
Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New

 ,
,  , and
, and
Abstract
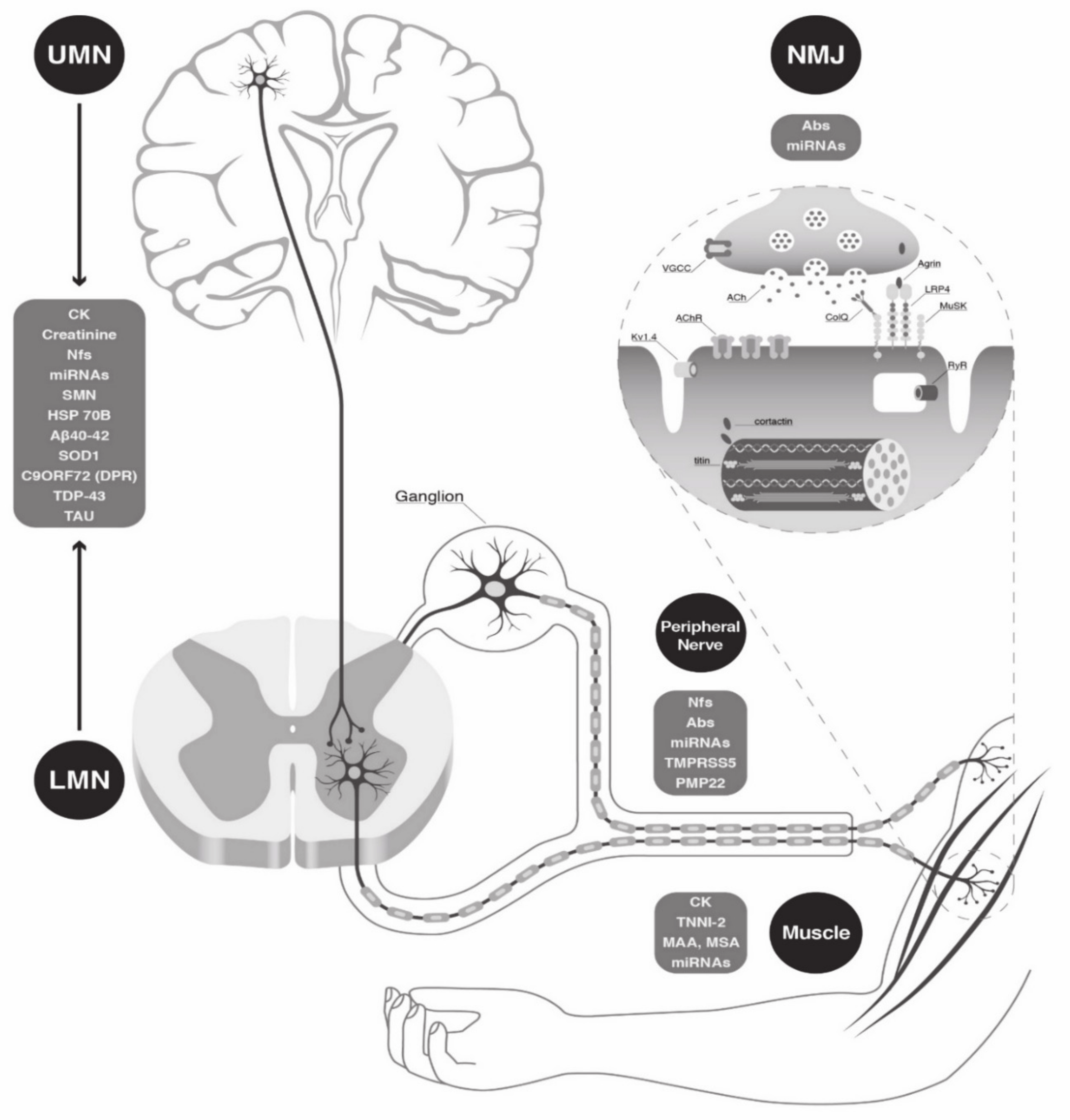
1. Introduction
2. Circulating Biomarkers
2.1. “Easy-to-Access” Biomarkers in Clinical Practice
2.1.1. CK in Muscular Dystrophies and in IIMs
2.1.2. CK in ALS
2.1.3. Creatinine in Motor Neuron Diseases
2.2. Antibodies
2.2.1. Antibodies in IIMs
2.2.2. Antibodies in NMJ Diseases
2.2.3. Antibodies in Dysimmune Neuropathies
2.3. Neurofilaments
2.3.1. Nfs in Dysimmune and Genetic Neuropathies
2.3.2. Nfs in Motor Neuron Diseases
2.4. microRNAs
2.4.1. microRNAs in Muscular Dystrophies
2.4.2. microRNAs in NMJ Diseases
2.4.3. microRNAs in Genetic Neuropathies
2.4.4. microRNAs in Motor Neuron Diseases
2.5. Gene Products
2.5.1. Gene Products in Genetic Neuropathies
2.5.2. Gene Products in Motor Neuron Diseases
2.6. Other Biomarker Proposals
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Aartsma-Rus, A.; Ferlini, A.; McNally, E.M.; Spitali, P.; Sweeney, H.L.; Aartsma-Rus, A.M.; Szigyarto, C.A.-K.; Bello, L.; Bronson, A.; Brown, K.; et al. 226th ENMC International Workshop: Towards validated and qualified biomarkers for therapy development for Duchenne muscular dystrophy 20-22 January 2017, Heemskerk, The Netherlands. Neuromuscul. Disord. 2018, 28, 77–86. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Ferlini, A.; Vroom, E. Biomarkers and surrogate endpoints in Duchenne: Meeting report. Neuromuscul. Disord. 2014, 24, 743–745. [Google Scholar] [CrossRef]
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Barthel, B.L.; Cox, D.; Barbieri, M.; Ziemba, M.; Straub, V.; Hoffman, E.P.; Russell, A.J. Elevation of fast but not slow troponin I in the circulation of patients with Becker and Duchenne muscular dystrophy. Muscle Nerve 2021, 64, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, I.T.; Calissano, M.; Scoto, M.; Preston, M.D.; Cirak, S.; Feng, L.; Collins, J.; Kole, R.; Guglieri, M.; Straub, V.; et al. Dystromirs as Serum Biomarkers for Monitoring the Disease Severity in Duchenne Muscular Dystrophy. PLoS ONE 2013, 8, e80263. [Google Scholar] [CrossRef] [PubMed]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef]
- Roberts, T.C.; Blomberg, K.E.M.; McClorey, G.; EL Andaloussi, S.; Godfrey, C.; Betts, C.; Coursindel, T.; Gait, M.J.; Smith, C.I.E.; Wood, M.J. Expression Analysis in Multiple Muscle Groups and Serum Reveals Complexity in the MicroRNA Transcriptome of the mdx Mouse with Implications for Therapy. Mol. Ther. Nucleic Acids 2012, 1, e39. [Google Scholar] [CrossRef]
- Cazzella, V.; Martone, J.; Pinnarò, C.; Santini, T.; Twayana, S.S.; Sthandier, O.; D’Amico, A.; Ricotti, V.; Bertini, E.; Muntoni, F.; et al. Exon 45 Skipping Through U1-snRNA Antisense Molecules Recovers the Dys-nNOS Pathway and Muscle Differentiation in Human DMD Myoblasts. Mol. Ther. 2012, 20, 2134–2142. [Google Scholar] [CrossRef]
- Ibrahim, G.A.; Zweber, B.A.; Awad, E.A. Muscle and serum enzymes and isoenzymes in muscular dystrophies. Arch. Phys. Med. Rehabilitation 1981, 62, 265–269. [Google Scholar]
- Pegoraro, V.; Angelini, C. Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies. Genes 2021, 12, 85. [Google Scholar] [CrossRef]
- Koutsoulidou, A.; Kyriakides, T.C.; Papadimas, G.K.; Christou, Y.; Kararizou, E.; Papanicolaou, E.Z.; Phylactou, L.A. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PLoS ONE 2015, 10, e0125341. [Google Scholar] [CrossRef] [PubMed]
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, V.; Cudia, P.; Baba, A.; Angelini, C. MyomiRNAs and myostatin as physical rehabilitation biomarkers for myotonic dystrophy. Neurol. Sci. 2020, 41, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Tymms, K.; Webb, J. Dermatopolymyositis and other connective tissue diseases: A review of 105 cases. J. Rheumatol. 1985, 12, 1140–1148. [Google Scholar] [PubMed]
- Pluk, H.; Van Hoeve, B.J.A.; Van Dooren, S.H.J.; Stammen-Vogelzangs, J.; Van Der Heijden, A.; Schelhaas, H.J.; Verbeek, M.; Badrising, U.A.; Arnardottir, S.; Gheorghe, K.; et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann. Neurol. 2012, 73, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Milone, M. Diagnosis and Management of Immune-Mediated Myopathies. Mayo Clin. Proc. 2017, 92, 826–837. [Google Scholar] [CrossRef]
- Palterer, B.; Vitiello, G.; Carraresi, A.; Giudizi, M.G.; Cammelli, D.; Parronchi, P. Bench to bedside review of myositis autoantibodies. Clin. Mol. Allergy 2018, 16, 1–17. [Google Scholar] [CrossRef]
- Benveniste, O.; Goebel, H.-H.; Stenzel, W. Biomarkers in Inflammatory Myopathies—An Expanded Definition. Front. Neurol. 2019, 10, 554. [Google Scholar] [CrossRef]
- Lucchini, M.; Maggi, L.; Pegoraro, E.; Filosto, M.; Rodolico, C.; Antonini, G.; Garibaldi, M.; Valentino, M.; Siciliano, G.; Tasca, G.; et al. Anti-cN1A Antibodies Are Associated with More Severe Dysphagia in Sporadic Inclusion Body Myositis. Cells 2021, 10, 1146. [Google Scholar] [CrossRef]
- Hochberg, M.C.; Feldman, D.; Stevens, M.B. Adult onset polymyositis/dermatomyositis: An analysis of clinical and laboratory features and survival in 76 patients with a review of the literature. Semin. Arthritis Rheum. 1986, 15, 168–178. [Google Scholar] [CrossRef]
- Kojima, Y.; Uzawa, A.; Ozawa, Y.; Yasuda, M.; Onishi, Y.; Akamine, H.; Kawaguchi, N.; Himuro, K.; Noto, Y.I.; Mizuno, T.; et al. Rate of change in acetylcholine receptor antibody levels predicts myasthenia gravis outcome. J. Neurol. Neurosurg. Psychiatry 2021, 92, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, H.; Limburg, P.C.; Hummel-Tappel, E.; The, T.H. Anti-acetylcholine receptor antibodies in myasthenia gravis: Part 2. Clinical and serological follow-up of individual patients. J. Neurol. Sci. 1983, 58, 371–385. [Google Scholar] [CrossRef]
- McConville, J.; Farrugia, M.E.; Beeson, D.; Kishore, U.; Metcalfe, R.; Newsom-Davis, J.; Vincent, A. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann. Neurol. 2004, 55, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.B.; El-Salem, K.; Massey, J.M.; McConville, J.; Vincent, A. Clinical aspects of MuSK antibody positive seronegative MG. Neurology 2003, 60, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Evoli, A.; Tonali, P.A.; Padua, L.; Monaco, M.L.; Scuderi, F.; Batocchi, A.P.; Marino, M.; Bartoccioni, E. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain 2003, 126, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Bartoccioni, E.; Scuderi, F.; Minicuci, G.M.; Marino, M.; Ciaraffa, F.; Evoli, A. Anti-MuSK antibodies: Correlation with myasthenia gravis severity. Neurology 2006, 67, 505–507. [Google Scholar] [CrossRef]
- Tandan, R.; Hehir, M.K.; Waheed, W.; Howard, D.B. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve 2017, 56, 185–196. [Google Scholar] [CrossRef]
- Zisimopoulou, P.; Evangelakou, P.; Tzartos, J.; Lazaridis, K.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; Deymeer, F.; et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J. Autoimmun. 2014, 52, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Romi, F.; Suzuki, S.; Suzuki, N.; Petzold, A.; Plant, G.T.; Gilhus, N.E. Anti-voltage-gated potassium channel Kv1.4 antibodies in myasthenia gravis. J. Neurol. 2012, 259, 1312–1316. [Google Scholar] [CrossRef]
- Buckley, C.; Newsom-Davis, J.; Willcox, N.; Vincent, A. Do titin and cytokine antibodies in MG patients predict thymoma or thymoma recurrence? Neurology 2001, 57, 1579–1582. [Google Scholar] [CrossRef]
- Suzuki, S.; Baba, A.; Kaida, K.; Utsugisawa, K.; Kita, Y.; Tsugawa, J.; Ogawa, G.; Nagane, Y.; Kuwana, M.; Suzuki, N. Cardiac involvements in myasthenia gravis associated with anti-Kv1.4 antibodies. Eur. J. Neurol. 2014, 21, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Punga, T.; Le Panse, R.; Andersson, M.; Truffault, F.; Berrih-Aknin, S.; Punga, A. Circulating mi RNA s in myasthenia gravis: miR-150-5p as a new potential biomarker. Ann. Clin. Transl. Neurol. 2013, 1, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Sabre, L.; Maddison, P.; Wong, S.H.; Sadalage, G.; Ambrose, P.A.; Plant, G.T.; Punga, A.R. miR-30e-5p as predictor of generalization in ocular myasthenia gravis. Ann. Clin. Transl. Neurol. 2019, 6, 243–251. [Google Scholar] [CrossRef]
- Punga, A.R.; Andersson, M.; Alimohammadi, M.; Punga, T. Disease specific signature of circulating miR-150-5p and miR-21-5p in myasthenia gravis patients. J. Neurol. Sci. 2015, 356, 90–96. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Griesmann, G.E.; O’Suilleabhain, P.E.; Windebank, A.J.; Woppmann, A.; Miljanich, G.P.; Lambert, E.H. Calcium-Channel Antibodies in the Lambert–Eaton Syndrome and Other Paraneoplastic Syndromes. New Engl. J. Med. 1995, 332, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Motomura, M.; Lang, B.; Johnston, I.; Palace, J.; Vincent, A.; Newsom-Davis, J. Incidence of serum anti-P/Q-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J. Neurol. Sci. 1997, 147, 35–42. [Google Scholar] [CrossRef]
- Sabater, L.; Titulaer, M.; Saiz, A.; Verschuuren, J.; Gure, A.O.; Graus, F. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology 2008, 70, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Maddison, P.; Gozzard, P.; Sadalage, G.; Ambrose, P.A.; Chapman, C.J.; Murray, A.; Thomsen, S.; Berretta, A.; Lang, B. Neuronal antibody detection and improved lung cancer prediction in Lambert-Eaton myasthenic syndrome. J. Neuroimmunol. 2020, 340, 577149. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; Klooster, R.; Potman, M.; Sabater, L.; Graus, F.; Hegeman, I.M.; Thijssen, P.E.; Wirtz, P.W.; Twijnstra, A.; Smitt, P.A.S.; et al. SOX Antibodies in Small-Cell Lung Cancer and Lambert-Eaton Myasthenic Syndrome: Frequency and Relation With Survival. J. Clin. Oncol. 2009, 27, 4260–4267. [Google Scholar] [CrossRef]
- Sandelius, Å.; Zetterberg, H.; Blennow, K.; Adiutori, R.; Malaspina, A.; Laura, M.; Reilly, M.M.; Rossor, A.M. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology 2018, 90, e518–e524. [Google Scholar] [CrossRef]
- Wang, H.; Davison, M.; Wang, K.; Xia, T.; Kramer, M.; Call, K.; Luo, J.; Wu, X.; Zuccarino, R.; Bacon, C.; et al. Transmembrane protease serine 5: A novel Schwann cell plasma marker for CMT1A. Ann. Clin. Transl. Neurol. 2020, 7, 69–82. [Google Scholar] [CrossRef]
- Matsunami, N.; Smith, B.; Ballard, L.; Lensch, M.W.; Robertson, M.; Albertsen, H.; Hanemann, C.O.; Müller, H.W.; Bird, T.D.; White, R.; et al. Peripheral myelin protein–22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot–Marie–Tooth 1A. Nat. Genet. 1992, 1, 176–179. [Google Scholar] [CrossRef]
- Pantera, H.; Shy, M.E.; Svaren, J. Regulating PMP22 expression as a dosage sensitive neuropathy gene. Brain Res. 2020, 1726, 146491. [Google Scholar] [CrossRef]
- Svaren, J.; Moran, J.J.; Wu, X.; Zuccarino, R.; Bacon, C.; Bai, Y.; Ramesh, R.; Gutmann, L.; Do, D.M.A.; Pavelec, D.; et al. Schwann cell transcript biomarkers for hereditary neuropathy skin biopsies. Ann. Neurol. 2019, 85, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Davison, M.; Wang, K.; Xia, T.-H.; Call, K.M.; Luo, J.; Wu, X.; Zuccarino, R.; Bacha, A.; Bai, Y.; et al. MicroRNAs as Biomarkers of Charcot-Marie-Tooth Disease Type 1A. Neurology 2021, 97, e489–e500. [Google Scholar] [CrossRef]
- Yuki, N.; Hartung, H.-P. Guillain–Barré Syndrome. New Engl. J. Med. 2012, 366, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Uncini, A.; Yuki, N.; Misawa, S.; Notturno, F.; Nasu, S.; Kanai, K.; Noto, Y.-I.; Fujimaki, Y.; Shibuya, K.; et al. Antiganglioside antibodies are associated with axonal Guillain–Barré syndrome: A Japanese–Italian collaborative study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 23–28. [Google Scholar] [CrossRef]
- Cats, E.A.; Jacobs, B.C.; Yuki, N.; Tio-Gillen, A.P.; Piepers, S.; Franssen, H.; Van Asseldonk, J.-T.; Berg, L.H.V.D.; Van Der Pol, W.-L. Multifocal motor neuropathy: Association of anti-GM1 IgM antibodies with clinical features. Neurology 2010, 75, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Koga, M.; Odaka, M.; Hirata, K.; Yuki, N. Continuous Spectrum of Pharyngeal-Cervical-Brachial Variant of Guillain-Barré Syndrome. Arch. Neurol. 2007, 64, 1519–1523. [Google Scholar] [CrossRef]
- Nobile-Orazio, E.; Manfredini, E.; Carpo, M.; Meucci, N.; Monaco, S.; Ferrari, S.; Bonetti, B.; Cavaletti, G.; Gemignani, F.; Durelli, L.; et al. Frequency and clinical correlates of anti-neural IgM antibodies in neuropathy associated with IgM monoclonal gammopathy. Ann. Neurol. 1994, 36, 416–424. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef]
- Breville, G.; Lascano, A.M.; Roux-Lombard, P.; Vuilleumier, N.; Lalive, P.H. Interleukin 8, a Biomarker to Differentiate Guillain-Barré Syndrome From CIDP. Neurol.-Neuroimmunol. Neuroinflammation 2021, 8, e1031. [Google Scholar] [CrossRef]
- Martín-Aguilar, L.; Camps-Renom, P.; Lleixà, C.; Pascual-Goñi, E.; Díaz-Manera, J.; Rojas-García, R.; De Luna, N.; Gallardo, E.; Cortés-Vicente, E.; Muñoz, L.; et al. Serum neurofilament light chain predicts long-term prognosis in Guillain-Barré syndrome patients. J. Neurol. Neurosurg. Psychiatry 2021, 92, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Tiziano, F.D.; Pinto, A.M.; Fiori, S.; Lomastro, R.; Messina, S.; Bruno, C.; Pini, A.; Pane, M.; D’Amico, A.; Ghezzo, A.; et al. SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur. J. Hum. Genet. 2010, 18, 52–58. [Google Scholar] [CrossRef]
- Brichta, L.; Hölker, I.; Haug, K.; Klockgether, T.; Wirth, B. In vivo activation ofSMNin spinal muscular atrophy carriers and patients treated with valproate. Ann. Neurol. 2006, 59, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Kolb, S.J.; Harmison, G.G.; Jeffries, N.O.; Schadt, K.; Finkel, R.S.; Dreyfuss, G.; Fischbeck, K.H. SMN mRNA and protein levels in peripheral blood. Neurology 2006, 66, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef]
- Tiziano, F.D.; Lomastro, R.; Abiusi, E.; Pasanisi, M.B.; Di Pietro, L.; Fiori, S.; Baranello, G.; Angelini, C.; Sorarù, G.; Gaiani, A.; et al. Longitudinal evaluation of SMN levels as biomarker for spinal muscular atrophy: Results of a phase IIb double-blind study of salbutamol. J. Med Genet. 2019, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.-K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef]
- Alves, C.R.; Zhang, R.; Johnstone, A.J.; Garner, R.; Nwe, P.H.; Siranosian, J.J.; Swoboda, K.J. Serum creatinine is a biomarker of progressive denervation in spinal muscular atrophy. Neurology 2020, 94, e921–e931. [Google Scholar] [CrossRef] [PubMed]
- Catapano, F.; Zaharieva, I.; Scoto, M.; Marrosu, E.; Morgan, J.; Muntoni, F.; Zhou, H. Altered Levels of MicroRNA-9, -206, and -132 in Spinal Muscular Atrophy and Their Response to Antisense Oligonucleotide Therapy. Mol. Ther.-Nucleic Acids 2016, 5, e331. [Google Scholar] [CrossRef]
- Bonanno, S.; Marcuzzo, S.; Malacarne, C.; Giagnorio, E.; Masson, R.; Zanin, R.; Arnoldi, M.T.; Andreetta, F.; Simoncini, O.; Venerando, A.; et al. Circulating MyomiRs as Potential Biomarkers to Monitor Response to Nusinersen in Pediatric SMA Patients. Biomedicines 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Eichelberger, E.J.; Alves, C.R.R.; Zhang, R.; Petrillo, M.; Cullen, P.; Farwell, W.; Hurt, J.A.; Staropoli, J.F.; Swoboda, K.J. Increased systemic HSP70B levels in spinal muscular atrophy infants. Ann. Clin. Transl. Neurol. 2021, 8, 1495–1501. [Google Scholar] [CrossRef]
- Introna, A.; Milella, G.; D’Errico, E.; Fraddosio, A.; Scaglione, G.; Ucci, M.; Bsc, M.R.; Simone, I.L. Is cerebrospinal fluid amyloid-β42 a promising biomarker of response to nusinersen in adult spinal muscular atrophy patients? Muscle Nerve 2021, 63, 905–909. [Google Scholar] [CrossRef]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef]
- Xu, Z.; Henderson, R.D.; David, M.; McCombe, P.A. Neurofilaments as Biomarkers for Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0164625. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Lombardi, V.; Jeromin, A.; Bowser, R.; Andersen, P.M.; Malaspina, A. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Huss, A.; Mayer, B.; Grehl, T.; Grosskreutz, J.; Borck, G.; Kuhle, J.; Lulé, D.; Meyer, T.; Oeckl, P.; et al. Diagnostic and prognostic significance of neurofilament light chain NF-L, but not progranulin and S100B, in the course of amyotrophic lateral sclerosis: Data from the German MND-net. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Oeckl, P.; Huss, A.; Müller, K.; Volk, A.E.; Kuhle, J.; Knehr, A.; Andersen, P.M.; Prudlo, J.; Steinacker, P.; et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann. Neurol. 2016, 79, 152–158. [Google Scholar] [CrossRef]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; Von Arnim, C.A.F.; Böhm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 12–20. [Google Scholar] [CrossRef]
- Rossi, D.; Volanti, P.; Brambilla, L.; Colletti, T.; Spataro, R.; La Bella, V. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J. Neurol. 2018, 265, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, R.; Copetti, M.; Panza, F.; Cortese, R.; Capozzo, R.; D’Errico, E.; Fontana, A.; Simone, I.L.; Logroscino, G. Time to generalisation as a predictor of prognosis in amyotrophic lateral sclerosis: Table 1. J. Neurol. Neurosurg. Psychiatry 2016, 87, 678–679. [Google Scholar] [CrossRef] [PubMed]
- Gille, B.; De Schaepdryver, M.; Goossens, J.; Dedeene, L.; De Vocht, J.; Oldoni, E.; Goris, A.; Bosch, L.V.D.; Depreitere, B.; Claeys, K.G.; et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2019, 45, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Thouvenot, E.; Demattei, C.; Lehmann, S.; Maceski-Maleska, A.; Hirtz, C.; Juntas-Morales, R.; Pageot, N.; Esselin, F.; Alphandéry, S.; Vincent, T.; et al. Serum neurofilament light chain at time of diagnosis is an independent prognostic factor of survival in amyotrophic lateral sclerosis. Eur. J. Neurol. 2020, 27, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Falzone, Y.M.; Domi, T.; Agosta, F.; Pozzi, L.; Schito, P.; Fazio, R.; Del Carro, U.; Barbieri, A.; Comola, M.; Leocani, L.; et al. Serum phosphorylated neurofilament heavy-chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J. Neurol. 2020, 267, 2272–2280. [Google Scholar] [CrossRef] [PubMed]
- Lehmer, C.; Oeckl, P.; Weishaupt, J.H.; Volk, A.E.; Diehl-Schmid, J.; Schroeter, M.L.; Lauer, M.; Kornhuber, J.; Levin, J.; Fassbender, K.; et al. Poly- GP in cerebrospinal fluid links C9orf72 -associated dipeptide repeat expression to the asymptomatic phase of ALS / FTD. EMBO Mol. Med. 2017, 9, 859–868. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bs, L.M.D.; Ms, M.G.H.; Bs, N.N.D.; ScM, J.W.; Miller, T.M.; Pastor, P.; Trojanowski, J.Q.; Grossman, M.; Berry, M.J.D.; et al. Phosphorylated neurofilament heavy chain: A biomarker of survival for C9ORF 72 -associated amyotrophic lateral sclerosis. Ann. Neurol. 2017, 82, 139–146. [Google Scholar] [CrossRef]
- Bourbouli, M.; Rentzos, M.; Bougea, A.; Zouvelou, V.; Constantinides, V.C.; Zaganas, I.; Evdokimidis, I.; Kapaki, E.; Paraskevas, G.P. Cerebrospinal Fluid TAR DNA-Binding Protein 43 Combined with Tau Proteins as a Candidate Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Spectrum Disorders. Dement. Geriatr. Cogn. Disord. 2017, 44, 144–152. [Google Scholar] [CrossRef]
- Ong, M.-L.; Tan, P.F.; Holbrook, J.D. Predicting functional decline and survival in amyotrophic lateral sclerosis. PLoS ONE 2017, 12, e0174925. [Google Scholar] [CrossRef]
- Rafiq, M.K.; Lee, E.; Bradburn, M.; McDermott, C.J.; Shaw, P.J. Creatine kinase enzyme level correlates positively with serum creatinine and lean body mass, and is a prognostic factor for survival in amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 1071–1078. [Google Scholar] [CrossRef]
- Lanznaster, D.; Hergesheimer, R.C.; Bakkouche, S.E.; Beltran, S.; Vourc’H, P.; Andres, C.R.; Dufour-Rainfray, D.; Corcia, P.; Blasco, H. Aβ1-42 and Tau as Potential Biomarkers for Diagnosis and Prognosis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2911. [Google Scholar] [CrossRef] [PubMed]
- Lange, D.J.; Shahbazi, M.; Silani, V.; Ludolph, A.C.; Weishaupt, J.H.; Ajroud-Driss, S.; Fields, K.G.; Remanan, R.; Appel, S.H.; Morelli, C.; et al. Pyrimethamine significantly lowers cerebrospinal fluid Cu/Zn superoxide dismutase in amyotrophic lateral sclerosis patients with SOD1 mutations. Ann. Neurol. 2017, 81, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Lange, D.J.; Andersen, P.M.; Remanan, R.; Marklund, S.; Benjamin, D. Pyrimethamine decreases levels of SOD1 in leukocytes and cerebrospinal fluid of ALS patients: A phase I pilot study. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 199–204. [Google Scholar] [CrossRef]
- Ravnik-Glavač, M.; Glavač, D. Circulating RNAs as Potential Biomarkers in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 1714. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimer’s Res. Ther. 2017, 9, 1–13. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Martone, J.; Lepore, E.; Casola, I.; Petrucci, A.; Inghilleri, M.; Morlando, M.; Colantoni, A.; Scicchitano, B.M.; Calvo, A.; et al. A longitudinal study defined circulating microRNAs as reliable biomarkers for disease prognosis and progression in ALS human patients. Cell Death Discov. 2021, 7, 1–11. [Google Scholar] [CrossRef]
- McLeish, M.J.; Kenyon, G.L. Relating Structure to Mechanism in Creatine Kinase. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 1–20. [Google Scholar] [CrossRef]
- Lang, H.; Würzburg, U. Creatine kinase, an enzyme of many forms. Clin. Chem. 1982, 28, 1439–1447. [Google Scholar] [CrossRef]
- Chakrabarty, T.; Tirupathi, S.; Thompson, A. How to use: Creatine kinase. Arch. Dis. Child. Educ. Pr. Ed. 2020, 105, 157–163. [Google Scholar] [CrossRef]
- Jackson, M.J.; Round, J.M.; Newham, D.J.; Edwards, R.H.T. An examination of some factors influencing creatine kinase in the blood of patients with muscular dystrophy. Muscle Nerve 1987, 10, 15–21. [Google Scholar] [CrossRef]
- Hou, L.; Jiao, B.; Xiao, T.; Zhou, L.; Zhou, Z.; Du, J.; Yan, X.; Wang, J.; Tang, B.; Shen, L. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci. Rep. 2016, 6, 32478. [Google Scholar] [CrossRef]
- Tai, H.; Cui, L.; Guan, Y.; Liu, M.; Li, X.; Shen, D.; Li, D.; Cui, B.; Fang, J.; Ding, Q.; et al. Correlation of Creatine Kinase Levels with Clinical Features and Survival in Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 322. [Google Scholar] [CrossRef]
- Sinaki, M.; Mulder, D.W. Amyotrophic lateral sclerosis: Relationship between serum creatine kinase level and patient survival. Arch. Phys. Med. Rehabil. 1986, 67, 169–171. [Google Scholar] [CrossRef]
- Felice, K.J.; North, W.A. Creatine kinase values in amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 160, S30–S32. [Google Scholar] [CrossRef]
- Sandilands, E.A.; Dhaun, N.; Dear, J.W.; Webb, D.J. Measurement of renal function in patients with chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 504–515. [Google Scholar] [CrossRef]
- Szunyogova, E.; Zhou, H.; Maxwell, G.K.; Powis, R.A.; Muntoni, F.; Gillingwater, T.H.; Parson, S.H. Survival Motor Neuron (SMN) protein is required for normal mouse liver development. Sci. Rep. 2016, 6, 34635. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.C.; Siranosian, J.J.; Rosales, I.; Deguise, M.-O.; Sharma, A.; Muhtaseb, A.W.; Nwe, P.; Johnstone, A.J.; Zhang, R.; Fatouraei, M.; et al. Impaired kidney structure and function in spinal muscular atrophy. Neurol. Genet. 2019, 5, e353. [Google Scholar] [CrossRef] [PubMed]
- Van Eijk, R.P.A.; Eijkemans, M.J.C.; Ferguson, T.A.; Nikolakopoulos, S.; Veldink, J.H.; Berg, L.H.V.D. Monitoring disease progression with plasma creatinine in amyotrophic lateral sclerosis clinical trials. J. Neurol. Neurosurg. Psychiatry 2018, 89, 156–161. [Google Scholar] [CrossRef]
- Targoff, I.N. Idiopathic inflammatory myopathy: Autoantibody update. Curr. Rheumatol. Rep. 2002, 4, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A. Cytoplasmic 5′-nucleotidase autoantibodies in inclusion body myositis: Isotypes and diagnostic utility. Muscle Nerve 2014, 50, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Limaye, V.S.; Lester, S.; Blumbergs, P.; Greenberg, S.A. Anti- C N1A antibodies in South Australian patients with inclusion body myositis. Muscle Nerve 2016, 53, 654–655. [Google Scholar] [CrossRef]
- Herbert, M.K.; Stammen-Vogelzangs, J.; Verbeek, M.M.; Rietveld, A.; Lundberg, I.E.; Chinoy, H.; Lamb, J.A.; Cooper, R.G.; Roberts, M.; Badrising, U.; et al. Disease specificity of autoantibodies to cytosolic 5′-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann. Rheum. Dis. 2016, 75, 696–701. [Google Scholar] [CrossRef]
- Lloyd, T.E.; Christopher-Stine, L.; Pinal-Fernandez, I.; Tiniakou, E.; Petri, M.; Baer, A.; Danoff, S.K.; Pak, K.; Casciola-Rosen, L.A.; Mammen, A.L. Cytosolic 5′-Nucleotidase 1A As a Target of Circulating Autoantibodies in Autoimmune Diseases. Arthritis Rheum. 2016, 68, 66–71. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Skeie, G.O.; Romi, N.E.G.G.O.S.F.; Lazaridis, K.; Zisimopoulou, P.; Tzartos, K.L.P.Z.S. Myasthenia gravis—Autoantibody characteristics and their implications for therapy. Nat. Rev. Neurol. 2016, 12, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul. Disord. 2006, 16, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J.G.M. Myasthenia gravis. Nat. Rev. Dis. Prim. 2019, 5, 30. [Google Scholar] [CrossRef]
- Leite, M.I.; Jacob, S.; Viegas, S.; Cossins, J.; Clover, L.; Morgan, B.P.; Beeson, D.; Willcox, N.; Vincent, A. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis†. Brain 2008, 131, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Adams, R.N.; Josifek, L.F.; Self, S.G. Functional Activities of Autoantibodies to Acetylcholine Receptors and the Clinical Severity of Myasthenia Gravis. New Engl. J. Med. 1982, 307, 769–775. [Google Scholar] [CrossRef]
- Romi, F.; Skeie, G.O.; Aarli, J.A.; Gilhus, N.E. The severity of myasthenia gravis correlates with the serum concentration of titin and ryanodine receptor antibodies. Arch. Neurol. 2000, 57, 1596–1600. [Google Scholar] [CrossRef]
- Kaminski, H.J.; Kusner, L.L.; Wolfe, G.I.; Aban, I.; Minisman, G.; Conwit, R.; Cutter, G. Biomarker development for myasthenia gravis. Ann. New York Acad. Sci. 2012, 1275, 101–106. [Google Scholar] [CrossRef]
- Zisimopoulou, P.; Brenner, T.; Trakas, N.; Tzartos, S.J. Serological diagnostics in myasthenia gravis based on novel assays and recently identified antigens. Autoimmun. Rev. 2013, 12, 924–930. [Google Scholar] [CrossRef]
- Koneczny, I.; Cossins, J.; Waters, P.; Beeson, D.; Vincent, A. MuSK Myasthenia Gravis IgG4 Disrupts the Interaction of LRP4 with MuSK but Both IgG4 and IgG1-3 Can Disperse Preformed Agrin-Independent AChR Clusters. PLoS ONE 2013, 8, e80695. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Zisimopoulou, P.; Rentzos, M.; Karandreas, N.; Zouvelou, V.; Evangelakou, P.; Tsonis, A.; Thomaidis, T.; Lauria, G.; Andreetta, F.; et al. LRP 4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2014, 1, 80–87. [Google Scholar] [CrossRef]
- Rivner, M.H.; Liu, S.; Quarles, B.; Fleenor, B.; Shen, C.; Pan, J.; Mei, L. Agrin and low-density lipoprotein-related receptor protein 4 antibodies in amyotrophic lateral sclerosis patients. Muscle Nerve 2017, 55, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, C.; Melms, A.; Schoser, B.; Zhang, Y.; Meltoranta, J.; Risson, V.; Schaeffer, L.; Schalke, B.; Kroger, S. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014, 82, 1976–1983. [Google Scholar] [CrossRef]
- Zhang, B.; Shen, C.; Bealmear, B.; Ragheb, S.; Xiong, W.-C.; Lewis, R.A.; Lisak, R.P.; Mei, L. Autoantibodies to Agrin in Myasthenia Gravis Patients. PLoS ONE 2014, 9, e91816. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, E.; Martinez-Hernandez, E.; Titulaer, M.; Huijbers, M.G.; Martínez, M.A.; Ramos-Fransi, A.; Querol, L.; Diaz-Manera, J.; Rojas-García, R.; Hayworth, C.R.; et al. Cortactin autoantibodies in myasthenia gravis. Autoimmun. Rev. 2014, 13, 1003–1007. [Google Scholar] [CrossRef]
- Labrador-Horrillo, M.; Martínez, M.Á.; Selva-O’Callaghan, A.; Trallero-Araguás, E.; Grau-Junyent, J.M.; Vilardell-Tarrés, M.; Juárez, C. Identification of a novel myositis-associated antibody directed against cortactin. Autoimmun. Rev. 2014, 13, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Katarzyna, M.Z.; Belaya, K.; Leite, M.; Patrick, W.; Vincent, A.; Beeson, D. Collagen Q – A potential target for autoantibodies in myasthenia gravis. J. Neurol. Sci. 2015, 348, 241–244. [Google Scholar] [CrossRef]
- Graus, F.; Lang, B.; Pozo-Rosich, P.; Saiz, A.; Casamitjana, R.; Vincent, A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002, 59, 764–766. [Google Scholar] [CrossRef]
- Zoccarato, M.; Gastaldi, M.; Zuliani, L.; Biagioli, T.; Brogi, M.; Bernardi, G.; Corsini, E.; Bazzigaluppi, E.; Fazio, R.; Giannotta, C.; et al. Diagnostics of paraneoplastic neurological syndromes. Neurol. Sci. 2017, 38, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, L.; Zoccarato, M.; Gastaldi, M.; Iorio, R.; Evoli, A.; Biagioli, T.; Casagrande, S.; Bazzigaluppi, E.; Fazio, R.; Giannotta, C.; et al. Diagnostics of autoimmune encephalitis associated with antibodies against neuronal surface antigens. Neurol. Sci. 2017, 38, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Chiba, A.; Kusunoki, S.; Obata, H.; Machinami, R.; Kanazawa, I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barre syndrome: Clinical and immunohistochemical studies. Neurology 1993, 43, 1911. [Google Scholar] [CrossRef] [PubMed]
- Franciotta, D.; Gastaldi, M.; Benedetti, L.; Pesce, G.; Biagioli, T.; Lolli, F.; Costa, G.; Melis, C.; Andreetta, F.; Simoncini, O.; et al. Diagnostics of dysimmune peripheral neuropathies. Neurol. Sci. 2017, 38, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Hirano, M.; Kuwahara, M.; Samukawa, M.; Takada, K.; Morise, J.; Yabuno, K.; Oka, S.; Kusunoki, S. Binding specificity of anti-HNK-1 IgM M-protein in anti-MAG neuropathy: Possible clinical relevance. Neurosci. Res. 2015, 91, 63–68. [Google Scholar] [CrossRef]
- Planche, V.; Arnaud, L.; Viala, K.; Hospital, M.A.; Miyara, M.; Leger, J.M.; Neil, J.; Leblond, V.; Musset, L. Heavy/Light Chain Ratio as a Biomarker for Monitoring Patients with IgM Monoclonal Gammopathy and Anti-MAG Neuropathy. J. Hematol. Thromboembolic Dis. 2014, 2, 142. [Google Scholar] [CrossRef]
- Liem, R.K.; Yen, S.H.; Salomon, G.D.; Shelanski, M.L. Intermediate filaments in nervous tissues. J. Cell Biol. 1978, 79, 637–645. [Google Scholar] [CrossRef]
- Hoffman, P.N.; Cleveland, D.W.; Griffin, J.W.; Landes, P.W.; Cowan, N.J.; Price, D.L. Neurofilament gene expression: A major determinant of axonal caliber. Proc. Natl. Acad. Sci. 1987, 84, 3472–3476. [Google Scholar] [CrossRef]
- Nixon, R.A.; Shea, T.B. Views and reviews dynamics of neuronal intermediate filaments: A developmental perspective. Cell Motil Cytoskeleton 1992, 22, 81–91. [Google Scholar] [CrossRef]
- Dale, J.M.; Garcia, M.L. Neurofilament Phosphorylation during Development and Disease: Which Came First, the Phosphorylation or the Accumulation? J. Amino Acids 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the Multiple Aspects of Neurofilament Functions, and their Possible Contribution to Neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, B.H.; Yip, W.; Chou, P.; Yip, B.-S. Neurofilament Proteins as Prognostic Biomarkers in Neurological Disorders. Curr. Pharm. Des. 2019, 25, 4560–4569. [Google Scholar] [CrossRef] [PubMed]
- Poesen, K.; Van Damme, P. Diagnostic and Prognostic Performance of Neurofilaments in ALS. Front. Neurol. 2019, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef]
- Wurster, C.D.; Steinacker, P.; Günther, R.; Koch, J.C.; Lingor, P.; Uzelac, Z.; Witzel, S.; Wollinsky, K.; Winter, B.; Osmanovic, A.; et al. Neurofilament light chain in serum of adolescent and adult SMA patients under treatment with nusinersen. J. Neurol. 2020, 267, 36–44. [Google Scholar] [CrossRef]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament Heavy Chain and Tau Protein Are Not Elevated in Cerebrospinal Fluid of Adult Patients with Spinal Muscular Atrophy during Loading with Nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef]
- Spicer, C.; Lu, C.-H.; Catapano, F.; Scoto, M.; Zaharieva, I.; Malaspina, A.; Morgan, J.E.; Greensmith, L.; Muntoni, F.; Zhou, H. The altered expression of neurofilament in mouse models and patients with spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2021, 8, 866–876. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Karlsson, J.-E.; Karlsson, J.-O.; Persson, L.I.; Wikkelsø, C. Patients with Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases Have Increased Levels of Neurofilament Protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Vacchiano, V.; Zenesini, C.; Polischi, B.; de Pasqua, S.; Fileccia, E.; Mammana, A.; di Stasi, V.; Capellari, S.; Salvi, F.; et al. Diagnostic-prognostic value and electrophysiological correlates of CSF biomarkers of neurodegeneration and neuroinflammation in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, D.; Faravelli, I.; Meneri, M.; Saccomanno, D.; Govoni, A.; Magri, F.; Ricci, G.; Siciliano, G.; Comi, G.P.; Corti, S. Diagnostic and prognostic value of CSF neurofilaments in a cohort of patients with motor neuron disease: A cross-sectional study. J. Cell. Mol. Med. 2021, 25, 3765–3771. [Google Scholar] [CrossRef]
- Menke, R.A.L.; Gray, E.; Lu, C.-H.; Kuhle, J.; Talbot, K.; Malaspina, A.; Turner, M.R. CSF neurofilament light chain reflects corticospinal tract degeneration in ALS. Ann. Clin. Transl. Neurol. 2015, 2, 748–755. [Google Scholar] [CrossRef]
- Gaiani, A.; Martinelli, I.; Bello, L.; Querin, G.; Puthenparampil, M.; Ruggero, S.; Toffanin, E.; Cagnin, A.; Briani, C.; Pegoraro, E.; et al. Diagnostic and Prognostic Biomarkers in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2017, 74, 525–532. [Google Scholar] [CrossRef]
- Dreger, M.; Steinbach, R.; Gaur, N.; Metzner, K.; Stubendorff, B.; Witte, O.W.; Grosskreutz, J. Cerebrospinal Fluid Neurofilament Light Chain (NfL) Predicts Disease Aggressiveness in Amyotrophic Lateral Sclerosis: An Application of the D50 Disease Progression Model. Front. Neurosci. 2021, 15, 264. [Google Scholar] [CrossRef]
- Huang, W. MicroRNAs: Biomarkers, Diagnostics, and Therapeutics. Methods Mol. Biol. 2017, 1617, 57–67. [Google Scholar] [CrossRef]
- Magri, F.; Vanoli, F.; Corti, S. miRNA in spinal muscular atrophy pathogenesis and therapy. J. Cell. Mol. Med. 2017, 22, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Viswambharan, V.; Thanseem, I.; Vasu, M.M.; Poovathinal, S.A.; Anitha, A. miRNAs as biomarkers of neurodegenerative disorders. Biomarkers Med. 2017, 11, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. 2007, 104, 17016–17021. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-H.; Chen, J.-A. Multifaceted roles of microRNAs: From motor neuron generation in embryos to degeneration in spinal muscular atrophy. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Renshaw, D.; Getting, S.; MacKenzie, R. The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol. 2012, 205, 324–340. [Google Scholar] [CrossRef] [PubMed]
- Amirouche, A.; Jahnke, V.E.; Lunde, J.A.; Koulmann, N.; Freyssenet, D.G.; Jasmin, B.J. Muscle-specific microRNA-206 targets multiple components in dystrophic skeletal muscle representing beneficial adaptations. Am. J. Physiol. Physiol. 2017, 312, C209–C221. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Williams, A.H.; Maxeiner, J.M.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Investig. 2012, 122, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Esser, K.A.; Andrade, F.H. MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am. J. Physiol. Physiol. 2007, 293, C451–C457. [Google Scholar] [CrossRef]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a Key Modulator of Skeletal Muscle Development and Disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as Biomarkers in Amyotrophic Lateral Sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Vignier, N.; Amor, F.; Fogel, P.; Duvallet, A.; Poupiot, J.; Charrier, S.; Arock, M.; Montus, M.; Nelson, I.; Richard, I.; et al. Distinctive Serum miRNA Profile in Mouse Models of Striated Muscular Pathologies. PLoS ONE 2013, 8, e55281. [Google Scholar] [CrossRef]
- Hu, J.; Kong, M.; Ye, Y.; Hong, S.; Cheng, L.; Jiang, L. Serum miR-206 and other muscle-specific microRNAs as non-invasive biomarkers for Duchenne muscular dystrophy. J. Neurochem. 2014, 129, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, Y.; Kishi, S.; Aoki, Y.; Komaki, H.; Oya, Y.; Takeda, S.-I.; Hashido, K. Three novel serum biomarkers, miR-1, miR-133a, and miR-206 for Limb-girdle muscular dystrophy, Facioscapulohumeral muscular dystrophy, and Becker muscular dystrophy. Environ. Heal. Prev. Med. 2014, 19, 452–458. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zhao, L.; Zhang, D.; Yao, X.; Zhang, H.; Wang, Y.-C.; Wang, X.-Y.; Xia, H.; Yan, J.; et al. Circulating Muscle-specific miRNAs in Duchenne Muscular Dystrophy Patients. Mol. Ther. - Nucleic Acids 2014, 3, e177. [Google Scholar] [CrossRef]
- Molin, C.J.; Sabre, L.; Weis, C.-A.; Punga, T.; Punga, A.R. Thymectomy lowers the myasthenia gravis biomarker miR-150-5p. Neurol.-Neuroimmunol. Neuroinflammation 2018, 5, e450. [Google Scholar] [CrossRef]
- Punga, T.; Bartoccioni, E.; Lewandowska, M.; Damato, V.; Evoli, A.; Punga, A.R. Disease specific enrichment of circulating let-7 family microRNA in MuSK+ myasthenia gravis. J. Neuroimmunol. 2016, 292, 21–26. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.-T.; Tan, L. Causes and Consequences of MicroRNA Dysregulation in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 51, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Yunusova, Y.; Van Blitterswijk, M.; Dib, S.; Ghani, M.; Moreno, D.; Sato, C.; Liang, Y.; Singleton, A.; Robertson, J.; et al. Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology 2014, 83, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xi, Z.; Ghani, M.; Jia, P.; Pal, M.; Werynska, K.; Moreno, D.; Sato, C.; Liang, Y.; Robertson, J.; et al. Genetic and epigenetic study of ALS-discordant identical twins with double mutations inSOD1andARHGEF28. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Božič, A.L.; Walter, M.; Bonin, M.; Mayer, B.; Von Arnim, C.A.F.; et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain 2014, 137, 2938–2950. [Google Scholar] [CrossRef]
- Jang, S.-W.; Lopez-Anido, C.; MacArthur, R.; Svaren, J.; Inglese, J. Identification of Drug Modulators Targeting Gene-Dosage Disease CMT1A. ACS Chem. Biol. 2012, 7, 1205–1213. [Google Scholar] [CrossRef]
- Lewis, R.A. High-Dosage Ascorbic Acid Treatment in Charcot-Marie-Tooth Disease Type 1A. JAMA Neurol. 2013, 70, 981–987. [Google Scholar] [CrossRef]
- Katona, I.; Wu, X.; Feely, S.M.E.; Sottile, S.; Siskind, C.E.; Miller, L.J.; Shy, M.E.; Li, J. PMP22 expression in dermal nerve myelin from patients with CMT1A. Brain 2009, 132, 1734–1740. [Google Scholar] [CrossRef]
- Nobbio, L.; Visigalli, D.; Radice, D.; Fiorina, E.; Solari, A.; Lauria, G.; Reilly, M.M.; Santoro, L.; Schenone, A.; Pareyson, D.; et al. PMP22 messenger RNA levels in skin biopsies: Testing the effectiveness of a Charcot-Marie-Tooth 1A biomarker. Brain 2014, 137, 1614–1620. [Google Scholar] [CrossRef]
- Simard, L.R.; Bélanger, M.-C.; Morissette, S.; Wride, M.; Prior, T.W.; Swoboda, K. Preclinical validation of a multiplex real-time assay to quantify SMN mRNA in patients with SMA. Neurology 2007, 68, 451–456. [Google Scholar] [CrossRef]
- Vezain, M.; Saugier-Veber, P.; Melki, J.; Toutain, A.; Bieth, E.; Husson, M.; Pedespan, J.-M.; Viollet, L.; Penisson-Besnier, I.; Fehrenbach, S.; et al. A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur. J. Hum. Genet. 2007, 15, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.T.; Olson, R.J.; Sly, L.; Swanson, C.J.; Chung, B.; Naryshkin, N.; Narasimhan, J.; Bhattacharyya, A.; Mullenix, M.; Chen, K.S. Utility of Survival Motor Neuron ELISA for Spinal Muscular Atrophy Clinical and Preclinical Analyses. PLoS ONE 2011, 6, e24269. [Google Scholar] [CrossRef]
- Kobayashi, D.T.; Decker, D.; Zaworski, P.; Klott, K.; McGonigal, J.; Ghazal, N.; Sly, L.; Chung, B.; VanderLugt, J.; Chen, K.S. Evaluation of Peripheral Blood Mononuclear Cell Processing and Analysis for Survival Motor Neuron Protein. PLoS ONE 2012, 7, e50763. [Google Scholar] [CrossRef]
- Czech, C.; Tang, W.; Bugawan, T.; Mano, C.; Horn, C.; Iglesias, V.A.; Fröhner, S.; Zaworski, P.G.; Paushkin, S.; Chen, K.; et al. Biomarker for Spinal Muscular Atrophy: Expression of SMN in Peripheral Blood of SMA Patients and Healthy Controls. PLoS ONE 2015, 10, e0139950. [Google Scholar] [CrossRef]
- Jong, Y.-J.; Chang, J.-G.; Lin, S.-P.; Yang, T.-Y.; Wang, J.-C.; Chang, C.-P.; Lee, C.-C.; Li, H.; Hsieh-Li, H.-M.; Tsai, C.-H. Analysis of the mRNA transcripts of the survival motor neuron (SMN) gene in the tissue of an SMA fetus and the peripheral blood mononuclear cells of normals, carriers and SMA patients. J. Neurol. Sci. 2000, 173, 147–153. [Google Scholar] [CrossRef]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Baseline results of the Neuro NEXT spinal muscular atrophy infant biomarker study. Ann. Clin. Transl. Neurol. 2016, 3, 132–145. [Google Scholar] [CrossRef]
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered Intracellular Localization of SOD1 in Leukocytes from Patients with Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e75916. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Highley, J.R.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.-Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a Biomarker and Lead Small Molecules to Target r(GGGGCC)-Associated Defects in c9FTD/ALS. Neuron 2014, 83, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.A.; Allsop, D.; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2008, 117, 55–62. [Google Scholar] [CrossRef]
- Junttila, A.; Kuvaja, M.; Hartikainen, P.; Siloaho, M.; Helisalmi, S.; Moilanen, V.; Kiviharju, A.; Jansson, L.; Tienari, P.J.; Remes, A.M.; et al. Cerebrospinal Fluid TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis Patients with and without the C9ORF72 Hexanucleotide Expansion. Dement. Geriatr. Cogn. Disord. Extra 2016, 6, 142–149. [Google Scholar] [CrossRef]
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 351–356. [Google Scholar] [CrossRef]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Sjögren, M.; Davidsson, P.; Wallin, A.; Granérus, A.-K.; Grundström, E.; Askmark, H.; Vanmechelen, E.; Blennow, K. Decreased CSF-β-Amyloid 42 in Alzheimer’s Disease and Amyotrophic Lateral Sclerosis May Reflect Mismetabolism of β-Amyloid Induced by Disparate Mechanisms. Dement. Geriatr. Cogn. Disord. 2002, 13, 112–118. [Google Scholar] [CrossRef]
- Paladino, P.; Valentino, F.; Piccoli, T.; La Bella, V.; Piccoli, F. Cerebrospinal fluidtauprotein is not a biological marker in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 257–261. [Google Scholar] [CrossRef]
- Lehnert, S.; Costa, J.; De Carvalho, M.; Kirby, J.; Kuzma-Kozakiewicz, M.; Morelli, C.; Robberecht, W.; Shaw, P.; Silani, V.; Steinacker, P.; et al. Multicentre quality control evaluation of different biomarker candidates for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. Biomarkers and surrogate endpoints. Br. J. Clin. Pharmacol. 2005, 59, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Fox, P.T.; Planer, G.J.; Brooke, M.H. Activity, creatine kinase, and myoglobin in Duchenne muscular dystrophy: A clue to etiology? Neurology 1985, 35, 758. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.; Passarelli, C.; Neri, M.; Ferlini, A. Biomarkers in rare neuromuscular diseases. Exp. Cell Res. 2014, 325, 44–49. [Google Scholar] [CrossRef]
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Henrotin, Y.; Lohmander, L.S.; Losina, E.; Onnerfjord, P.; Persiani, S. OARSI Clinical Trials Recommendations: Soluble biomarker assessments in clinical trials in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Kraus, V.B. Biomarkers as drug development tools: Discovery, validation, qualification and use. Nat. Rev. Rheumatol. 2018, 14, 354–362. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J. So, You Want to Look for Biomarkers (Introduction to the Special Biomarkers Issue). J. Proteome Res. 2005, 4, 1053–1059. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| NMD | Therapeutic Biomarkers | Ref. | |||
|---|---|---|---|---|---|
| Diagnostic Biomarkers | Prognostic Biomarkers | Predictive Biomarkers | Surrogate Endpoint | ||
| DMD | CK TTNI-2 | miR-1, -31, 29c, -133, -206 TTNI-2 | [3,4,5,6,7,8] | ||
| LGMDs | CK | miR-206 | [9,10] | ||
| DM1 | miR-1, -133a, -133b, -206, -140, -454, -574 | miR-1,-133a, -133b, -206 | [11,12,13] | ||
| IIMs | CK MSA (anti-cN1A) MAA | MSA (anti-cN1A) MAA | CK | [14,15,16,17,18,19,20] | |
| MG | AChR Abs MuSK Abs LRP4 Abs miR150-5p, 21-5p, 27a3p |
MuSK Abs Titin and RyR Abs LRP4 Abs Kv1.4 Abs miR150-5p, -30e-5p | MuSK Abs | AChR Abs titer MuSK Abs titer miR-150-5p, 21-5p | [21,22,23,24,25,26,27,28,29,30,31,32,33,34] |
| LEMS | P/Q-type VGCC Abs | SOX1 Abs N-type VGCC Abs Onconeural Abs GABAB receptor Abs | [35,36,37,38,39] | ||
| CMT | NfL TMPRSS5 PMP22 miR133a, -206, -223 | TMPRSS5 PMP22 | [40,41,42,43,44,45] | ||
| Dysimmune Neuropathies | Gangliosides Abs MAG Abs IL-8 |
MAG Abs Gangliosides Abs NfL | MAG Abs | MAG Abs | [46,47,48,49,50,51,52,53] |
| SMA | SMN pNfH miR9, -206 | SMN Creatinine | SMN pNfH HSP70B Aβ40-42 miR-133a, -133b, -206 and -1 | [54,55,56,57,58,59,60,61,62,63,64,65] | |
| ALS | NfL, pNfH C9ORF72 (DPR) TDP-43 TAU miR-206, -133-b, -27a, -338-p, -183, -451, let-7, -125, -9, -129-3p, -335-5p, -199a-5p, -423-3p | CK Creatinine NfL, pNfH | Creatinine Aβ-42 SOD1 C9ORF72 (DPR) miR-206, -133a, -151a-5p | [66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barp, A.; Ferrero, A.; Casagrande, S.; Morini, R.; Zuccarino, R. Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New. Biomolecules 2021, 11, 1246. https://doi.org/10.3390/biom11081246
Barp A, Ferrero A, Casagrande S, Morini R, Zuccarino R. Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New. Biomolecules. 2021; 11(8):1246. https://doi.org/10.3390/biom11081246
Chicago/Turabian StyleBarp, Andrea, Amanda Ferrero, Silvia Casagrande, Roberta Morini, and Riccardo Zuccarino. 2021. "Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New" Biomolecules 11, no. 8: 1246. https://doi.org/10.3390/biom11081246
APA StyleBarp, A., Ferrero, A., Casagrande, S., Morini, R., & Zuccarino, R. (2021). Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New. Biomolecules, 11(8), 1246. https://doi.org/10.3390/biom11081246