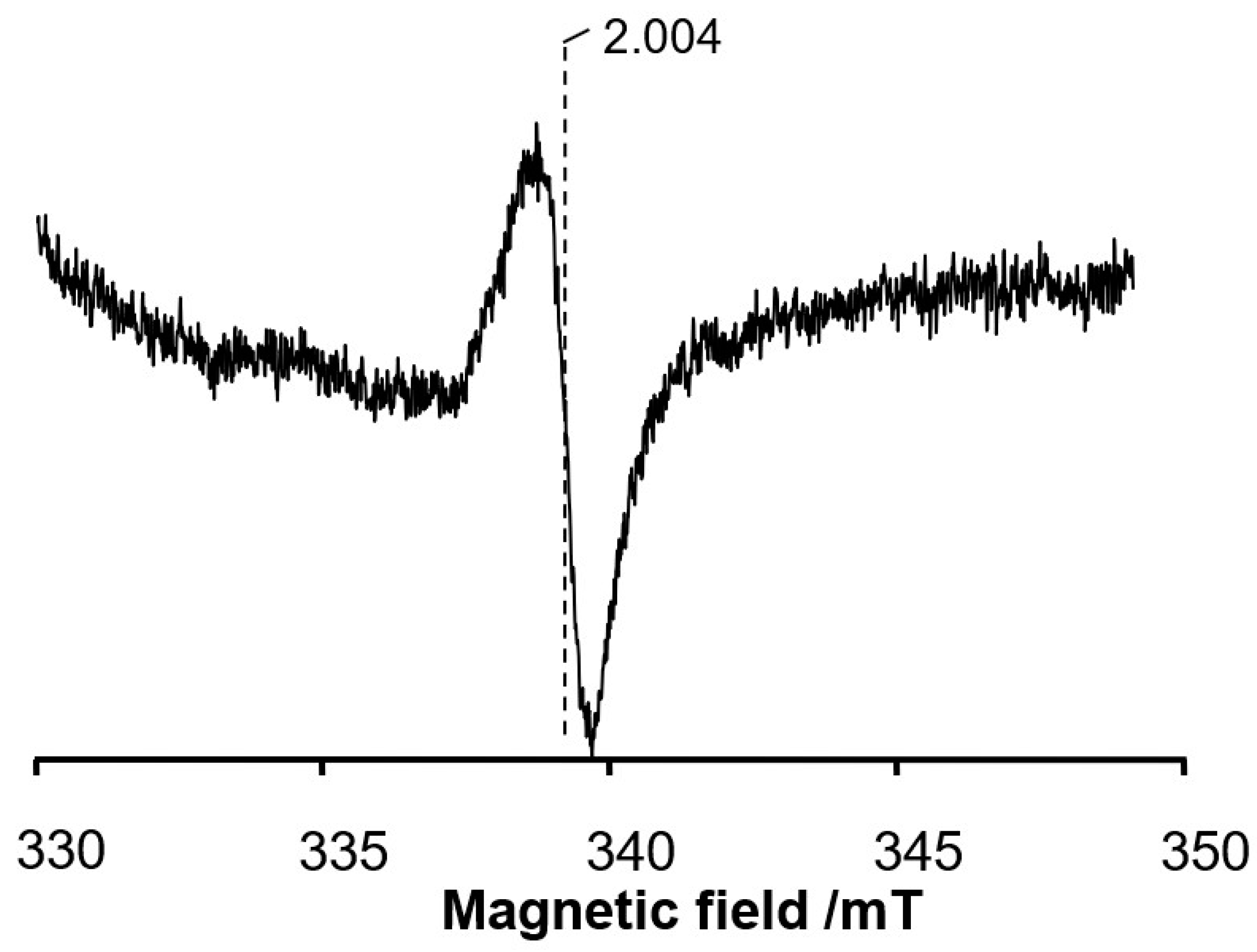
3.4. EPR Signal Indicates the Involvement of Cysteine 11 Residue in Covalent Linkage Formation
3.5. Oxidation State in a Metal Ion Which Affects the Linkage Formation
4. Materials and Methods
4.1. Chemicals
4.2. Expression and Purification of rC552 C14A Mutant in E. coli
4.3. Spectroscopy
4.4. Heme Staining
4.5. Reaction with Radical Generators
4.6. EPR Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mac Munn, C.A. VI. Researches on myohamatin and the histohæmatins. Philos. Trans. R. Soc. London 1886, 177, 267–298. [Google Scholar]
- Keilin, D. A comparative study of turacin and haematin and its bearing on cytochrome. Proc. R. Soc. Lond. 1926, 100B, 129–151. [Google Scholar]
- Bowman, S.E.J.; Bren, K.L. The chemistry and biochemistry of heme c: Functional bases for covalent attachment. Nat. Prod. Rep. 2008, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Kranz, R.G.; Richard-Fogal, C.; Taylor, J.-S.; Frawley, E.R. Cytochrome c biogenesis: Mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 2009, 73, 510–528. [Google Scholar] [CrossRef]
- Barker, P.D.; Ferguson, S.J. Still a puzzle: Why is haem covalently attached in c-type cytochromes? Structure 1999, 7, R281–R290. [Google Scholar] [CrossRef]
- Schulz, H.; Hennecke, H.; Thöny-Meyer, L. Prototype of a heme chaperone essential for cytochrome c maturation. Science 1998, 281, 1197–1200. [Google Scholar] [CrossRef]
- Kleingardner, J.G.; Bren, K.L. Biological significance and applications of heme c proteins and peptides. Acc. Chem. Res. 2015, 48, 1845–1852. [Google Scholar] [CrossRef]
- Pettigrew, G.W.; Moore, G.R. Cytochromes C: Biological Aspects; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Fisher, W.R.; Taniuchi, H.; Anfinsen, C.B. On the role of heme in the formation of the structure of cytochrome c. J. Bio. Chem. 1973, 248, 3188–3195. [Google Scholar] [CrossRef]
- Sawyer, E.B.; Stephens, E.; Ferguson, S.J.; Allen, J.W.; Barker, P.D. Aberrant attachment of heme to cytochrome by the Ccm system results in a cysteine persulfide linkage. J. Am. Chem. Soc. 2010, 132, 4974–4975. [Google Scholar] [CrossRef]
- Barker, P.D.; Nerou, E.P.; Freund, S.M.V.; Fearnley, I.M. Conversion of cytochrome b562 to c-type cytochromes. Biochemistry 1995, 34, 15191–15203. [Google Scholar] [CrossRef]
- Lin, Y.-W.; Wang, W.-H.; Zhang, Q.; Lu, H.-J.; Yang, P.-Y.; Xie, Y.; Huang, Z.-X.; Wu, H.-M. Converting Cytochrome b5 into Cytochrome c-Like Protein. ChemBioChem 2005, 6, 1356–1359. [Google Scholar] [CrossRef]
- Daltrop, O.; Allen, J.W.A.; Willis, A.C.; Ferguson, S.J. In vitro formation of a c-type cytochrome. Proc. Natl. Acad. Sci. USA 2002, 99, 7872–7876. [Google Scholar] [CrossRef]
- Daltrop, O.; Ferguson, S.J. The in Vitro Reactions Of Horse Heart Apocytochromec And Paracoccus Denitrificans Apocytochromec550 with Heme. J. Biol. Chem. 2003, 278, 4404–4409. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.M.; Daltrop, O.; Allen, J.W.A.; Ferguson, S.J. C-type cytochrome formation: Chemical and biological enigmas. Acc. Chem. Res. 2004, 37, 999–1007. [Google Scholar] [CrossRef]
- Metcafle, C.L.; Daltrop, O.; Ferguson, S.J.; Raven, E.L. Tuning the formation of a covalent haem–protein link by selection of reductive or oxidative conditions as exemplified by ascorbate peroxidase. Biochem. J. 2007, 408, 355–361. [Google Scholar]
- Ibrahim, S.M.; Nakajima, H.; Ohta, T.; Ramanathan, K.; Takatani, N.; Naruta, Y.; Watanabe, Y. Cytochrome c 552 from Thermus thermophilus engineered for facile substitution of prosthetic group. Biochemistry 2011, 50, 9826–9835. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol–ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Daltrop, O.; Smith, K.M.; Ferguson, S.J. Stereoselective in vitro formation of c-type cytochrome variants from Hydrogenobacter thermophilus containing only a single thioether bond. J. Biol. Chem. 2003, 278, 24308–24313. [Google Scholar] [CrossRef] [PubMed]
- Barker, P.D.; Ferrer, J.C.; Mylrajan, M.; Loehr, T.M.; Feng, R.; Konishi, Y.; Funk, W.D.; Macgillivray, R.T.A.; Mauk, A.G. Transmutation of a heme protein. Proc. Natl. Acad. Sci. USA 1993, 90, 6542–6546. [Google Scholar] [CrossRef]
- Daltrop, O.; Ferguson, S.J. In vitro studies on thioether bond formation between Hydrogenobacter thermophilus apocytochrome c552 with metalloprotoporphyrin derivatives. J. Biol. Chem. 2004, 279, 45347–45353. [Google Scholar] [CrossRef] [PubMed]
- Griesbaum, K. Problems and possibilities of the free-radical addition of thiols to unsaturated compounds. Angew. Chem. Int. Ed. 1970, 9, 273–287. [Google Scholar] [CrossRef]
- Zard, S.Z. Radical Reactionsin Organic Synthesis; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- López-Alarcón, C.; Fuentes-Lemus, E.; Figueroa, J.D.; Dorta, E.; Schoeneich, C.; Davies, M.J. Azocompounds as generators of defined radical species: Contributions and challenges for free radical research. Free Radic. Biol. Med. 2020, 160, 78–91. [Google Scholar] [CrossRef]
- Miner, K.D.; Pfister, T.D.; Hosseinzadeh, P.; Karaduman, N.; Donald, L.J.; Loewen, P.C.; Lu, Y.; Ivancich, A. Identifying the elusive sites of tyrosyl radicals in cytochrome c peroxidase: Implications for oxidation of substrates bound at a site remote from the heme. Biochemistry 2014, 53, 3781–3789. [Google Scholar] [CrossRef]
- Stubbe, J.; Van Der Donk, W.A. Protein radicals in enzyme catalysis. Chem. Rev. 1998, 98, 705–762. [Google Scholar] [CrossRef]
- Bolman, P.S.H.; Safarik, I.; Stiles, D.A.; Tyerman, W.J.R.; Strausz, O.P. Electron paramagnetic resonance spectra of some sulfur-containing radicals. Can. J. Chem. 1970, 48, 3872–3876. [Google Scholar] [CrossRef]
- Posner, T. Beiträge zur Kenntniss der ungesättigten Verbindungen. II. Ueber die Addition von Mercaptanen an ungesättigte Kohlenwasserstoffe. Chem. Ber. 1905, 38, 646–657. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Lowe, A.B.; Bowman, C. Thiol-click chemistry: A multifaceted toolbox for small molecule and polymer synthesis. Chem. Soc. Rev. 2009, 39, 1355–1587. [Google Scholar] [CrossRef] [PubMed]
- Killops, K.L.; Campos, L.M.; Hawker, C.J. Robust, efficient, and orthogonal synthesis of dendrimers via thiol-ene “click” chemistry. J. Am. Chem. Soc. 2008, 130, 5062–5064. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Commentary on “Azocompounds as generators of defined radical species: Contributions and challenges for free radical research” by López-Alarcón et al. Free Radic. Biol. Med. 2021, 164, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.A.; Daltrop, O.; Stevens, J.M.; Ferguson, S.J. C-type cytochromes: Diverse structures and biogenesis systems pose evolutionary problems. Philos. Trans. R. Soc. London B 2002, 358, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Harvat, E.M.; Daltrop, O.; Sobott, F.; Moreau, M.; Barker, P.D.; Stevens, J.M.; Ferguson, S.J. Metal and redox selectivity of protoporphyrin binding to the heme chaperone CcmE. Metallomics 2011, 3, 363–368. [Google Scholar] [CrossRef]
- Thöny-Meyer, L. Cytochrome c maturation: A complex pathway for a simple task? Biochem. Soc. Trans. 2002, 30, 633–638. [Google Scholar] [CrossRef]
- Lassmann, G.; Kolberg, M.; Bleifuss, G.; Gräslund, A.; Sjöberg, B.M.; Lubitz, W. Protein thiyl radicals in disordered systems: A comparative EPR study at low temperature. Phys. Chem. Chem. Phys. 2003, 5, 2442–2453. [Google Scholar] [CrossRef]
- Kolberg, M.; Bleifuss, G.; Gräslund, A.; Sjöberg, B.M.; Lubitz, W.; Lendzian, F.; Lassmann, G. Protein thiyl radicals directly observed by EPR spectroscopy. Arch. Biochem. Biophys. 2002, 403, 141–144. [Google Scholar] [CrossRef]
- McCaslin, T.G.; Pagba, C.V.; Hwang, H.; Gumbart, J.C.; Chi, S.H.; Perry, J.W.; Barry, B.A. Tyrosine, cysteine, and proton coupled electron transfer in a ribonucleotide reductase-inspired beta hairpin maquette. Chem. Commun. 2019, 55, 9399–9402. [Google Scholar] [CrossRef]
- Sevilla, M.D.; Yan, M.Y.; Becker, D. Thiol peroxyl radical formation from the reaction of cysteine thiyl radical with molecular oxygen: An ESR investigation. Biochem. Biophys. Res. Commun. 1988, 155, 405–410. [Google Scholar] [CrossRef]
- Sevilla, M.D.; Becker, D.; Swarts, S.; Herrington, J. Sulfinyl radical formation from the reaction of cysteine and glutathione thiyl radicals with molecular oxygen. Biochem. Biophys. Res. Commun. 1987, 144, 1037–1042. [Google Scholar] [CrossRef]
- Nauser, T.; Koppenol, W.H.; Schöneich, C. Protein thiyl radical reactions and product formation: A kinetic simulation. Free Radic. Biol. Med. 2015, 80, 158–163. [Google Scholar] [CrossRef]
- Francis, R.T.; Becker, R.R. Specific indication of hemoproteins in polyacrylamide gels using a double-staining process. Anal. Biochem. 1984, 136, 509–514. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
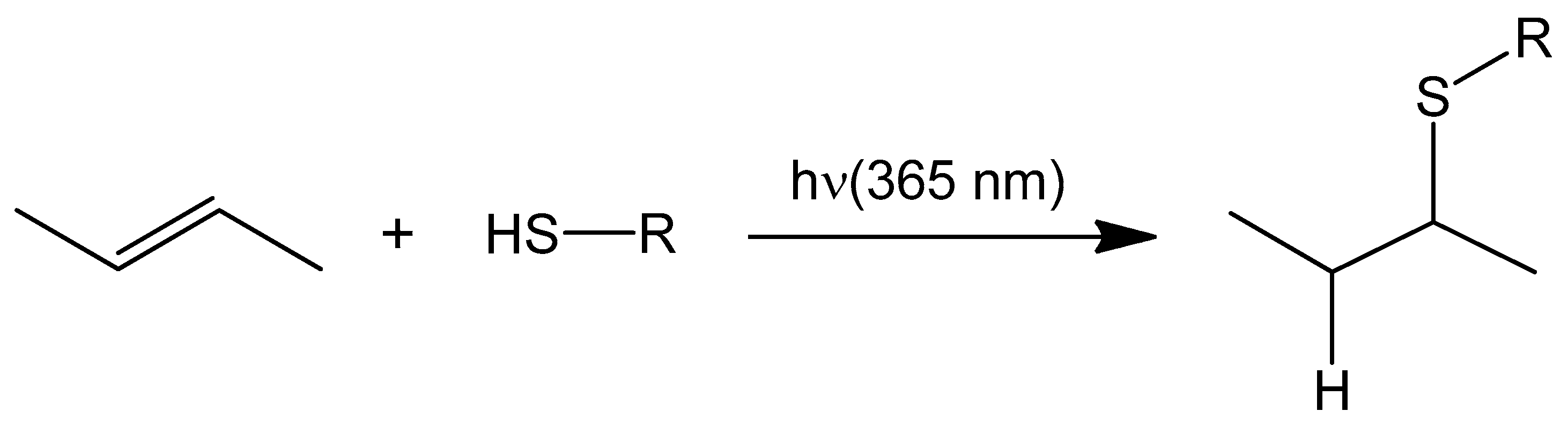{kind=link}
{kind=link}
| λmax/nm (e/cm−1·mM−1) | ||
|---|---|---|
| Ferric Heme b | Ferrous Heme b | |
| Immediate after the reconstitution | 413 (99) | 424 (128) |
| 532 (11), 564(8) | 528 (13), 558 (17) | |
| After forming the covalent linkage | 411 (98) | 421 (137) |
| 527 (9) | 524 (13), 559 (17) | |
| λmax/nm | ||
|---|---|---|
| Soret Band | Q-Band | |
| Immediate after the reconstitution | 438 | 564 |
| After forming the covalent linkage | 435 | 559 |
| Reconstituted Protein/Metal Ions | O2/Light | Radical |
|---|---|---|
| Ferric heme-C14A | ✓ | ✓ |
| Ferrous heme-C14A | ✓ | ✓ |
| Zn-C14A | ✓ | ✓ |
| Mn(II)-C14A | ✓ | ✓ |
| Mn(III)-C14A | 🗶 | 🗶 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, S.M.; Ben Aoun, S.; Nakajima, H.; Kooli, F.; Watanabe, Y. Radical Mediated Rapid In Vitro Formation of c-Type Cytochrome. Biomolecules 2022, 12, 1329. https://doi.org/10.3390/biom12101329
Ibrahim SM, Ben Aoun S, Nakajima H, Kooli F, Watanabe Y. Radical Mediated Rapid In Vitro Formation of c-Type Cytochrome. Biomolecules. 2022; 12(10):1329. https://doi.org/10.3390/biom12101329
Chicago/Turabian StyleIbrahim, Sheikh Muhammad, Sami Ben Aoun, Hiroshi Nakajima, Fethi Kooli, and Yoshihito Watanabe. 2022. "Radical Mediated Rapid In Vitro Formation of c-Type Cytochrome" Biomolecules 12, no. 10: 1329. https://doi.org/10.3390/biom12101329
APA StyleIbrahim, S. M., Ben Aoun, S., Nakajima, H., Kooli, F., & Watanabe, Y. (2022). Radical Mediated Rapid In Vitro Formation of c-Type Cytochrome. Biomolecules, 12(10), 1329. https://doi.org/10.3390/biom12101329