
The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation


Abstract
1. Introduction
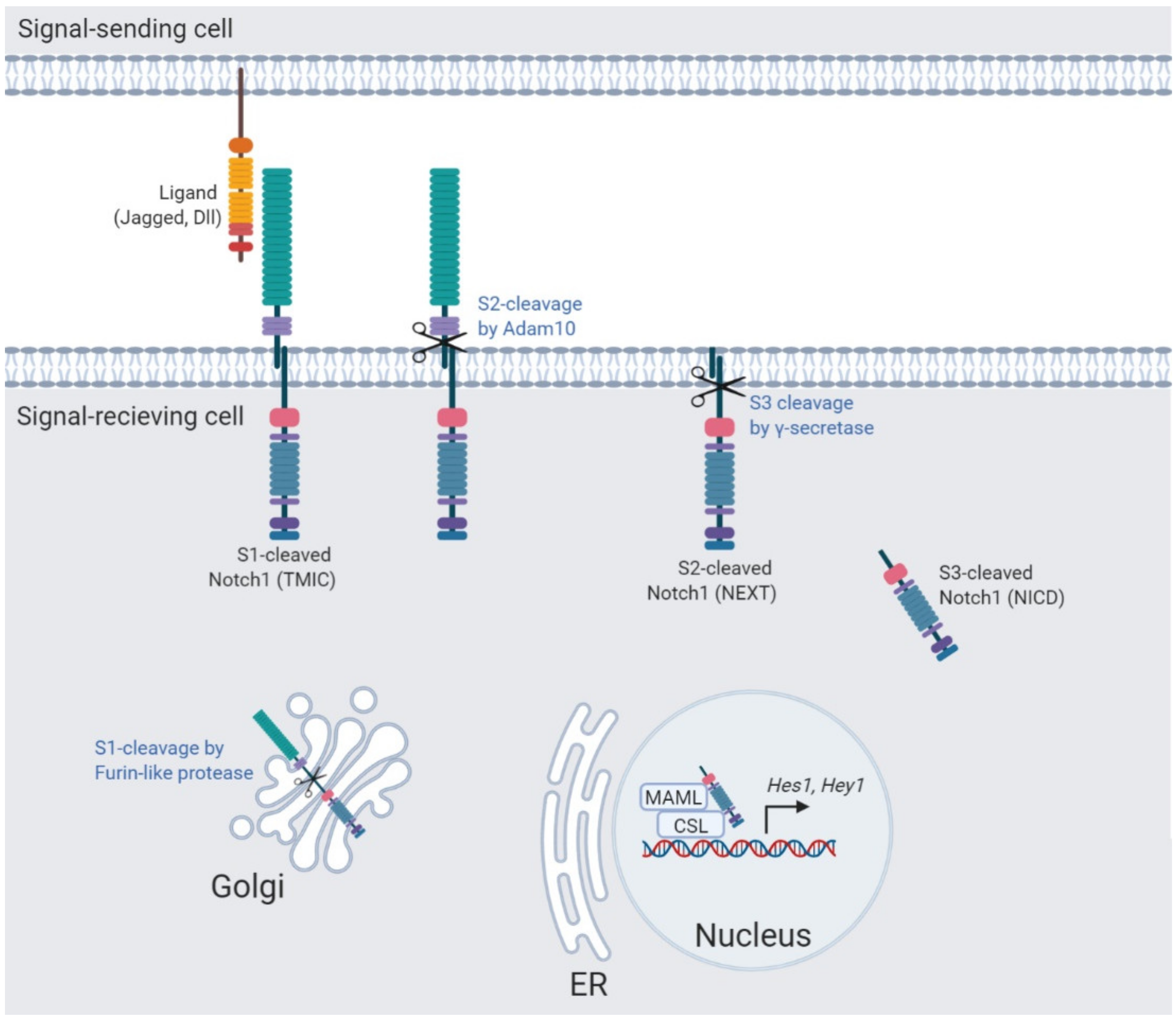
2. The Notch Signaling Pathway
3. Intracellular Trafficking of Transmembrane Proteins
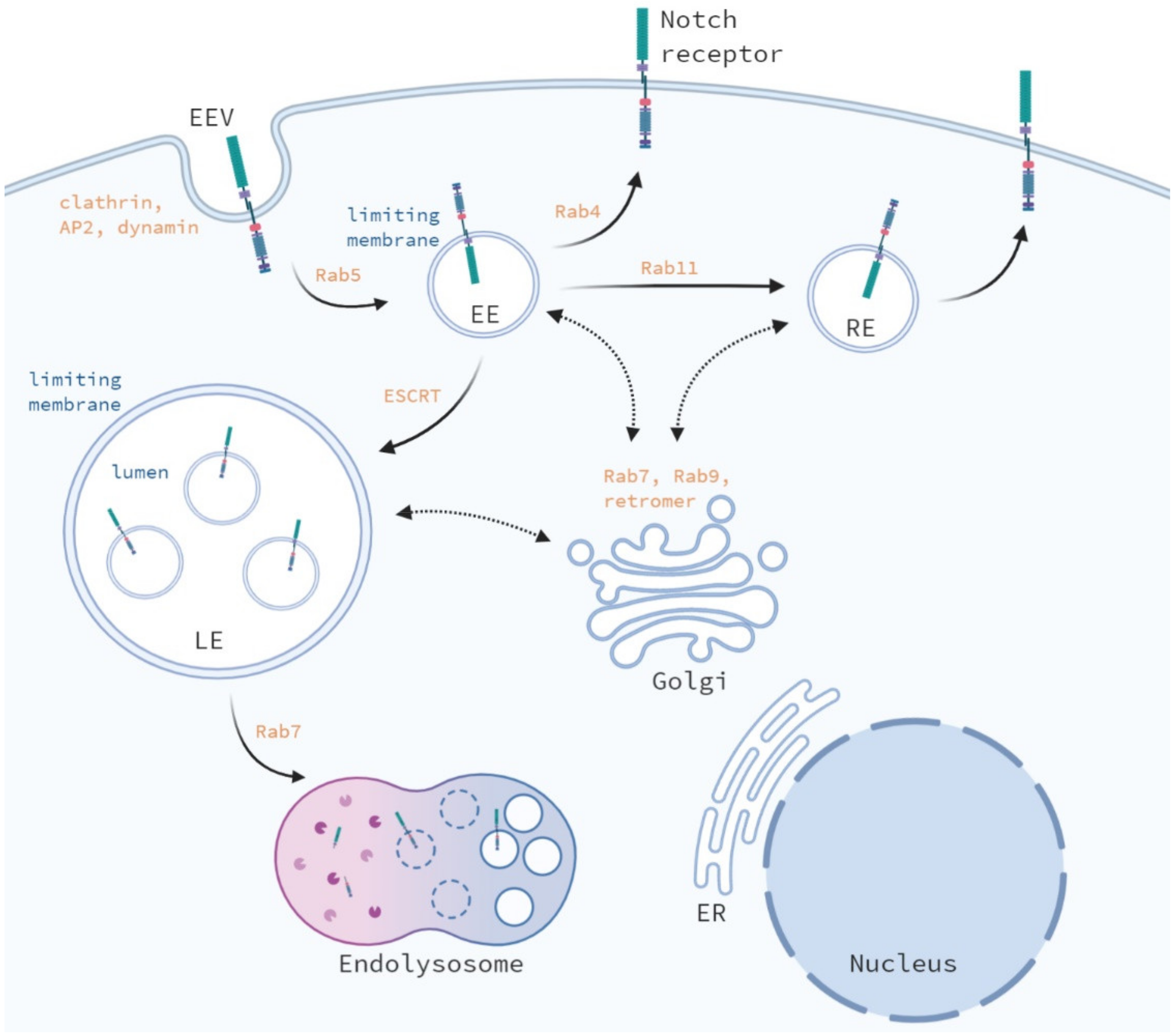
4. Endosomal Trafficking of the Notch Receptor; A Comparison between Drosophila and Mammals
4.1. Internalization of the Notch Receptor
4.2. Recycling of the Notch Receptor
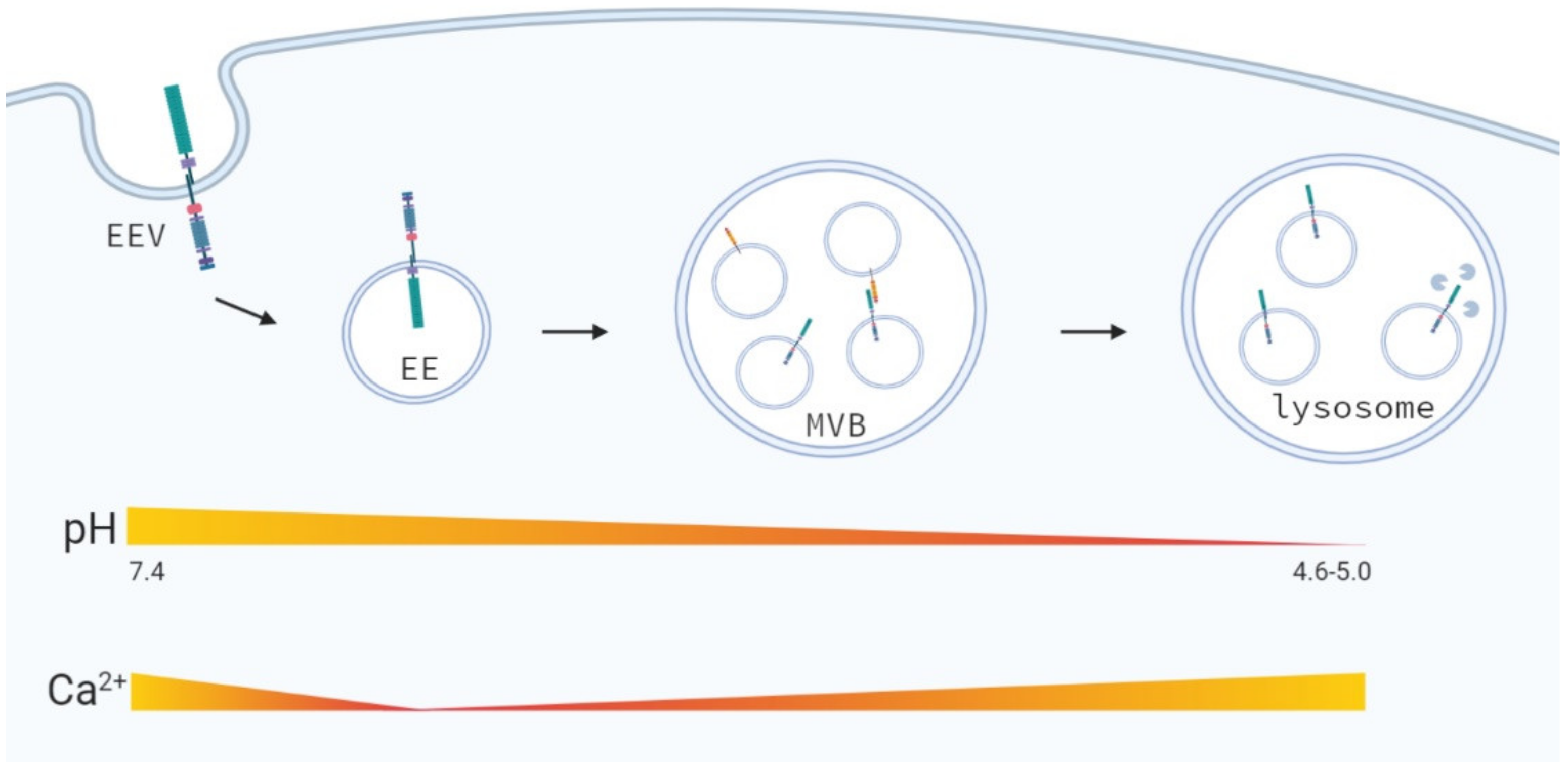
4.3. Early Endosomal Trafficking
4.4. Secretion via Exosomes?
4.5. Late Endosomal and Lysosomal Trafficking
4.6. Nuclear Translocation of Nicd and Notch Activation
5. Ligand-Independent Notch Signaling in Cancer
6. Physiological Ligand-Independent Signaling in Mammals?
7. Mechanisms of Ligand-Independent Notch Signaling
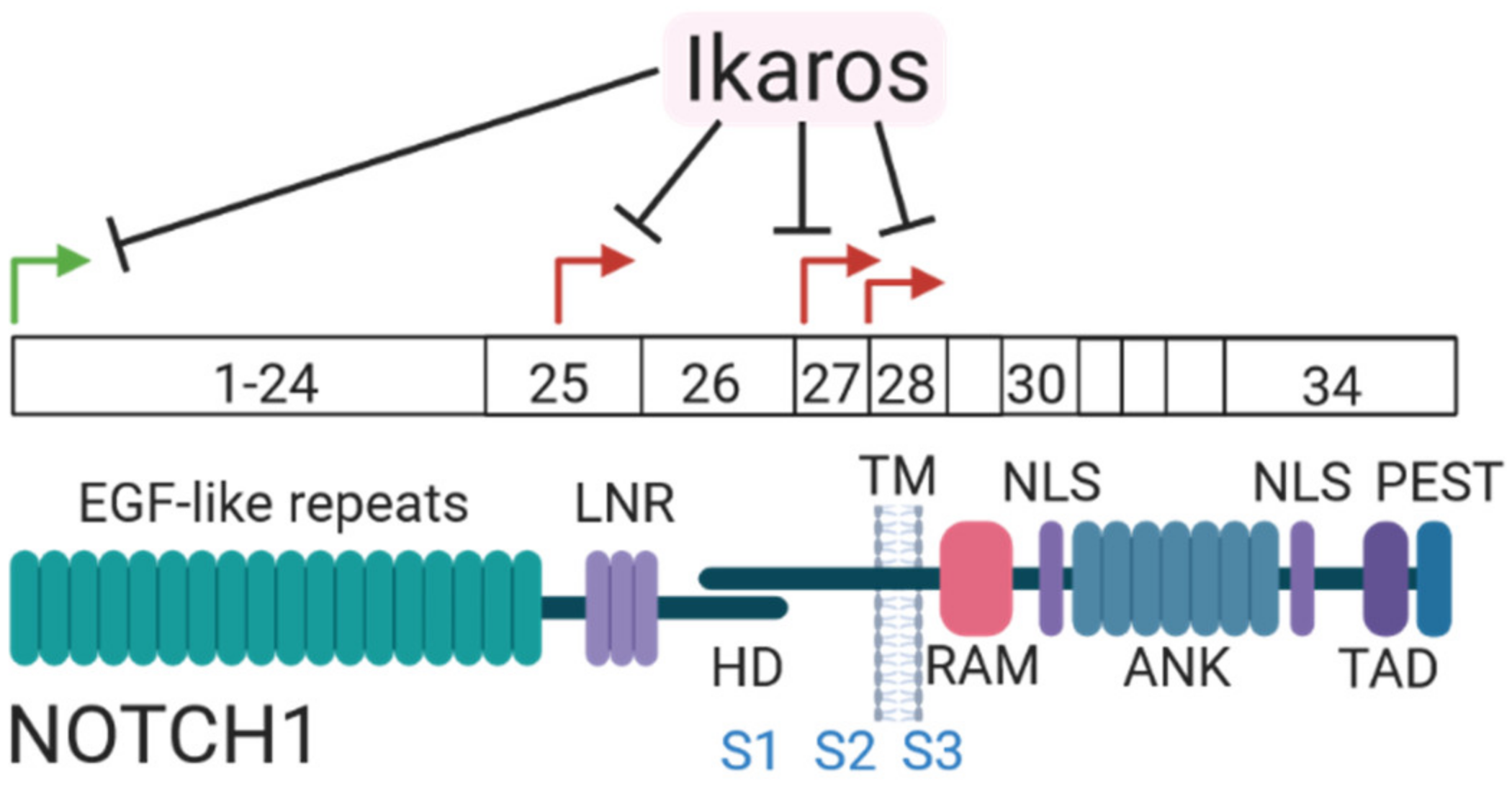
7.1. Mutations in the Notch Receptor
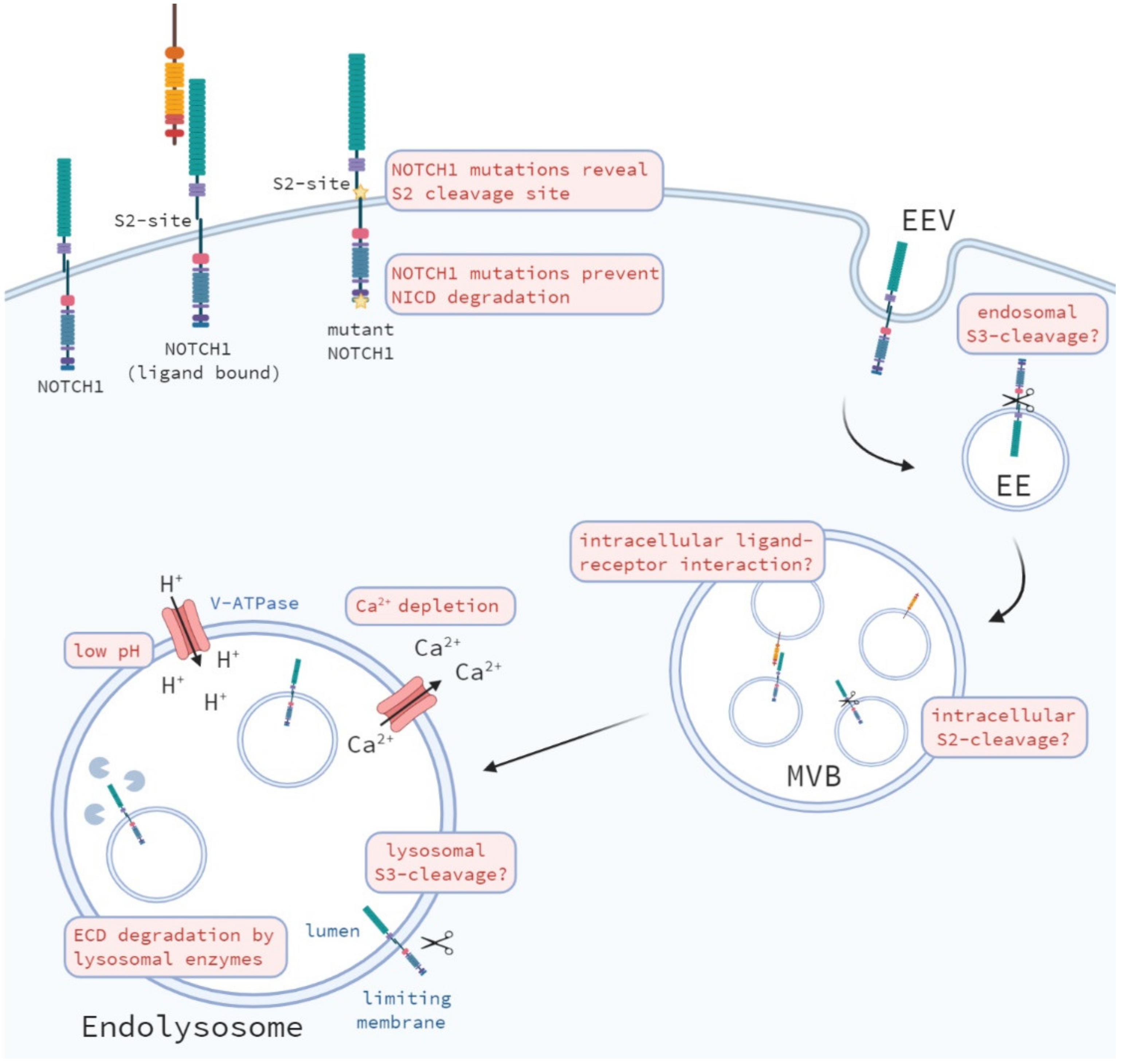
7.2. Activation during Endocytic and Lysosomal Trafficking of the Notch Receptor
7.3. Changes in the Endosomal-Lysosomal Environment
7.4. Alternative S2 Cleavage?
7.5. Alternative S3 Cleavage?
8. Therapeutic Targeting of Ligand-Independent Notch Signaling
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Gordon, W.; Roy, M.; Ulu, D.V.; Garfinkel, M.; Mansour, M.; Aster, J.C.; Blacklow, S.C. Structure of the Notch1-negative regulatory region: Implications for normal activation and pathogenic signaling in T-ALL. Blood 2009, 113, 4381–4390. [Google Scholar] [CrossRef]
- Van Tetering, G.; van Diest, P.; Verlaan, I.; van der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef]
- Mumm, J.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.; Ray, W.; Kopan, R. A Ligand-Induced Extracellular Cleavage Regulates γ-Secretase-like Proteolytic Activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J. Biol. Chem. 1974, 249, 5153–5162. [Google Scholar] [CrossRef]
- Merrifield, C.J.; Kaksonen, M. Endocytic Accessory Factors and Regulation of Clathrin-Mediated Endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016733. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Yamashiro, D.J. Endosome acidification and the pathways of receptor-mediated endocytosis. In Immunobiology of Proteins and Peptides IV; Springer: Berlin/Heidelberg, Germany, 1987; pp. 189–198. [Google Scholar]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Multiple routes of protein transport from endosomes to the trans Golgi network. FEBS Lett. 2009, 583, 3811–3816. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Parks, A.; Klueg, K.; Stout, J.; Muskavitch, M. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development 2000, 127, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Meloty-Kapella, L.; Shergill, B.; Kuon, J.; Botvinick, E.; Weinmaster, G. Notch Ligand Endocytosis Generates Mechanical Pulling Force Dependent on Dynamin, Epsins, and Actin. Dev. Cell 2012, 22, 1299–1312. [Google Scholar] [CrossRef]
- Montagne, C.; Gonzalez-Gaitan, M. Sara endosomes and the asymmetric division of intestinal stem cells. Development 2014, 141, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Lieber, T. Furin cleavage is not a requirement for Drosophila Notch function. Mech. Dev. 2002, 115, 41–51. [Google Scholar] [CrossRef]
- Gordon, W.R.; Vardar-Ulu, D.; L’Heureux, S.; Ashworth, T.; Malecki, M.J.; Sanchez-Irizarry, C.; McArthur, D.G.; Histen, G.; Mitchell, J.L.; Aster, J.C.; et al. Effects of S1 Cleavage on the Structure, Surface Export, and Signaling Activity of Human Notch1 and Notch2. PLoS ONE 2009, 4, e6613. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Kim, S.-H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial Segregation of γ-Secretase and Substrates in DistinctMembraneDomains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef]
- Okamura, Y.; Saga, Y. Pofut1 is required for the proper localization of the Notch receptor during mouse development. Mech. Dev. 2008, 125, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Hsu, W.M.; Wang, B.J.; Wu, G.H.; Lin, T.Y.; Lee, S.J.; Yen, C.T.; Liang, S.M.; Liao, Y.F. Caveolin-1 regulates γ-secretase-mediated AβPP processing by modulating spatial distribution of γ-secretase in membrane. J. Alzheimers Dis. 2010, 22, 423–442. [Google Scholar] [CrossRef] [PubMed]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V. Restricted location of PSEN2/γ-secretase determines substrate specificity and generates an intracellular Aβ pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Seugnet, L.; Simpson, P.; Haenlin, M. Requirement for Dynamin during Notch Signaling inDrosophilaNeurogenesis. Dev. Biol. 1997, 192, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, T.; Lu, H.; Kanwar, R.; Fortini, M.E.; Bilder, D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J. Cell Biol. 2008, 180, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Chapman, G.; Major, J.; Iyer, K.; James, A.; Pursglove, S.; Moreau, J.L.M.; Dunwoodie, S. Notch1 endocytosis is induced by ligand and is required for signal transduction. Biochim. et Biophys. Acta (BBA)–Bioenerg. 2016, 1863, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, E.B.; Conner, S.D. γ-Secretase-Dependent Cleavage Initiates Notch Signaling from the Plasma Membrane. Traffic 2010, 11, 1234–1245. [Google Scholar] [CrossRef]
- Gupta-Rossi, N.; Six, E.; LeBail, O.; Logeat, F.; Chastagner, P.; Olry, A.; Israël, A.; Brou, C. Monoubiquitination and endocytosis direct γ-secretase cleavage of activated Notch receptor. J. Cell Biol. 2004, 166, 73–83. [Google Scholar] [CrossRef]
- Park, R.; Shen, H.; Liu, L.; Liu, X.; Ferguson, S.M.; De Camilli, P. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J. Cell Sci. 2013, 126, 5305–5312. [Google Scholar] [CrossRef] [PubMed]
- Nemetschke, L.; Knust, E. Drosophila Crumbs prevents ectopic Notch activation in developing wings by inhibiting ligand-independent endocytosis. Development 2016, 143, 4543–4553. [Google Scholar] [CrossRef]
- Herranz, H.; Stamataki, E.; Feiguin, F.; Milán, M. Self-refinement of Notch activity through the transmembrane protein Crumbs: Modulation of γ-Secretase activity. EMBO Rep. 2006, 7, 297–302. [Google Scholar] [CrossRef]
- Ohata, S.; Aoki, R.; Kinoshita, S.; Yamaguchi, M.; Tsuruoka-Kinoshita, S.; Tanaka, H.; Wada, H.; Watabe, S.; Tsuboi, T.; Masai, I.; et al. Dual Roles of Notch in Regulation of Apically Restricted Mitosis and Apicobasal Polarity of Neuroepithelial Cells. Neuron 2011, 69, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Jovic, M.; Sharma, M.; Rahajeng, J.; Caplan, S. The early endosome: A busy sorting station for proteins at the crossroads. Histol. Histopathol. 2010, 25, 99–112. [Google Scholar] [PubMed]
- Fujibayashi, A.; Taguchi, T.; Misaki, R.; Ohtani, M.; Dohmae, N.; Takio, K.; Yamada, M.; Gu, J.; Yamakami, M.; Fukuda, M.; et al. Human RME-8 Is Involved in Membrane Trafficking through Early Endosomes. Cell Struct. Funct. 2008, 33, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Poupon, V.; Blondeau, F.; McPherson, P.S. The DnaJ-domain protein RME-8 functions in endosomal trafficking. J. Biol. Chem. 2005, 280, 40135–40143. [Google Scholar] [CrossRef]
- Shi, A.; Sun, L.; Banerjee, R.; Tobin, M.; Zhang, Y.; Grant, B.D. Regulation of endosomal clathrin and retromer-mediated endosome to Golgi retrograde transport by the J-domain protein RME-8. EMBO J. 2009, 28, 3290–3302. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lamarca, M.J.; Snowdon, L.A.; Seib, E.; Klein, T.; Bray, S.J. Rme-8 depletion perturbs Notch recycling and predisposes to pathogenic signaling. J. Cell Biol. 2015, 210, 303–318. [Google Scholar] [CrossRef]
- Johnson, S.A.; Zitserman, D.; Roegiers, F. Numb regulates the balance between Notch recycling and late-endosome targeting in Drosophila neural progenitor cells. Mol. Biol. Cell 2016, 27, 2857–2866. [Google Scholar] [CrossRef]
- Luo, Z.; Mu, L.; Zheng, Y.; Shen, W.; Li, J.; Xu, L.; Zhong, B.; Liu, Y.; Zhou, Y. NUMB enhances Notch signaling by repressing ubiquitination of NOTCH1 intracellular domain. J. Mol. Cell Biol. 2019, 12, 345–358. [Google Scholar] [CrossRef]
- Berdnik, D.; Török, T.; González-Gaitán, M.; Knoblich, J.A. The endocytic protein α-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev. Cell 2002, 3, 221–231. [Google Scholar] [CrossRef]
- Langevin, J.; Le Borgne, R.; Rosenfeld, F.; Gho, M.; Schweisguth, F.; Bellaïche, Y. Lethal Giant Larvae Controls the Localization of Notch-Signaling Regulators Numb, Neuralized, and Sanpodo in Drosophila Sensory-Organ Precursor Cells. Curr. Biol. 2005, 15, 955–962. [Google Scholar] [CrossRef]
- McGill, M.A.; McGlade, C.J. Mammalian Numb Proteins Promote Notch1 Receptor Ubiquitination and Degradation of the Notch1 Intracellular Domain. J. Biol. Chem. 2003, 278, 23196–23203. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef]
- Klezovitch, O.; Fernandez, T.E.; Tapscott, S.J.; Vasioukhin, V. Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 2004, 18, 559–571. [Google Scholar] [CrossRef]
- Li, H.; Koo, Y.; Mao, X.; Sifuentes-Dominguez, L.; Morris, L.; Jia, D.; Miyata, N.; Faulkner, R.A.; Van Deursen, J.M.; Vooijs, M.; et al. Endosomal sorting of Notch receptors through COMMD9-dependent pathways modulates Notch signaling. J. Cell Biol. 2015, 211, 605–617. [Google Scholar] [CrossRef]
- Dobrowolski, R.; De Robertis, E.M. Endocytic control of growth factor signalling: Multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Biol. 2011, 13, 53–60. [Google Scholar] [CrossRef]
- Hori, K.; Sen, A.; Kirchhausen, T.; Artavanis-Tsakonas, S. Synergy between the ESCRT-III complex and Deltex defines a ligand-independent Notch signal. J. Cell Biol. 2011, 195, 1005–1015. [Google Scholar] [CrossRef]
- Wilkin, M.; Tongngok, P.; Gensch, N.; Clemence, S.; Motoki, M.; Yamada, K.; Hori, K.; Taniguchi-Kanai, M.; Franklin, E.; Matsuno, K.; et al. Drosophila HOPS and AP-3 Complex Genes Are Required for a Deltex-Regulated Activation of Notch in the Endosomal Trafficking Pathway. Dev. Cell 2008, 15, 762–772. [Google Scholar] [CrossRef]
- Schneider, M.; Troost, T.; Grawe, F.; Martinez-Arias, A.; Klein, T. Activation of Notch in lgd mutant cells requires the fusion of late endosomes with the lysosome. J. Cell Sci. 2012, 126, 645–656. [Google Scholar] [CrossRef]
- Shimizu, H.; Woodcock, S.A.; Wilkin, M.B.; Trubenová, B.; Monk, N.; Baron, M. Compensatory Flux Changes within an Endocytic Trafficking Network Maintain Thermal Robustness of Notch Signaling. Cell 2014, 157, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, M.B.; Carbery, A.-M.; Fostier, M.; Aslam, H.; Mazaleyrat, S.L.; Higgs, J.; Myat, A.; Evans, D.A.; Cornell, M.; Baron, M. Regulation of Notch Endosomal Sorting and Signaling by Drosophila Nedd4 Family Proteins. Curr. Biol. 2004, 14, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, T.J.; Hori, K.; Sasamura, T.; Higgs, J.; Baron, M.; Matsuno, K. The first deltex null mutant indicates tissue-specific deltex-dependent Notch signaling in Drosophila. Mol. Genet. Genom. 2006, 275, 251–263. [Google Scholar] [CrossRef]
- Hori, K.; Fostier, M.; Ito, M.; Fuwa, T.J.; Go, M.J.; Okano, H.; Baron, M.; Matsuno, K. DrosophilaDeltex mediates Suppressor of Hairless-independent and late-endosomal activation of Notch signaling. Development 2004, 131, 5527–5537. [Google Scholar] [CrossRef]
- Mukherjee, A.; Veraksa, A.; Bauer, A.; Rosse, C.; Camonis, J.; Artavanis-Tsakonas, S. Regulation of Notch signalling by non-visual β-arrestin. Nat. Cell Biol. 2005, 7, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Kishi, N.; Tang, Z.; Maeda, Y.; Hirai, A.; Mo, R.; Ito, M.; Suzuki, S.; Nakao, K.; Kinoshita, T.; Kadesch, T. Murine homologs of deltex define a novel gene family involved in vertebrate Notch signaling and neurogenesis. Int. J. Dev. Neurosci. 2001, 19, 21–35. [Google Scholar] [CrossRef]
- Chastagner, P.; Rubinstein, E.; Brou, C. Ligand-activated Notch undergoes DTX4-mediated ubiquitylation and bilateral endocytosis before ADAM10 processing. Sci. Signal. 2017, 10, eaag2989. [Google Scholar] [CrossRef]
- Lehar, S.M.; Bevan, M.J. T Cells Develop Normally in the Absence of both Deltex1 and Deltex2. Mol. Cell. Biol. 2006, 26, 7358–7371. [Google Scholar] [CrossRef]
- Yamamoto, N.; Yamamoto, S.-I.; Inagaki, F.; Kawaichi, M.; Fukamizu, A.; Kishi, N.; Matsuno, K.; Nakamura, K.; Weinmaster, G.; Okano, H.; et al. Role of Deltex-1 as a Transcriptional Regulator Downstream of the Notch Receptor. J. Biol. Chem. 2001, 276, 45031–45040. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Yamada, K.; Fuwa, T.J.; Ayukawa, T.; Tanaka, T.; Nakamura, A.; Wilkin, M.B.; Baron, M.; Matsuno, K. Roles of Drosophila Deltex in Notch receptor endocytic trafficking and activation. Genes Cells 2011, 16, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Gardner, P. Notching up another pathway. Bioessays 2002, 24, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, R.; Fortini, M.E. Notch Signaling: A Different Sort Makes the Cut. Curr. Biol. 2004, 14, R1043–R1045. [Google Scholar] [CrossRef] [PubMed]
- Sakata, T.; Sakaguchi, H.; Tsuda, L.; Higashitani, A.; Aigaki, T.; Matsuno, K.; Hayashi, S. Drosophila Nedd4 Regulates Endocytosis of Notch and Suppresses Its Ligand-Independent Activation. Curr. Biol. 2004, 14, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, P.; Israël, A.; Brou, C. AIP4/Itch Regulates Notch Receptor Degradation in the Absence of Ligand. PLoS ONE 2008, 3, e2735. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, P.; Israël, A.; Brou, C. Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 2006, 7, 1147–1153. [Google Scholar] [CrossRef]
- Sheldon, H.; Heikamp, E.; Turley, H.; Dragovic, R.; Thomas, P.; Oon, C.E.; Leek, R.; Edelmann, M.; Kessler, B.; Sainson, R.C.A.; et al. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood 2010, 116, 2385–2394. [Google Scholar] [CrossRef]
- Cruz, L.; Romero, J.A.A.; Prado, M.B.; Santos, T.G.; Lopes, M.H. Evidence of Extracellular Vesicles Biogenesis and Release in Mouse Embryonic Stem Cells. Stem Cell Rev. Rep. 2017, 14, 262–276. [Google Scholar] [CrossRef]
- Wang, X.; Jiao, Y.; Pan, Y.; Zhang, L.; Gong, H.; Qi, Y.; Wang, M.; Gong, H.; Shao, M.; Wang, X.; et al. Fetal Dermal Mesenchymal Stem Cell-Derived Exosomes Accelerate Cutaneous Wound Healing by Activating Notch Signaling. Stem Cells Int. 2019, 2019, 2402916. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, Q. Plasma membrane-derived extracellular microvesicles mediate non-canonical intercellular NOTCH signaling. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Childress, J.L.; Acar, M.; Tao, C.; Halder, G. Lethal Giant Discs, a Novel C2-Domain Protein, Restricts Notch Activation during Endocytosis. Curr. Biol. 2006, 16, 2228–2233. [Google Scholar] [CrossRef]
- Drusenheimer, N.; Migdal, B.; Jäckel, S.; Tveriakhina, L.; Scheider, K.; Schulz, K.; Gröper, J.; Köhrer, K.; Klein, T. The mammalian orthologs of Drosophila Lgd, CC2D1A and CC2D1B, function in the endocytic pathway, but their individual loss of function does not affect notch signalling. PLoS Genet. 2015, 11, e1005749. [Google Scholar] [CrossRef]
- Leitch, C.C.; Lodh, S.; Prieto-Echagüe, V.; Badano, J.; Zaghloul, N.A. Basal body proteins regulate Notch signaling via endosomal trafficking. J. Cell Sci. 2014, 127, 2407–2419. [Google Scholar] [CrossRef]
- Zheng, L.; Saunders, C.A.; Sorensen, E.B.; Waxmonsky, N.C.; Conner, S.D. Notch signaling from the endosome requires a conserved dileucine motif. Mol. Biol. Cell 2013, 24, 297–307. [Google Scholar] [CrossRef]
- Vaccari, T.; Duchi, S.; Cortese, K.; Tacchetti, C.; Bilder, D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 2010, 137, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Kobia, F.M.; Duchi, S.; Deflorian, G.; Vaccari, T. Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol. Oncol. 2013, 8, 207–220. [Google Scholar] [CrossRef]
- Yan, Y.; Denef, N.; Schüpbach, T. The Vacuolar Proton Pump, V-ATPase, Is Required for Notch Signaling and Endosomal Trafficking in Drosophila. Dev. Cell 2009, 17, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Fortini, M.E.; Bilder, D. Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev. 2009, 19, 323–328. [Google Scholar] [CrossRef]
- Portela, M.; Yang, L.; Paul, S.; Li, X.; Veraksa, A.; Parsons, L.M.; Richardson, H.E. Lgl reduces endosomal vesicle acidification and Notch signaling by promoting the interaction between Vap33 and the V-ATPase complex. Sci. Signal. 2018, 11, eaar1976. [Google Scholar] [CrossRef] [PubMed]
- Justice, N.; Roegiers, F.; Jan, L.; Jan, Y.N. Lethal Giant Larvae Acts Together with Numb in Notch Inhibition and Cell Fate Specification in the Drosophila Adult Sensory Organ Precursor Lineage. Curr. Biol. 2003, 13, 778–783. [Google Scholar] [CrossRef][Green Version]
- Parsons, L.M.; Portela, M.; Grzeschik, N.A.; Richardson, H.E. Lgl regulates Notch signaling via endocytosis, independently of the apical aPKC-Par6-Baz polarity complex. Curr. Biol. 2014, 24, 2073–2084. [Google Scholar] [CrossRef]
- Portela, M.; Parsons, L.; Grzeschik, N.A.; Richardson, H.E. Regulation of Notch signaling and endocytosis by the Lgl neoplastic tumor suppressor. Cell Cycle 2015, 14, 1496–1506. [Google Scholar] [CrossRef]
- Hounjet, J.; Habets, R.; Schaaf, M.B.; Hendrickx, T.C.; Barbeau, L.M.O.; Yahyanejad, S.; Rouschop, K.M.; Groot, A.J.; Vooijs, M. The anti-malarial drug chloroquine sensitizes oncogenic NOTCH1 driven human T-ALL to γ-secretase inhibition. Oncogene 2019, 38, 5457–5468. [Google Scholar] [CrossRef]
- Sachan, N.; Mishra, A.K.; Mutsuddi, M.; Mukherjee, A. The Drosophila Importin-α3 Is Required for Nuclear Import of Notch In Vivo and It Displays Synergistic Effects with Notch Receptor on Cell Proliferation. PLoS ONE 2013, 8, e68247. [Google Scholar] [CrossRef]
- Huenniger, K.; Krämer, A.; Soom, M.; Chang, I.; Köhler, M.; Depping, R.; Kehlenbach, R.H.; Kaether, C. Notch1 signaling is mediated by importins alpha 3, 4, and 7. Experientia 2010, 67, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Sjöqvist, M.; Antfolk, D.; Ferraris, S.; Rraklli, V.; Haga, C.; Antila, C.; Mutvei, A.; Imanishi, S.Y.; Holmberg, J.; Jin, S.; et al. PKCζ regulates Notch receptor routing and activity in a Notch signaling-dependent manner. Cell Res. 2014, 24, 433–450. [Google Scholar] [CrossRef]
- Weng, A.; Ferrando, A.A.; Lee, W.; Iv, J.P.M.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.D.; Ciaccio, P.J. Modulation of Notch Processing by γ-Secretase Inhibitors Causes Intestinal Goblet Cell Metaplasia and Induction of Genes Known to Specify Gut Secretory Lineage Differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.-J.J. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.; Iyer, M.; Chinnaiyan, A.M.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef]
- Parr, C.; Watkins, G.; Jiang, W.G. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int. J. Mol. Med. 2004, 14, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Sharma, A.; Balaji, S.A.; Gowda, M.C.; Dighe, R.R.; Kumar, R.V.; Rangarajan, A. Coordinate Hyperactivation of Notch1 and Ras/MAPK Pathways Correlates with Poor Patient Survival: Novel Therapeutic Strategy for Aggressive Breast Cancers. Mol. Cancer Ther. 2014, 13, 3198–3209. [Google Scholar] [CrossRef]
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008, 451, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Rennstam, K.; McMichael, N.; Berglund, P.; Honeth, G.; Hegardt, C.; Rydén, L.; Luts, L.; Bendahl, P.-O.; Hedenfalk, I. Numb protein expression correlates with a basal-like phenotype and cancer stem cell markers in primary breast cancer. Breast Cancer Res. Treat. 2009, 122, 315–324. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Mukherjee, T.; Kim, W.S.; Mandal, L.; Banerjee, U. Interaction between Notch and Hif-α in development and survival of Drosophila blood cells. Science 2011, 332, 1210–1213. [Google Scholar] [CrossRef]
- Arco, P.G.-D.; Kashiwagi, M.; Jackson, A.F.; Naito, T.; Zhang, J.; Liu, F.; Kee, B.; Vooijs, M.; Radtke, F.; Redondo, J.M.; et al. Alternative Promoter Usage at the Notch1 Locus Supports Ligand-Independent Signaling in T Cell Development and Leukemogenesis. Immunity 2010, 33, 685–698. [Google Scholar] [CrossRef]
- Struhl, G.; Adachi, A. Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins. Mol. Cell 2000, 6, 625–636. [Google Scholar] [CrossRef]
- Struhl, G.; Greenwald, I. Presenilin-mediated transmembrane cleavage is required for Notch signal transduction in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 229–234. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef]
- McMillan, B.J.; Zimmerman, B.; Egan, E.; Lofgren, M.; Xu, X.; Hesser, A.; Blacklow, S.C. Structure of human POFUT1, its requirement in ligand-independent oncogenic Notch signaling, and functional effects of Dowling-Degos mutations. Glycobiology 2017, 27, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Chammaa, M.; Malysa, A.; Redondo, C.; Jang, H.; Chen, W.; Bepler, G.; Fernandez-Valdivia, R. RUMI is a novel negative prognostic marker and therapeutic target in non–small-cell lung cancer. J. Cell. Physiol. 2018, 233, 9548–9562. [Google Scholar] [CrossRef]
- Leonardi, J.; Fernandez-Valdivia, R.; Li, Y.-D.; Simcox, A.A.; Jafar-Nejad, H. Multiple O-glucosylation sites on Notch function as a buffer against temperature-dependent loss of signaling. Development 2011, 138, 3569–3578. [Google Scholar] [CrossRef]
- Nichols, J.; Miyamoto, A.; Olsen, S.L.; D’Souza, B.; Yao, C.; Weinmaster, G. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J. Cell Biol. 2007, 176, 445–458. [Google Scholar] [CrossRef]
- Gordon, W.R.; Zimmerman, B.; He, L.; Miles, L.J.; Huang, J.; Tiyanont, K.; McArthur, D.G.; Aster, J.C.; Perrimon, N.; Loparo, J.J.; et al. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev. Cell 2015, 33, 729–736. [Google Scholar] [CrossRef]
- Pan, D.; Rubin, G.M. Kuzbanian Controls Proteolytic Processing of Notch and Mediates Lateral Inhibition during Drosophila and Vertebrate Neurogenesis. Cell 1997, 90, 271–280. [Google Scholar] [CrossRef]
- Bozkulak, E.C.; Weinmaster, G. Selective Use of ADAM10 and ADAM17 in Activation of Notch1 Signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [PubMed]
- Sulis, M.L.; Saftig, P.; Ferrando, A.A. Redundancy and specificity of the metalloprotease system mediating oncogenic NOTCH1 activation in T-ALL. Leukemia 2011, 25, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Malecki, M.J.; Sanchez-Irizarry, C.; Mitchell, J.L.; Histen, G.; Xu, M.L.; Aster, J.C.; Blacklow, S.C. Leukemia-Associated Mutations within the NOTCH1 Heterodimerization Domain Fall into at Least Two Distinct Mechanistic Classes. Mol. Cell. Biol. 2006, 26, 4642–4651. [Google Scholar] [CrossRef] [PubMed]
- Habets, R.A.; Groot, A.J.; Yahyanejad, S.; Tiyanont, K.; Blacklow, S.C.; Vooijs, M. Human NOTCH2 Is Resistant to Ligand-independent Activation by Metalloprotease Adam17. J. Biol. Chem. 2015, 290, 14705–14716. [Google Scholar] [CrossRef]
- Xu, X.; Choi, S.H.; Hu, T.; Tiyanont, K.; Habets, R.; Groot, A.J.; Vooijs, M.; Aster, J.C.; Chopra, R.; Fryer, C.; et al. Insights into Autoregulation of Notch3 from Structural and Functional Studies of Its Negative Regulatory Region. Structure 2015, 23, 1227–1235. [Google Scholar] [CrossRef]
- Groot, A.J.; Habets, R.; Yahyanejad, S.; Hodin, C.M.; Reiss, K.; Saftig, P.; Theys, J.; Vooijs, M. Regulated Proteolysis of NOTCH2 and NOTCH3 Receptors by ADAM10 and Presenilins. Mol. Cell. Biol. 2014, 34, 2822–2832. [Google Scholar] [CrossRef]
- Scott, C.C.; Gruenberg, J. Ion flux and the function of endosomes and lysosomes: pH is just the start: The flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 2011, 33, 103–110. [Google Scholar] [CrossRef]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef]
- Gerasimenko, J.; Tepikin, A.; Petersen, O.; Gerasimenko, O. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 1998, 8, 1335–1338. [Google Scholar] [CrossRef]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; Di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.R.; Vardar-Ulu, D.; Histen, G.; Sanchez-Irizarry, C.; Aster, J.C.; Blacklow, S.C. Structural basis for autoinhibition of Notch. Nat. Struct. Mol. Biol. 2007, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Rand, M.D.; Grimm, L.M.; Artavanis-Tsakonas, S.; Patriub, V.; Blacklow, S.C.; Sklar, J.; Aster, J.C. Calcium Depletion Dissociates and Activates Heterodimeric Notch Receptors. Mol. Cell. Biol. 2000, 20, 1825–1835. [Google Scholar] [CrossRef]
- Vardar, D.; North, C.L.; Sanchez-Irizarry, C.; Aster, J.C.; Blacklow, S.C. Nuclear Magnetic Resonance Structure of a Prototype Lin12-Notch Repeat Module from Human Notch1†. Biochemistry 2003, 42, 7061–7067. [Google Scholar] [CrossRef]
- López-Schier, H.; Johnston, D.S. Drosophila Nicastrin Is Essential for the Intramembranous Cleavage of Notch. Dev. Cell 2002, 2, 79–89. [Google Scholar] [CrossRef]
- Ebsen, H.; Schröder, A.; Kabelitz, D.; Janssen, O. Differential Surface Expression of ADAM10 and ADAM17 on Human T Lymphocytes and Tumor Cells. PLoS ONE 2013, 8, e76853. [Google Scholar] [CrossRef]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef]
- Dornier, E.; Coumailleau, F.; Ottavi, J.-F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Eschenbrenner, E.; Dornier, E.; Boucheix, C.; Charrin, S.; Rubinstein, E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem. Soc. Trans. 2017, 45, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Skovronsky, D.M.; Moore, D.B.; Milla, M.E.; Doms, R.W.; Lee, V.M.-Y. Protein Kinase C-dependent α-Secretase Competes with β-Secretase for Cleavage of Amyloid-β Precursor Protein in the Trans-Golgi Network. J. Biol. Chem. 2000, 275, 2568–2575. [Google Scholar] [CrossRef]
- Small, S.A.; Gandy, S. Sorting through the cell biology of Alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 2006, 52, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, S.H.; Bagshaw, R.D.; Guiral, M.; Zhang, S.; Ackerley, C.A.; Pak, B.J.; Callahan, J.W.; Mahuran, D.J. Presenilin-1, nicastrin, amyloid precursor protein, and γ-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 2003, 278, 26687–26694. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, A.; Okochi, M.; Tagami, S.; Jiang, J.; Itoh, N.; Nakayama, T.; Yanagida, K.; Ishizuka-Katsura, Y.; Morihara, T.; Kamino, K. Presenilin-dependent γ-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry 2006, 45, 4907–4914. [Google Scholar] [CrossRef]
- Tagami, S.; Okochi, M.; Yanagida, K.; Ikuta, A.; Fukumori, A.; Matsumoto, N.; Ishizuka-Katsura, Y.; Nakayama, T.; Itoh, N.; Jiang, J.; et al. Regulation of Notch Signaling by Dynamic Changes in the Precision of S3 Cleavage of Notch-1. Mol. Cell. Biol. 2008, 28, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef]
- Okochi, M.; Fukumori, A.; Jiang, J.; Itoh, N.; Kimura, R.; Steiner, H.; Haass, C.; Tagami, S.; Takeda, M. Secretion of the Notch-1 Abeta-like peptide during Notch signaling. J. Biol. Chem. 2006, 281, 7890–7898. [Google Scholar] [CrossRef]
- Okochi, M.; Steiner, H.; Fukumori, A.; Tanii, H.; Tomita, T.; Tanaka, T.; Iwatsubo, T.; Kudo, T.; Takeda, M.; Haass, C. Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. EMBO J. 2002, 21, 5408–5416. [Google Scholar] [CrossRef]
- Chandu, D.; Huppert, S.S.; Kopan, R. Analysis of transmembrane domain mutants is consistent with sequential cleavage of Notch by gamma-secretase. J. Neurochem. 2006, 96, 228–235. [Google Scholar] [CrossRef]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Messersmith, W.A.; Mikulski, S.M.; Papadopoulos, K.P.; Kwak, E.L.; Gibbon, D.G.; Patnaik, A.; Falchook, G.S.; Dasari, A.; Shapiro, G.I.; et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. 2012, 30, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Tiyanont, K.; Wales, T.E.; Siebel, C.W.; Engen, J.R.; Blacklow, S.C. Insights into Notch3 Activation and Inhibition Mediated by Antibodies Directed against Its Negative Regulatory Region. J. Mol. Biol. 2013, 425, 3192–3204. [Google Scholar] [CrossRef][Green Version]
- Li, K.; Li, Y.; Wu, W.; Gordon, W.; Chang, D.W.; Lu, M.; Scoggin, S.; Fu, T.; Vien, L.; Histen, G.; et al. Modulation of Notch Signaling by Antibodies Specific for the Extracellular Negative Regulatory Region of NOTCH3. J. Biol. Chem. 2008, 283, 8046–8054. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2018, 37, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019, 8, 5148–5157. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor Vessel Normalization by Chloroquine Independent of Autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Drosophila/ Mammalian | Function | Loss of Function Drosophila | Loss of Function Mammals | |
|---|---|---|---|---|
| Internalization | Shibire/Dynamin | Endosomal vesicle formation | ↓Notch signaling ↑Notch receptor expression at the plasma membrane | Dynamin 1 =↓NotchΔE internalization, no effect on Val1744 levels in the nucleus (HeLa cells), γ-secretase-mediated Next cleavage Dynamin 2 =↓γ-secretase cleavage of Notch1-ΔE-TM |
| Clathrin heavy chain | Endosomal vesicle formation | ↓Ligand-dependent Notch signaling | ↓NotchΔE internalization, no effect on Val1744 levels in the nucleus | |
| Crumbs/Crb1 | Prevents Notch receptor internalization and activation | ↑Internalization of Notch ↑Ectopic ligand-independent Notch signaling | ||
| Recycling | Rme-8/ Dnajc13 | Endosomal sorting and recycling | ↑Notch in enlarged Rab4+ endosomes ↓Notch signaling | |
| Numb | Stimulates Notch receptor trafficking towards MVBs (degradation) instead of recycling endosomes | ↓Notch in late endosomes ↑Notch at the plasma membrane ↓Ubiquitination of Nicd ↑Notch signaling | ↑Notch signaling ~50% of all human breast cancers ~30% of all human NSCLC | |
| Lethal giant larvae/ Lgl1-4 | Inhibits Notch signaling by controlling the asymmetric localization of Numb | ↑Notch signaling ↑Nicd | ↑Notch signaling ↑Nicd | |
| Early endosomes | Rab5, Avl/Syntaxin 7 | Entry into early endosomes | ↓Notch signaling ↑Notch receptor expression at the plasma membrane | |
| Deltex (dx) | E3-ligase, mono-ubiquitination of Notch-icd Endosomal stabilization and activation of Notch | ↓Notch signaling ↓Notch internalization | ↑Notch signaling DTX1−/−2−/− mutant mice = no defects in Notch dependent T-cell development | |
| Suppressor of Deltex (Su (dx))/Itch | E3-ligase, poly-ubiquitination and lysosomal degradation of Notch | ↑Notch signaling | Itch mutant mice = severe autoimmune disease Itch deficiency in humans = severe autoimmune disease and morphologic and developmental abnormalities | |
| Late endosomes | ESCRT | Maturation of early endosomes into MVBs | ↑Notch in endosomes ↑Ligand-independent Notch signaling | CHMP5 (ESCRT-III)—No activation of Notch signaling Vps25 (ESCRT-II)—No activation of Notch signaling Tsg101 (ESCRT-I)—No activation of Notch signaling (in vitro), early embryonic lethal (in vivo) BBS1/3/4—↑ligand-independent Notch signaling, accumulation of Notch in late endosomes |
| Lgd/Cc2d1a | Maturation of early endosomes into MVBs | ↑Notch in early endosomes ↑Ligand-independent Notch signaling | ↑Endosomal size in intestinal Cc2d1a mutant mice, ~ Notch signaling Loss of both orthologs is not validated yet | |
| Hrs/Hgs | Component ESCRT-0, recognizes ubiquitinated proteins and facilitates transport from early to late endosome | ↑Notch in early endosomes ↓Notch signaling | ||
| Nuclear translocation | Importin α Importin β1 | Nuclear import of Nicd | ↓Nuclear localization of Notch ↑Notch in the cytoplasm | ↓Nuclear localization of Notch |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hounjet, J.; Vooijs, M. The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation. Biomolecules 2021, 11, 1369. https://doi.org/10.3390/biom11091369
Hounjet J, Vooijs M. The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation. Biomolecules. 2021; 11(9):1369. https://doi.org/10.3390/biom11091369
Chicago/Turabian StyleHounjet, Judith, and Marc Vooijs. 2021. "The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation" Biomolecules 11, no. 9: 1369. https://doi.org/10.3390/biom11091369
APA StyleHounjet, J., & Vooijs, M. (2021). The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation. Biomolecules, 11(9), 1369. https://doi.org/10.3390/biom11091369