
How Far Are We from the Completion of the Human Protein Interactome Reconstruction?




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. The PICKLE PPI Meta-Database
2.2. Network Analysis and Visualization—Protein Functional Analysis
2.3. Clustering Analysis
3. Results
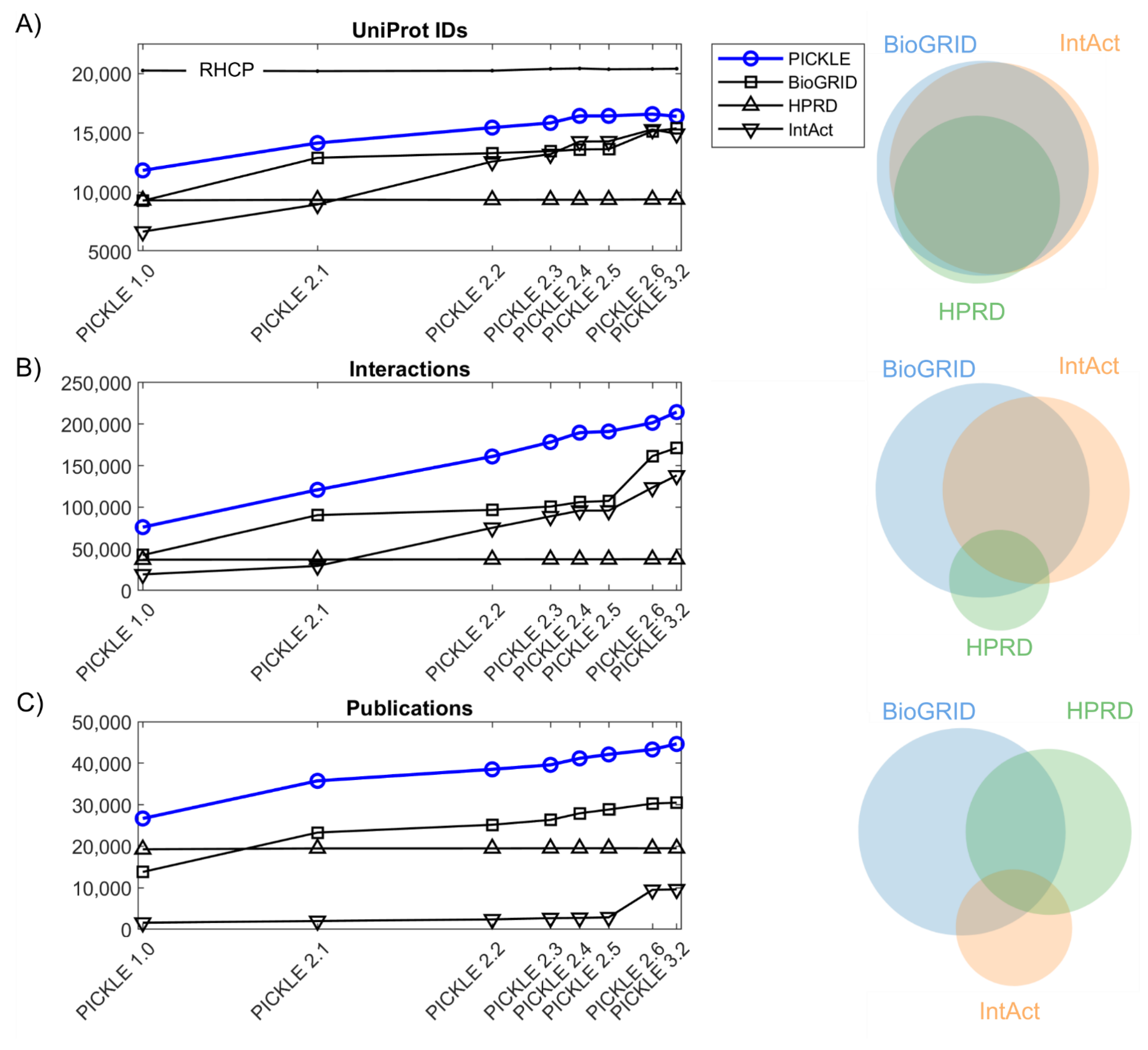
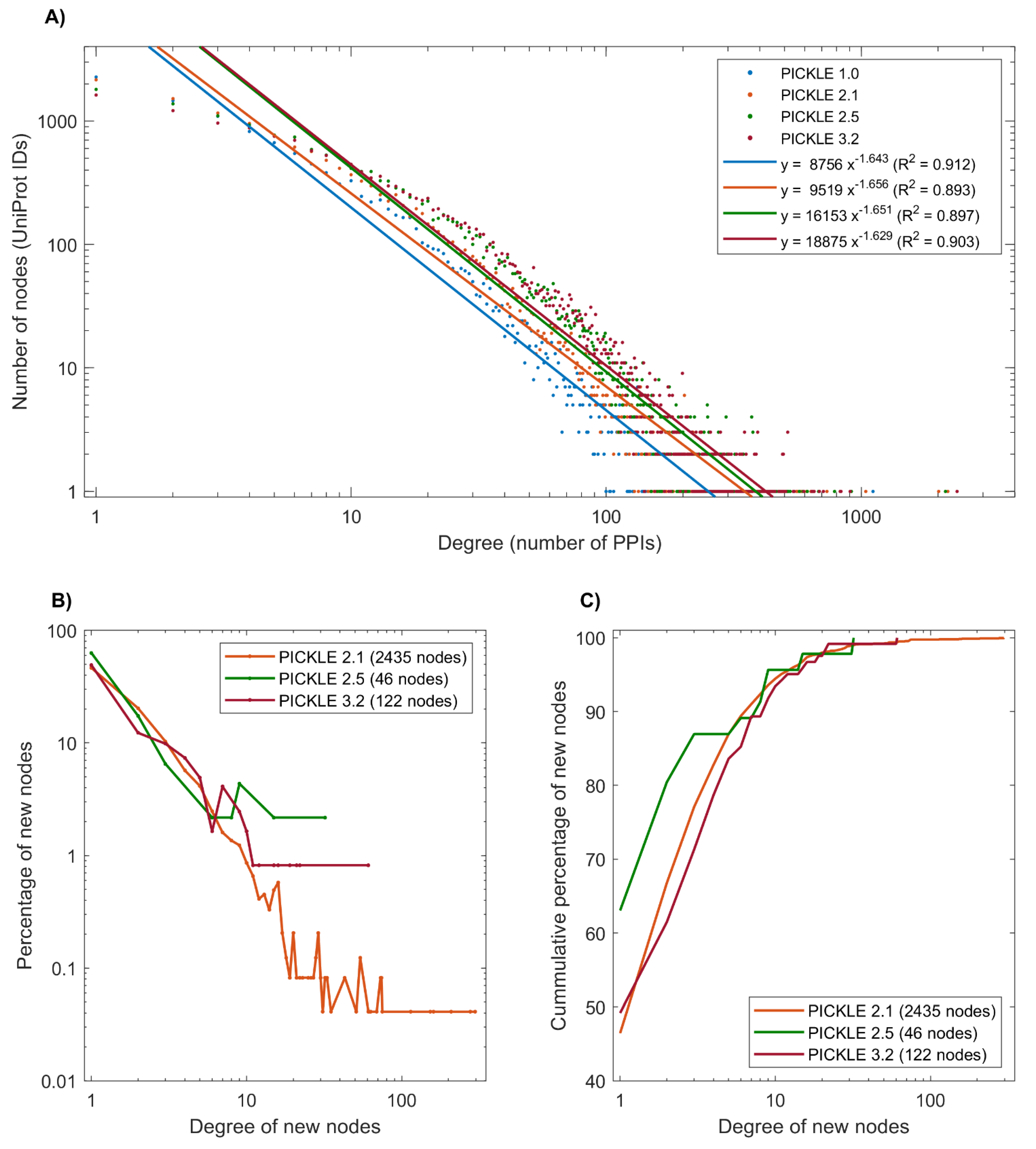
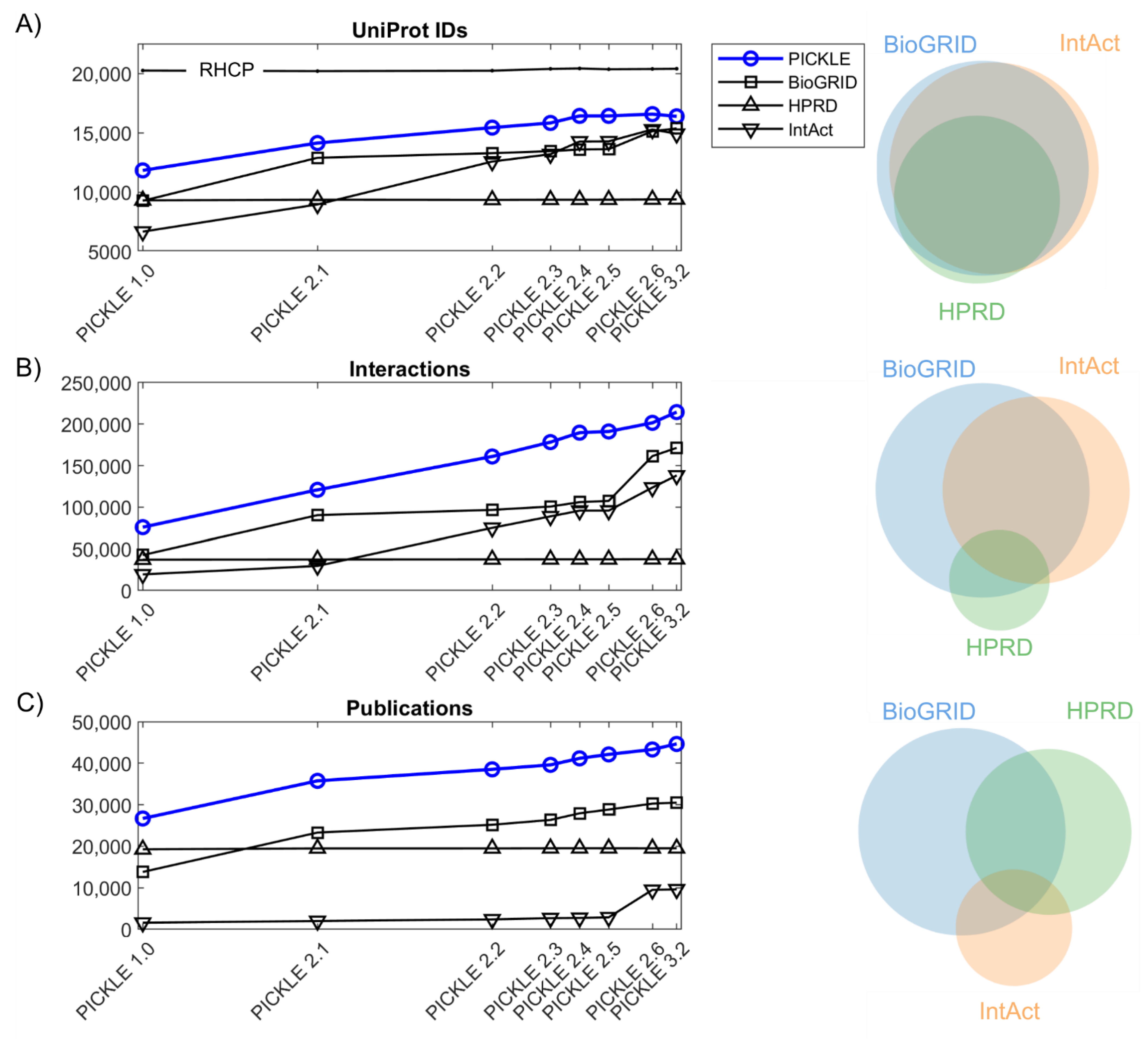
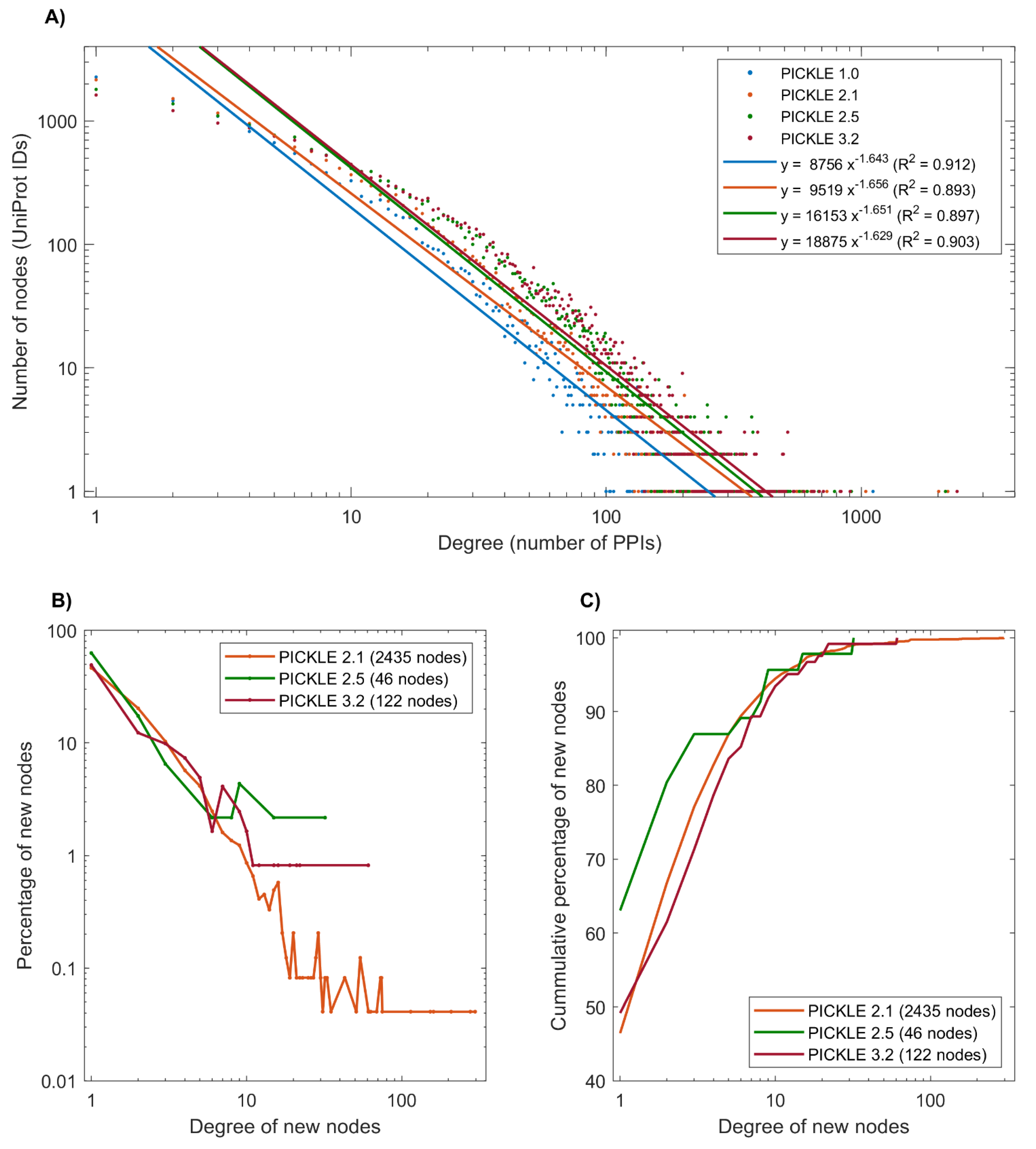
3.1. The Expansion of the Direct Human PPI Network
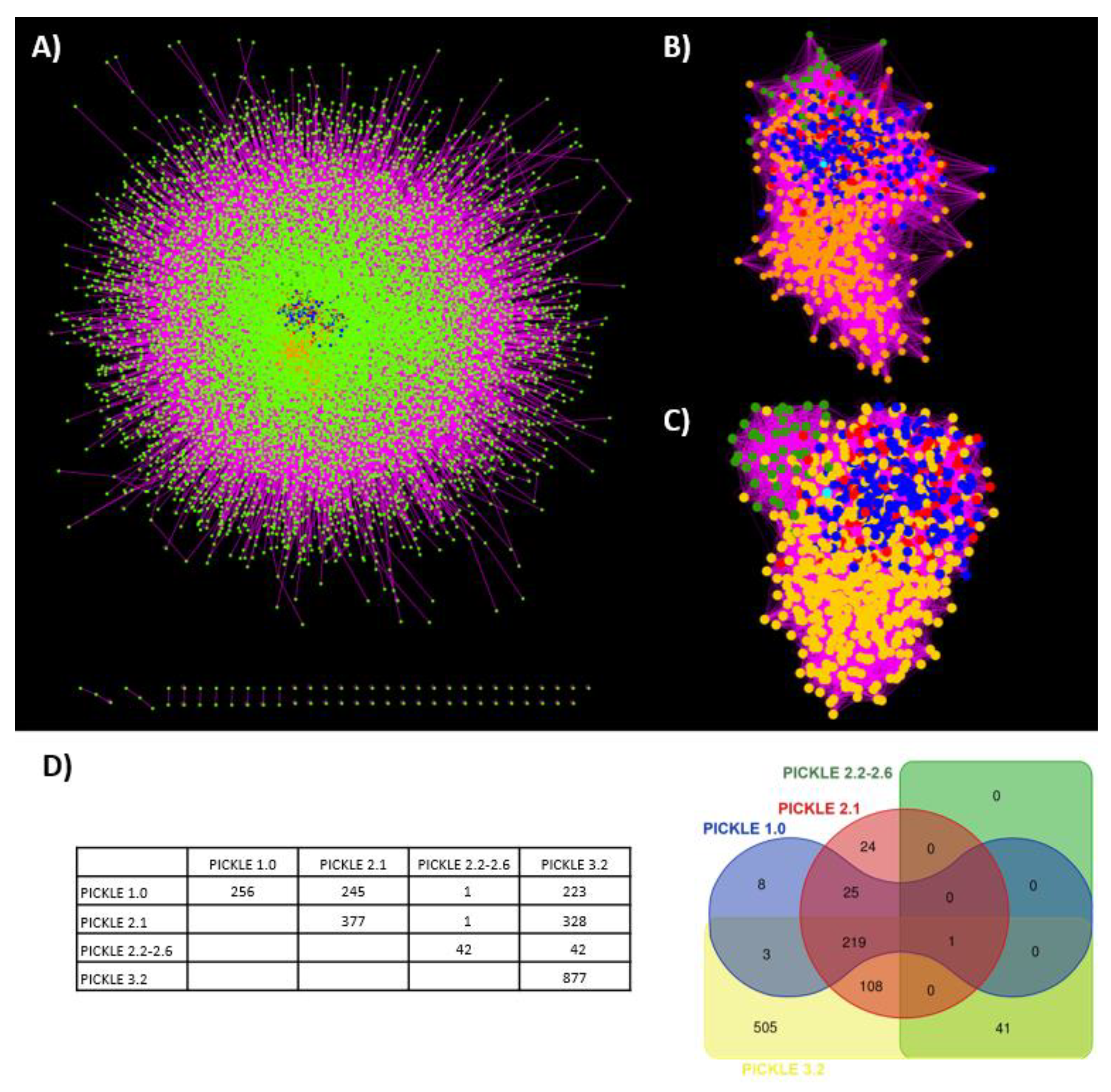
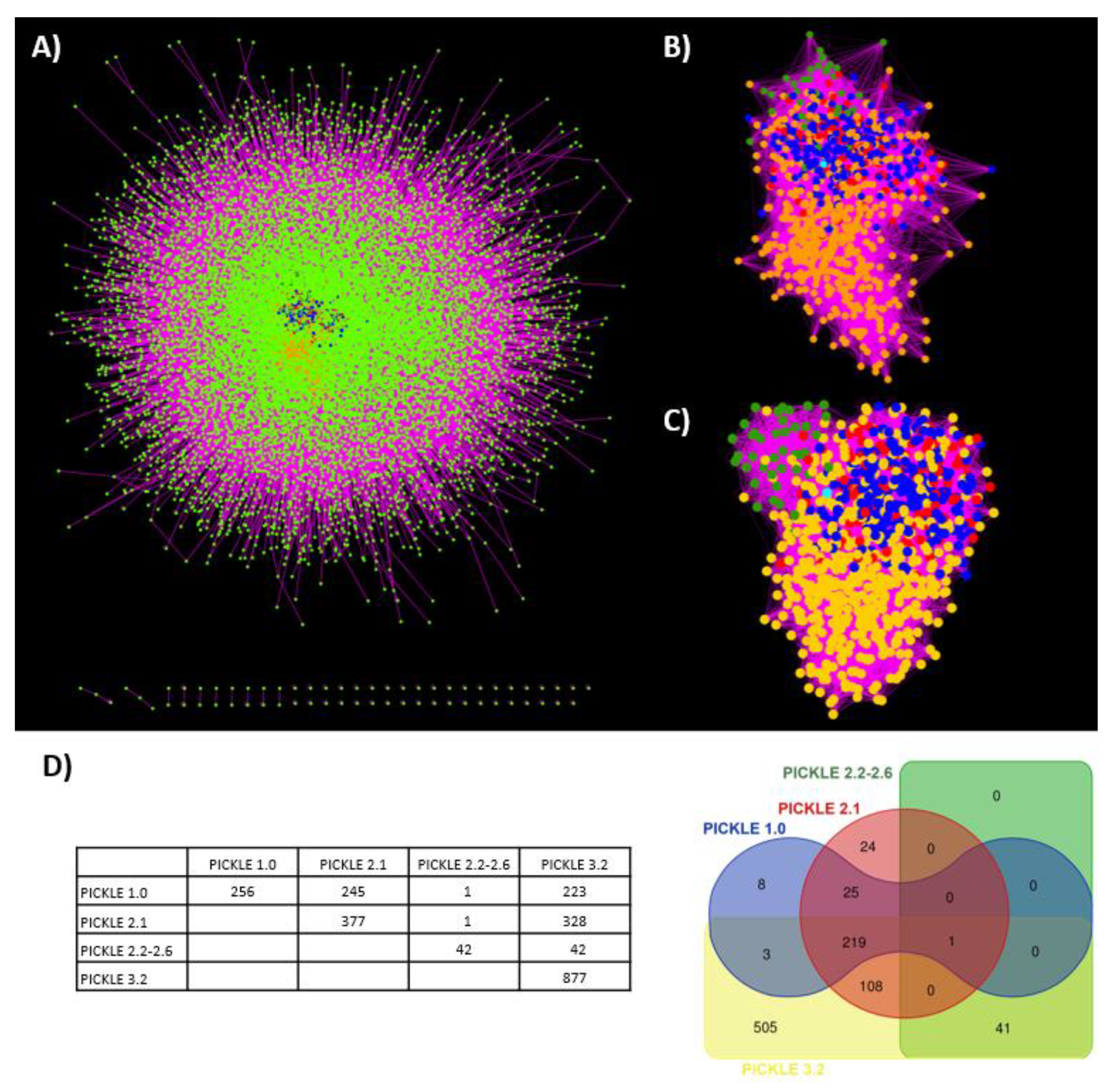
3.2. Proteins without Experimentally Detected Interactions
3.3. Network Analysis
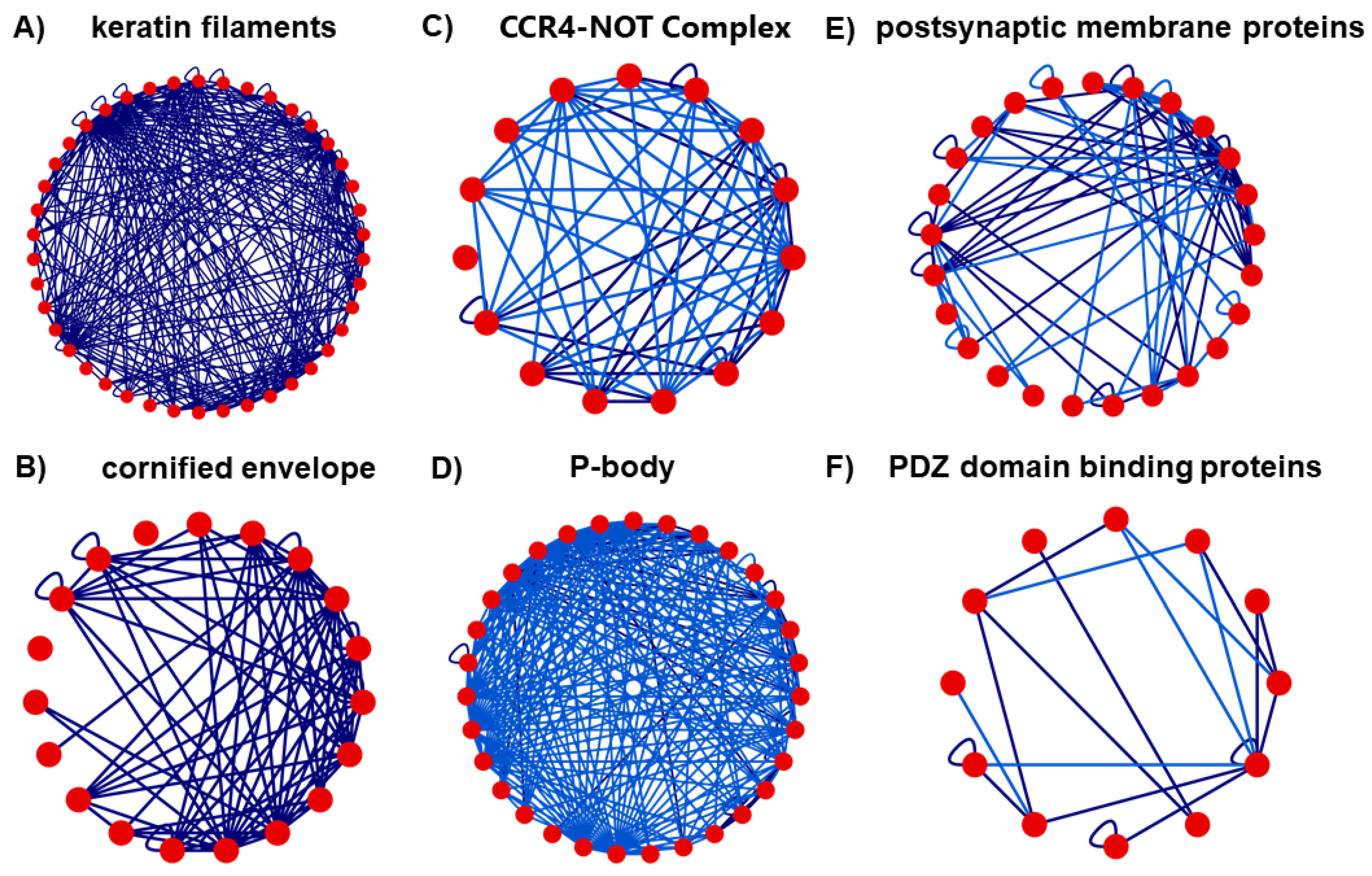
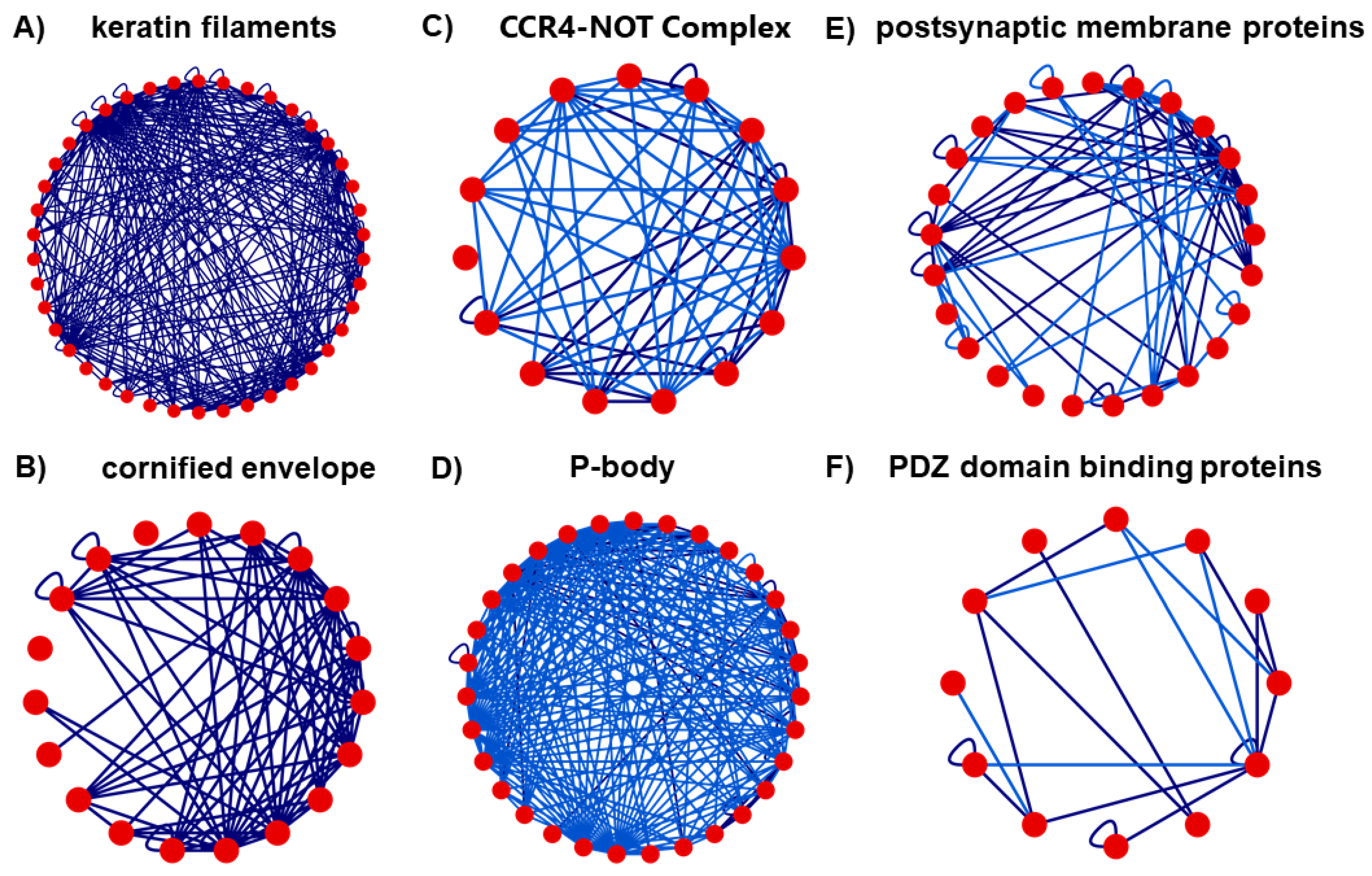
3.4. Clustering Analysis
3.4.1. Intersection 1: Between RW Cluster 5 and N2V-HC Cluster 2
3.4.2. Intersection 2: Between RW Cluster 8 and N2V-HC Cluster 3
3.4.3. Intersection 3: Between RW Cluster 10 and N2V-HC Cluster 8
3.4.4. Intersection 4: Between RW Cluster 13 and N2V-HC Cluster 10
4. Discussion
4.1. A Structurally Defined Reconstruction of the Human Protein Interactome Has Now Been Reached
4.2. The Fraction of RHCP Proteins without Identified Interactions
4.3. Exploring the Full Human Protein Interactome Topology Can Lead to Useful Biological Insights
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Miryala, S.K.; Anbarasu, A.; Ramaiah, S. Discerning molecular interactions: A comprehensive review on biomolecular interaction databases and network analysis tools. Gene 2018, 642, 84–94. [Google Scholar] [CrossRef]
- Kovács, I.A.; Luck, K.; Spirohn, K.; Wang, Y.; Pollis, C.; Schlabach, S.; Bian, W.; Kim, D.K.; Kishore, N.; Hao, T.; et al. Network-based prediction of protein interactions. Nat. Commun. 2019, 10, 1240. [Google Scholar] [CrossRef] [Green Version]
- Malod-Dognin, N.; Pržulj, N. Functional geometry of protein interactomes. Bioinformatics 2019, 35, 3727–3734. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Klingström, T.; Plewczynski, D. Protein–protein interaction and pathway databases, a graphical review. Brief Bioinform. 2011, 12, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Klapa, M.I.; Tsafou, K.; Theodoridis, E.; Tsakalidis, A.; Moschonas, N.K. Reconstruction of the experimentally supported human protein interactome: What can we learn? BMC Syst. Biol. 2013, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Gioutlakis, A.; Klapa, M.I.; Moschonas, N.K. PICKLE 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS ONE 2017, 12, e0186039. [Google Scholar] [CrossRef] [Green Version]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 Update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef]
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017, 45, D408–D414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razick, S.; Magklaras, G.; Donaldson, I.M. iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinform. 2008, 9, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-López, D.; Campos-Laborie, F.J.; Gutiérrez, M.A.; Lambourne, L.; Calderwood, M.A.; Vidal, M.; De Las Rivas, J. APID database: Redefining protein-protein interaction experimental evidences and binary interactomes. Database 2019, 2019, baz005. [Google Scholar] [CrossRef] [Green Version]
- Clerc, O.; Deniaud, M.; Vallet, S.D.; Naba, A.; Rivet, A.; Perez, S.; Thierry-Mieg, N.; Ricard-Blum, S. MatrixDB: Integration of new data with a focus on glycosaminoglycan interactions. Nucleic Acids Res. 2019, 47, D376–D381. [Google Scholar] [CrossRef]
- Dimitrakopoulos, G.N.; Klapa, M.I.; Moschonas, N.K. PICKLE 3.0: Enriching the human meta-database with the mouse protein interactome extended via mouse–human orthology. Bioinformatics 2021, 37, 145–146. [Google Scholar] [CrossRef]
- Omenn, G.S.; Lane, L.; Lundberg, E.K.; Beavis, R.C.; Overall, C.M.; Deutsch, E.W. Metrics for the Human Proteome Project 2016: Progress on Identifying and Characterizing the Human Proteome, Including Post-Translational Modifications. J. Proteome Res. 2016, 15, 3951–3960. [Google Scholar] [CrossRef] [Green Version]
- Zahn-Zabal, M.; Lane, L. What will neXtProt help us achieve in 2020 and beyond? Expert Rev. Proteomics 2020, 17, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Omenn, G.S. Reflections on the hupo human proteome project, the flagship project of the human proteome organization, at 10 years. Mol. Cell. Proteomics 2021, 20, 100062. [Google Scholar] [CrossRef]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Cristea, I.M.; Corrales, F.J.; Lindskog, C.; Paik, Y.K.; Van Eyk, J.E.; Liu, S.; Pennington, S.R.; et al. Research on the Human Proteome Reaches a Major Milestone: >90% of Predicted Human Proteins Now Credibly Detected, According to the HUPO Human Proteome Project. J. Proteome Res. 2020, 19, 4735–4746. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 Update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pons, P.; Latapy, M. Computing communities in large networks using random walks. J. Graph Algorithms Appl. 2006, 10, 191–218. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Peng, Q.; Liu, B.; Liu, Y.; Wang, Y. Disease Module Identification Based on Representation Learning of Complex Networks Integrated From GWAS, eQTL Summaries, and Human Interactome. Front. Bioeng. Biotechnol. 2020, 8, 418. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Inter. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Haenig, C.; Atias, N.; Taylor, A.K.; Mazza, A.; Schaefer, M.H.; Russ, J.; Riechers, S.P.; Jain, S.; Coughlin, M.; Fontaine, J.F.; et al. Interactome Mapping Provides a Network of Neurodegenerative Disease Proteins and Uncovers Widespread Protein Aggregation in Affected Brains. Cell Rep. 2020, 32, 105080. [Google Scholar] [CrossRef]
- Kang, H.J.; Lee, M.H.; Kang, H.L.; Kim, S.H.; Ahn, J.R.; Na, H.; Na, T.Y.; Kim, Y.N.; Seong, J.K.; Lee, M.O. Differential regulation of estrogen receptor a expression in breast cancer cells by metastasis-associated protein 1. Cancer Res. 2014, 74, 1484–1494. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.R.; Aldred, M.A.; Der Kaloustian, V.M.; Halal, F.; Gowans, G.; McLeod, D.R.; Zondag, S.; Toriello, H.V.; Magenis, R.E.; Elsea, S.H. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 2010, 87, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, P.G.; Huang, D.; Dai, Z. Haploinsufficiency of HDAC4 does not cause intellectual disability in all affected individuals. Am. J. Med. Genet. A 2014, 164, 1826–1829. [Google Scholar] [CrossRef]
- Ernst, A.; Appleton, B.A.; Ivarsson, Y.; Zhang, Y.; Gfeller, D.; Wiesmann, C.; Sidhu, S.S. A structural portrait of the PDZ domain family. J. Mol. Biol. 2014, 426, 3509–3519. [Google Scholar] [CrossRef]
- Oláh, J.; Vincze, O.; Virók, D.; Simon, D.; Bozsó, Z.; Tokési, N.; Horváth, I.; Hlavanda, E.; Kovács, J.; Magyar, A.; et al. Interactions of pathological hallmark proteins: Tubulin polymerization promoting protein/p25,β-amyloid, and α-synuclein. J. Biol. Chem. 2011, 286, 34088–34100. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Madahar, V.; Dang, R.; Jiang, L. Quantitative FRET (qFRET) Technology for the Determination of Protein–Protein Interaction Affinity in Solution. Molecules 2021, 26, 6339. [Google Scholar] [CrossRef]
- Duarte, J.G.; Blackburn, J.M. Advances in the development of human protein microarrays. Expert Rev. Proteomics 2017, 14, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Syu, G.D.; Dunn, J.; Zhu, H. Developments and applications of functional protein microarrays. Mol. Cell. Proteomics 2020, 19, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, A.L.; Eckhardt, M.; Krogan, N.J. Mass spectrometry-based protein–protein interaction networks for the study of human diseases. Mol. Syst. Biol. 2021, 17, e8792. [Google Scholar] [CrossRef]
- Abascal, F.; Juan, D.; Jungreis, I.; Martinez, L.; Rigau, M.; Rodriguez, J.M.; Vazquez, J.; Tress, M.L. Loose ends: Almost one in five human genes still have unresolved coding status. Nucleic Acids Res. 2018, 46, 7070–7084. [Google Scholar] [CrossRef] [Green Version]
- Menashe, I.; Aloni, R.; Lancet, D. A probabilistic classifier for olfactory receptor pseudogenes. BMC Bioinform. 2006, 7, 393. [Google Scholar] [CrossRef] [Green Version]
- Andjus, S.; Morillon, A.; Wery, M. From yeast to mammals, the nonsense-mediated mrna decay as a master regulator of long non-coding rnas functional trajectory. Non-Coding RNA 2021, 7, 44. [Google Scholar] [CrossRef]
- Yeramian, A.; Martin, L.; Serrat, N.; Arpa, L.; Soler, C.; Bertran, J.; McLeod, C.; Palacín, M.; Modolell, M.; Lloberas, J.; et al. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J. Immunol. 2006, 176, 5918–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervé, J.C.; Derangeon, M. Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 2013, 352, 21–31. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Niehues, H.; Tsoi, L.C.; van der Krieken, D.A.; Jansen, P.A.M.; Oortveld, M.A.W.; Rodijk-Olthuis, D.; van Vlijmen, I.M.J.J.; Hendriks, W.J.A.J.; Helder, R.W.; Bouwstra, J.A.; et al. Psoriasis-Associated Late Cornified Envelope (LCE) Proteins Have Antibacterial Activity. J. Investig. Dermatol. 2017, 137, 2380–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Gao, J.; Yin, X.; Sheng, Y.; Sun, L.; Cui, Y.; Zhang, X. Association of the Late Cornified Envelope-3 Genes with Psoriasis and Psoriatic Arthritis: A Systematic Review. J. Genet. Genomics 2015, 42, 49–56. [Google Scholar] [CrossRef]
- Raisch, T.; Chang, C.T.; Levdansky, Y.; Muthukumar, S.; Raunser, S.; Valkov, E. Reconstitution of recombinant human CCR4-NOT reveals molecular insights into regulated deadenylation. Nat. Commun. 2019, 10, 3173. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Fuentes, E.J. Emerging Themes in PDZ Domain Signaling: Structure, Function, and Inhibition. Int. Rev. Cell Mol. Biol. 2019, 343, 129–218. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Tsubata, S.; Bowles, K.R.; Haneda, N.; Uese, K.; Miyawaki, T.; Dreyer, W.J.; Messina, J.; Li, H.; Bowles, N.E.; et al. Novel Gene Mutations in Patients With Left Ventricular Noncompaction or Barth Syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikopoulos, K.; Venselaar, H.; Collin, R.W.J.; Riveiro-Alvarez, R.; Boonstra, F.N.; Hooymans, J.M.M.; Mukhopadhyay, A.; Shears, D.; Van Bers, M.; De Wijs, I.J.; et al. Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum. Mutat. 2010, 31, 656–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitrakopoulos, G.N.; Klapa, M.I.; Moschonas, N.K. How Far Are We from the Completion of the Human Protein Interactome Reconstruction? Biomolecules 2022, 12, 140. https://doi.org/10.3390/biom12010140
Dimitrakopoulos GN, Klapa MI, Moschonas NK. How Far Are We from the Completion of the Human Protein Interactome Reconstruction? Biomolecules. 2022; 12(1):140. https://doi.org/10.3390/biom12010140
Chicago/Turabian StyleDimitrakopoulos, Georgios N., Maria I. Klapa, and Nicholas K. Moschonas. 2022. "How Far Are We from the Completion of the Human Protein Interactome Reconstruction?" Biomolecules 12, no. 1: 140. https://doi.org/10.3390/biom12010140
APA StyleDimitrakopoulos, G. N., Klapa, M. I., & Moschonas, N. K. (2022). How Far Are We from the Completion of the Human Protein Interactome Reconstruction? Biomolecules, 12(1), 140. https://doi.org/10.3390/biom12010140