
Preparation of Affinity Purified Antibodies against ε-Glutaryl-Lysine Residues in Proteins for Investigation of Glutarylated Proteins in Animal Tissues

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Glutarylation of Proteins In Vitro
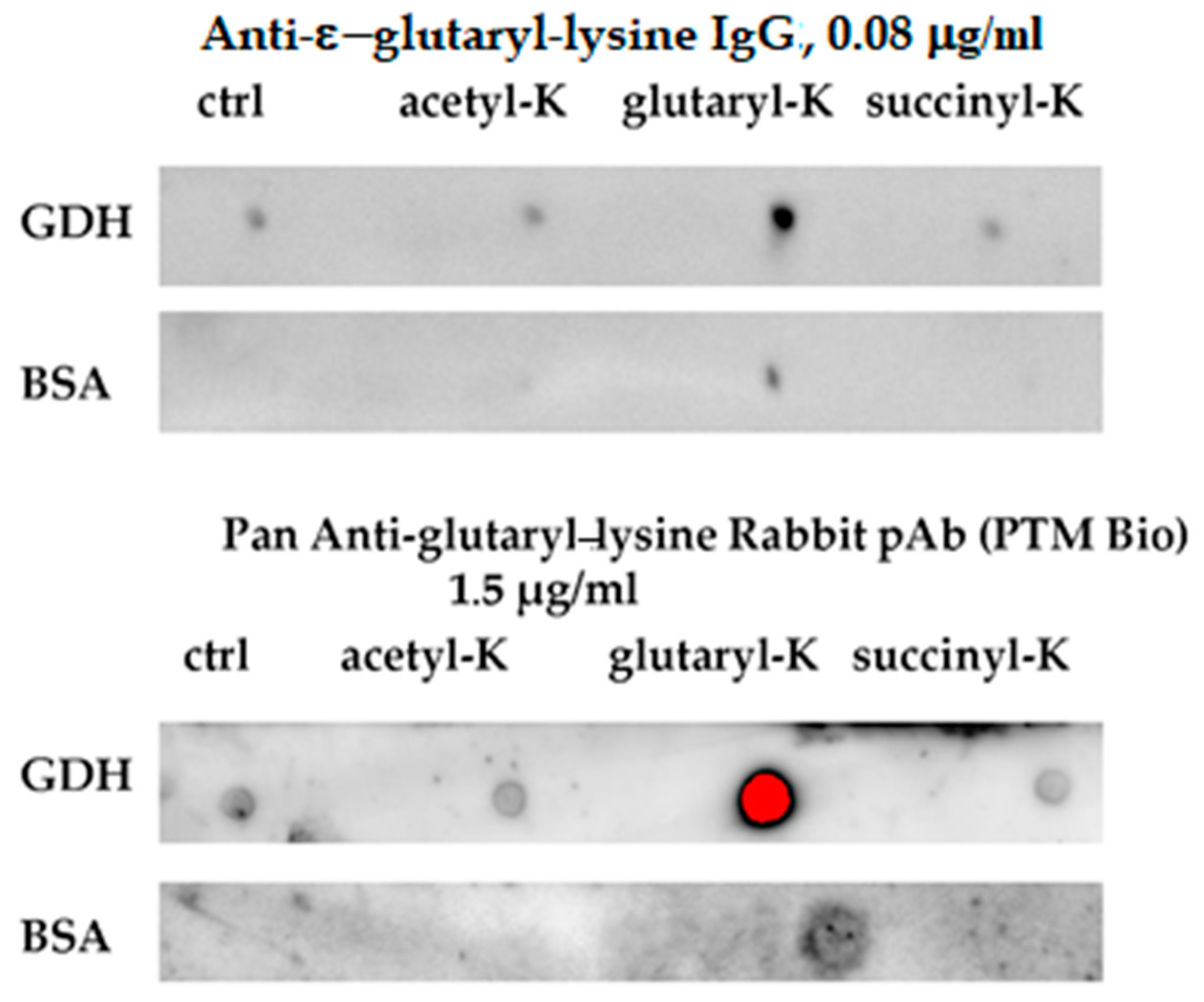
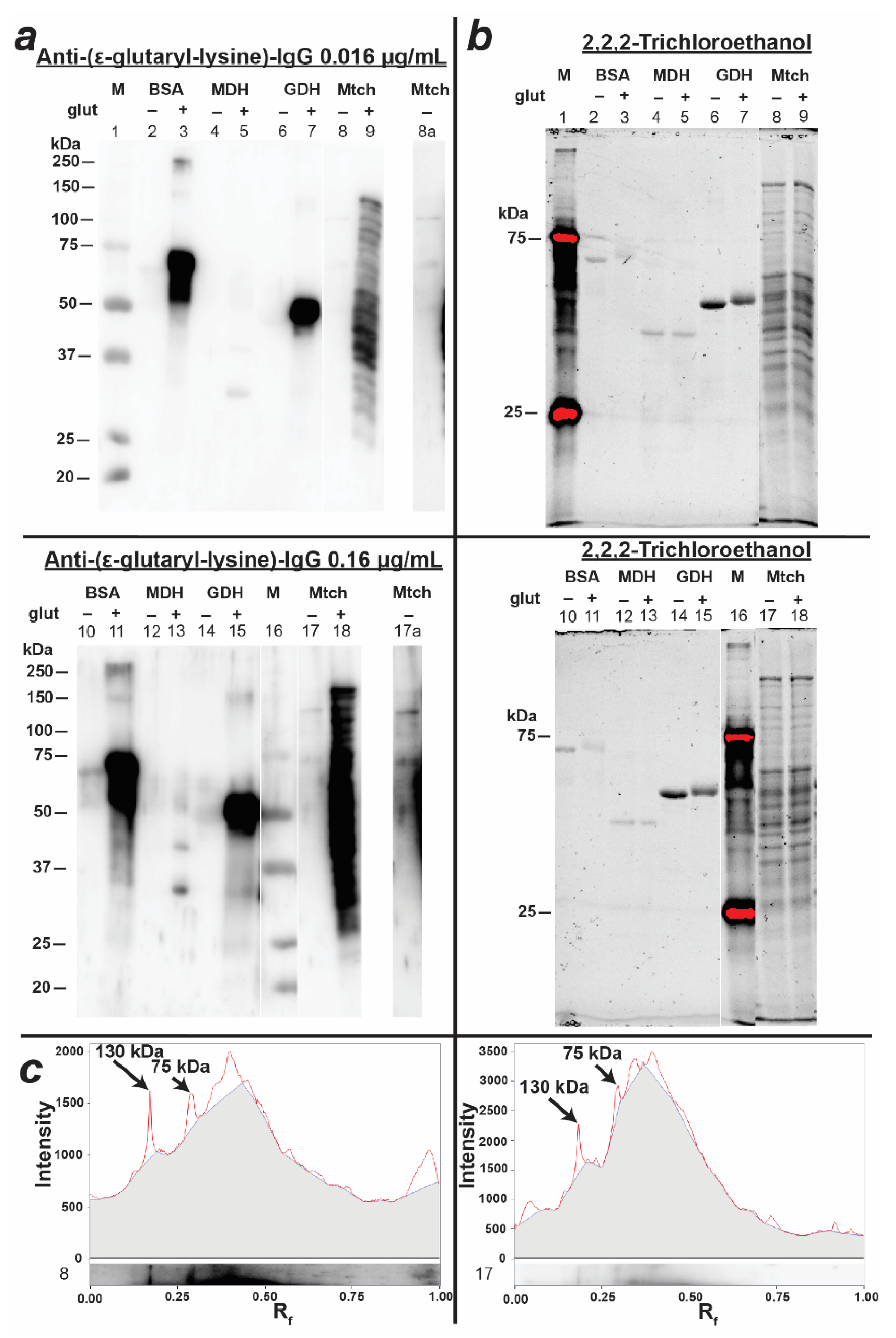
3.2. Preparation and Characterization of Antibodies against Glutarylated Proteins
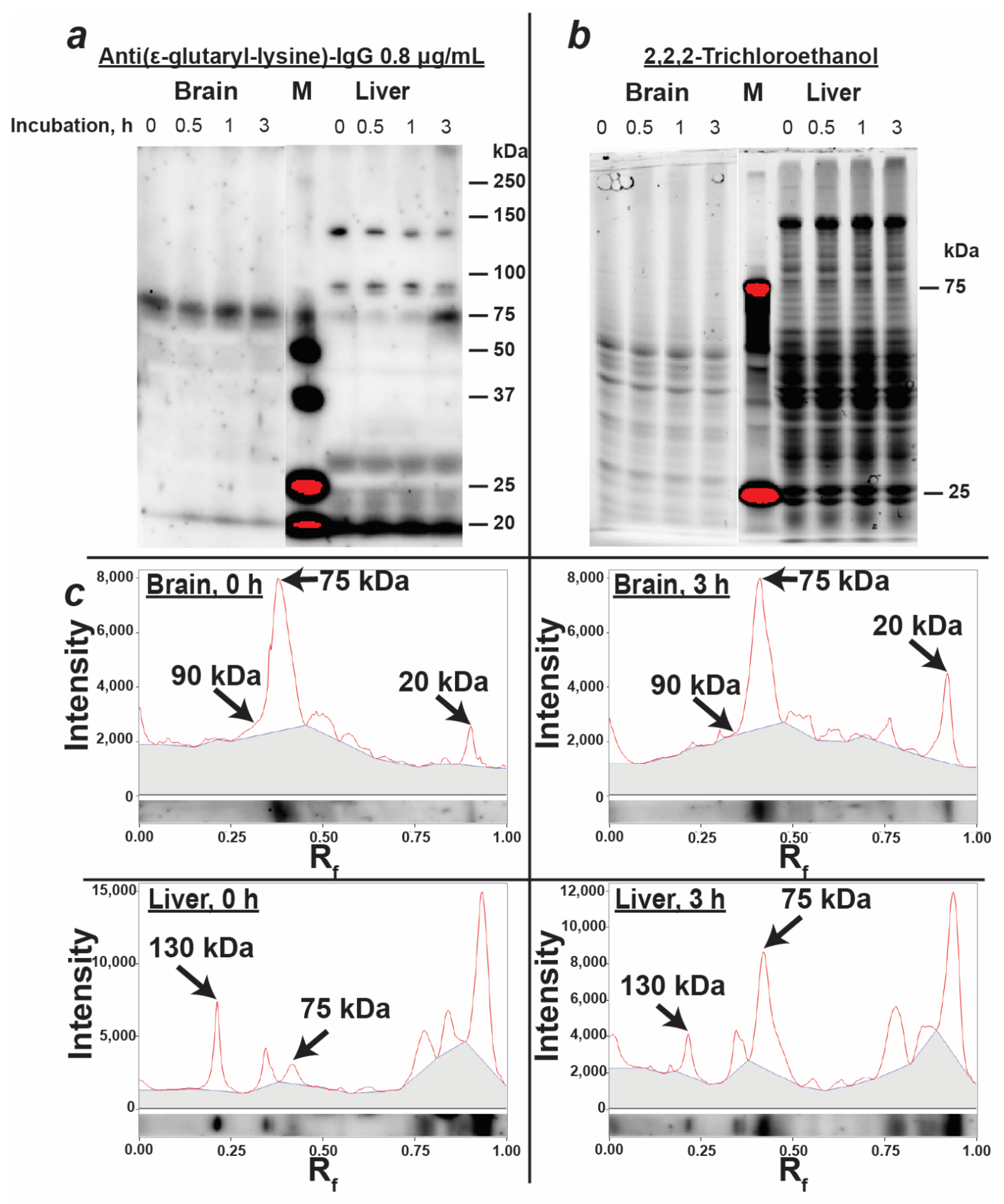
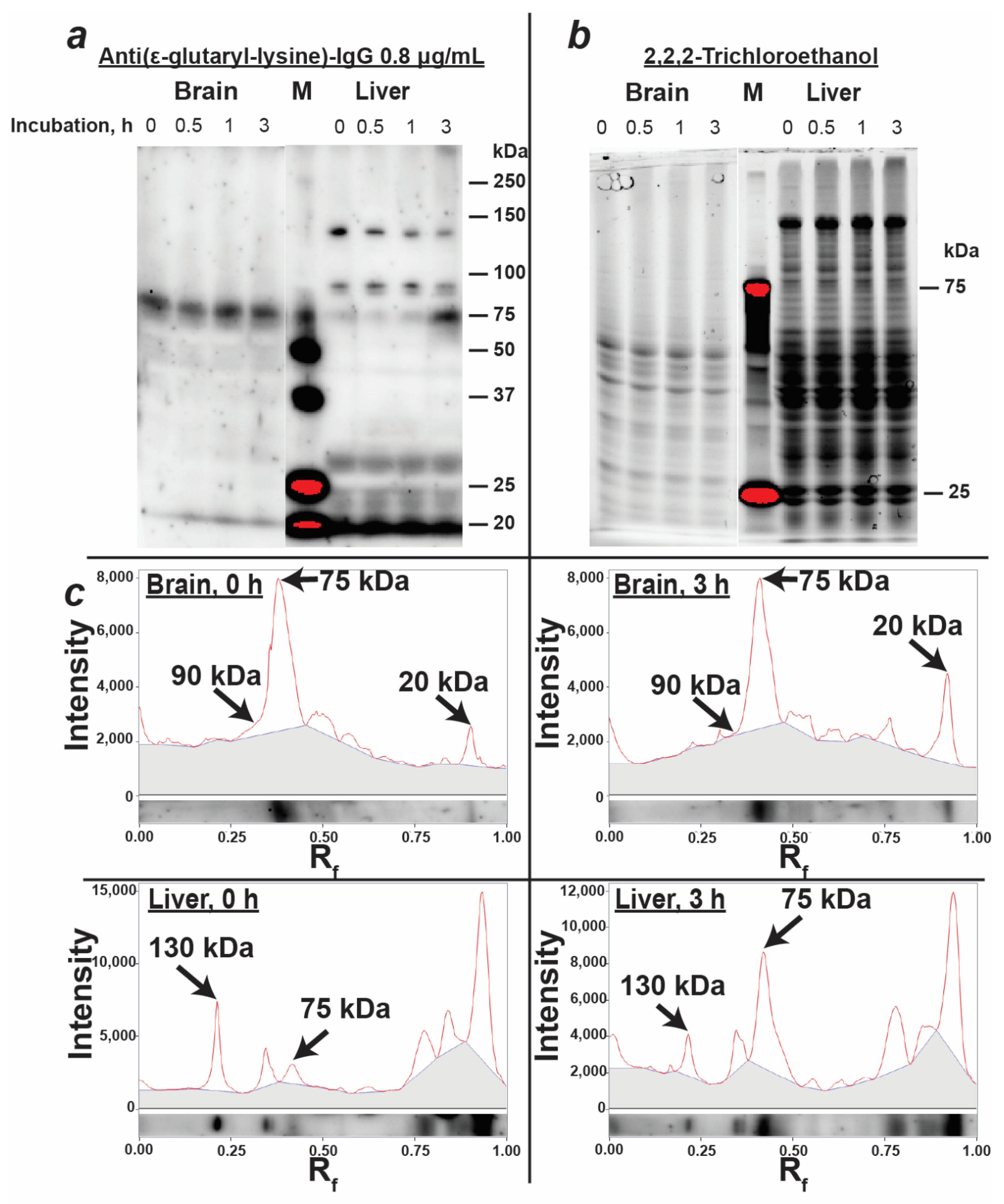
3.3. Characterization of Protein Glutarylation in the Rat Brain and Liver Homogenates
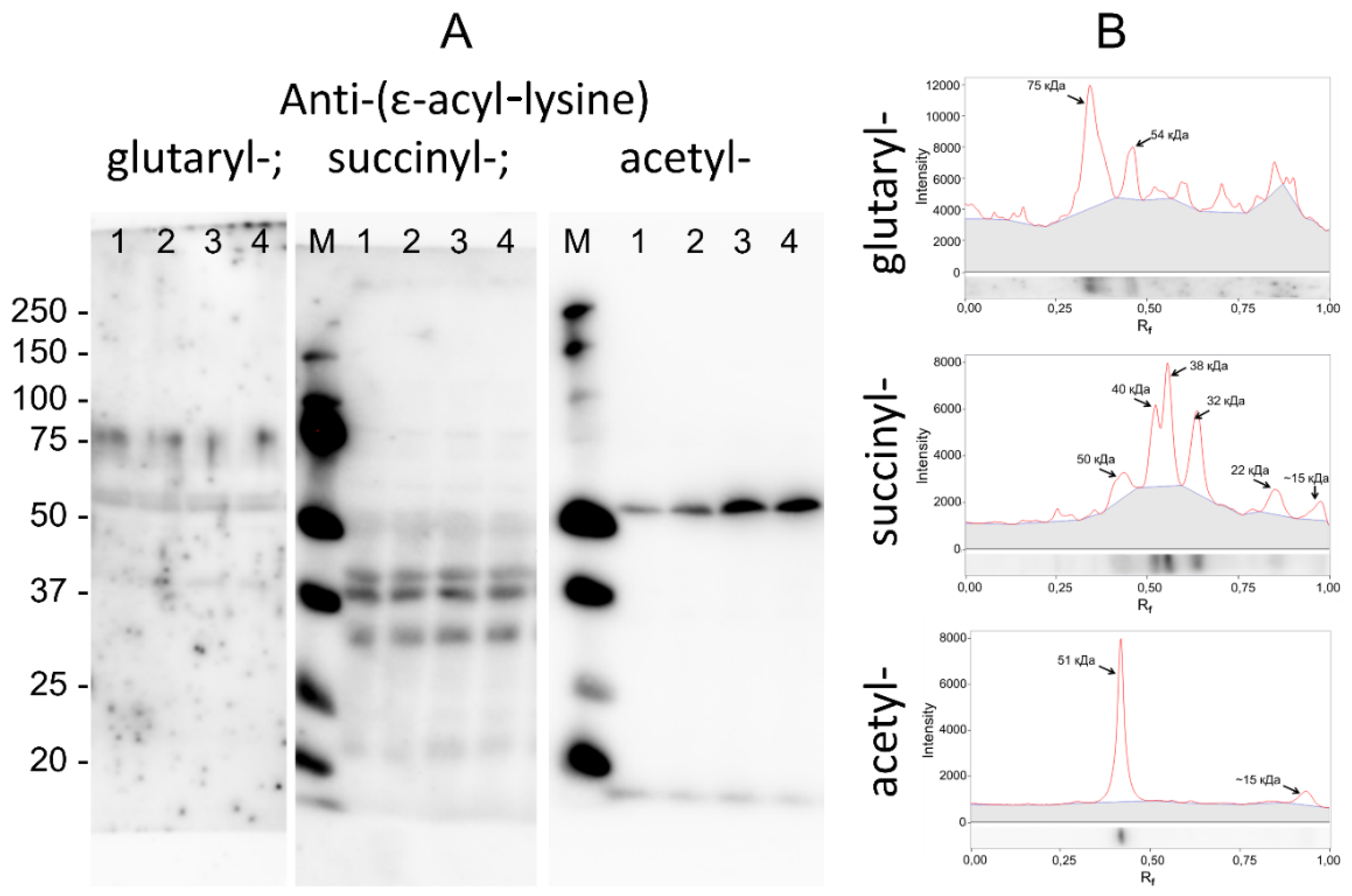
3.4. Specific Patterns of Glutarylation, Succinylation and Acetylation in Biological Preparations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Wang, G.; Yu, Z.; Zhou, M.; Li, Q.; Huang, H.; Xie, J. Proteome-wide lysine glutarylation profiling of the Mycobacterium tuberculosis H37Rv. J. Proteome Res. 2016, 15, 1379–1385. [Google Scholar] [CrossRef]
- Bao, X.; Liu, Z.; Zhang, W.; Gladysz, K.; Fung, Y.M.E.; Tian, G.; Xiong, Y.; Wong, J.W.H.; Yuen, K.W.Y.; Li, X.D. Glutarylation of histone H4 lysine 91 regulates chromatin dynamics. Mol. Cell 2019, 76, 660–675. [Google Scholar] [CrossRef]
- Green, S.R.; Storey, K.B. Purification of carbamoyl phosphate synthetase 1 (CPS1) from wood frog (Rana sylvatica) liver and its regulation in response to ice-nucleation and subsequent whole-body freezing. Mol. Cell. Biochem. 2019, 455, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Simic, Z.; Weiwad, M.; Schierhom, A.; Steegbom, C.; Schutkowski, M. The ε-amino group of protein lysine residues is highly susceptible to nonenzymatic acylation by several physiological acyl-CoA thioesters. ChemBioChem 2015, 16, 2337–2347. [Google Scholar] [CrossRef]
- Schmiesing, J.; Storch, S.; Dörfler, A.-C.; Schweizer, M.; Macrypidi-Fraune, G.; Thelen, M.; Sylvester, M.; Gieselman, V.; Meyer-Schwezinger, C.; Koch-Nolte, F.; et al. Disease-linked glutarylation impairs function and interactions of mitochondrial proteins and contributes to mitochondrial heterogeneity. Cell Rep. 2018, 24, 2946–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Wang, F.; Sun, R.; Chen, X.; Zhang, M.; Xu, Q.; Wang, Y.; Wang, S.; Xiong, Y.; Guan, K.L.; et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016, 17, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The mitochondrial acylome emerges: Proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Du, Y.; Xue, Y.; Miao, G.; Wei, T.; Zhang, P. Identification of malonylation, succinylation, and glutarylation in serum proteins of acute myocardial infarction patients. Proteom. Clin. Appl. 2020, 14, 1900103. [Google Scholar] [CrossRef]
- Cheng, Y.M.; Hu, X.N.; Peng, Z.; Pan, T.T.; Wang, F.; Chen, H.Y.; Chen, W.Q.; Zhang, Y.; Zeng, X.H.; Luo, T. Lysine glutarylation in human sperm is associated with progressive motility. Hum. Reprod. 2019, 34, 1186–1194. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Olp, M.D.; Zhu, N.; Smith, B.C. Metabolically derived lysine acylations and neighboring modifications tune the binding of the BET bromodomains to histone H4. Biochemistry 2017, 56, 5485–5495. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, B.; Molema, F.; Williams, M.; Schmiesing, J.; Mühlhausen, C.; Baumgartner, M.R.; Schumann, A.; Kohlker, S. Organic acidurias: Major gaps, new challenges, and a yet unfulfilled promise. J. Inherit. Metab. Dis. 2021, 44, 9–21. [Google Scholar] [CrossRef]
- Harmel, R.; Fiedler, D. Features and regulation of non-enzymatic posttranslational modifications. Nat. Chem. Biol. 2018, 14, 244–252. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Jörg, A.R.; Lopaschuk, G.D. Allosteric, transcriptional and post-translational control of mitochondrial energy metabolism. Biochem. J. 2019, 476, 1695–1712. [Google Scholar] [CrossRef] [PubMed]
- Hirshey, M.D.; Zhao, Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol. Cell. Proteom. 2015, 14, 2308–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.; Sun, R.; Tao, X.; Wang, X.; Kang, Q.; Wang, H.; Zhang, L.; Liu, P.; Zhang, J.; et al. SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS ONE 2019, 14, 0211796. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.F.; Freeden, C. On the role of amino groups in the structure and function of glutamate dehydrogenase I. Effect of acetylation on catalytic and regulatory properties. J. Biol. Chem. 1966, 241, 3652–3660. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Kaehne, T.; Graf, A.V.; Maslova, M.V.; Bunik, V.I. Diurnal regulation of the function of the rat brain glutamate dehydrogenase by acetylation and its dependence on thiamine administration. J. Neurochem. 2019, 153, 80–102. [Google Scholar] [CrossRef] [PubMed]
- Kolesanova, E.F.; Sanzhakov, M.A.; Kharybin, O.N. Development of the schedule for multiple parallel “difficult” peptide synthesis on pins. Int. J. Pept. 2013, 2013, 197317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aletras, A.; Barlos, K.; Gatos, D.; Koutsogianni, S.; Mamos, P. Preparation of the very acid-sensitive Fmoc-Lys(Mtt)-OH. Application in the synthesis of side-chain to side-chain cyclic peptides and oligolysine cores suitable for the solid-phase assembly f MAPs and TASPs. Int. J. Pept. Protein Res. 1995, 45, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Kolesanova, E.F.; Farafonova, T.E.; Aleshina, E.Y.; Pyndyk, N.V.; Veremieva, M.V.; Novosylnaya, A.V.; Kovalenko, M.I.; Shalak, V.F.; Negrutskii, B.S. Preparation of Monospecific Antibodies against Isoform 2 of Translation Elongation Factor 1A (eEF1A2). Biochem. Suppl. Ser. B Biomed. Chem. 2013, 7, 62–69. [Google Scholar] [CrossRef]
- Shalak, V.F.; Vislovukh, A.A.; Novosylna, O.V.; Khoruzhenko, A.I.; Kovalenko, M.I.; Kolesanova, E.F.; Egorova, E.A.; Mishin, A.A.; Krotevych, M.S.; Skoroda, L.V.; et al. Characterization of novel peptide-specific antibodies against the translation elongation factor eEF1A2 and their application for cancer research. Biopolym. Cell 2014, 30, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Artiukhov, A.V.; Grabarska, A.; Gumbarewicz, E.; Aleshin, V.A.; Kahne, T.; Obata, T.; Kazantsev, A.V.; Lukashev, N.V.; Stepulak, A.; Fernie, A.R.; et al. Synthetic analogues of 2-oxo acids discriminate metabolic contribution of the 2-oxoglutarate and 2-oxoadipate dehydrogenases in mammalian cells and tissues. Sci. Rep. 2020, 10, 1886. [Google Scholar] [CrossRef] [PubMed]
- Ladner, C.L.; Yang, J.; Turner, R.J.; Edwards, R.A. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004, 326, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.; Willmore, W.G.; Biggar, K.K. Protein quantification and visualization via ultraviolet-dependent labeling with 2,2,2-trichloroethanol. Sci. Rep. 2019, 9, 13923. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Spector, T. Refinement of the coomassie blue method of protein quantitation. A simple and linear spectrophotometric assay for less than or equal to 0.5 to 50 microgram of protein. Anal. Biochem. 1978, 86, 142–146. [Google Scholar] [CrossRef]
- Wagner, G.R.; Bhatt, D.P.; O’Connell, T.M.; Thompson, J.W.; Dubois, L.G.; Backos, D.S.; Yang, H.; Mitchell, G.A.; Ilkaeva, O.R.; Stevens, R.D.; et al. A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab. 2017, 25, 823–837. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glutarylated Lys Residue of GDH | Modification with 0.16 mM Glutaric Anhydride | Modification with 0.5 mM Glutaryl-CoA [6] |
|---|---|---|
| K90 | - | + |
| K171 | + | + |
| K183 | + | + |
| K187 | + | - |
| K191 | + | + |
| K200 | - | + |
| K352 | - | + |
| K365 | - | + |
| K386 | - | + |
| K390 | + | - |
| K457 | + | - |
| K477 | + | + |
| K480 * | + | + |
| K503 | + | + |
| K527 | - | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artiukhov, A.V.; Kolesanova, E.F.; Boyko, A.I.; Chashnikova, A.A.; Gnedoy, S.N.; Kaehne, T.; Ivanova, D.A.; Kolesnichenko, A.V.; Aleshin, V.A.; Bunik, V.I. Preparation of Affinity Purified Antibodies against ε-Glutaryl-Lysine Residues in Proteins for Investigation of Glutarylated Proteins in Animal Tissues. Biomolecules 2021, 11, 1168. https://doi.org/10.3390/biom11081168
Artiukhov AV, Kolesanova EF, Boyko AI, Chashnikova AA, Gnedoy SN, Kaehne T, Ivanova DA, Kolesnichenko AV, Aleshin VA, Bunik VI. Preparation of Affinity Purified Antibodies against ε-Glutaryl-Lysine Residues in Proteins for Investigation of Glutarylated Proteins in Animal Tissues. Biomolecules. 2021; 11(8):1168. https://doi.org/10.3390/biom11081168
Chicago/Turabian StyleArtiukhov, Artem V., Ekaterina F. Kolesanova, Aleksandra I. Boyko, Anastasiya A. Chashnikova, Sergei N. Gnedoy, Thilo Kaehne, Daria A. Ivanova, Alyona V. Kolesnichenko, Vasily A. Aleshin, and Victoria I. Bunik. 2021. "Preparation of Affinity Purified Antibodies against ε-Glutaryl-Lysine Residues in Proteins for Investigation of Glutarylated Proteins in Animal Tissues" Biomolecules 11, no. 8: 1168. https://doi.org/10.3390/biom11081168
APA StyleArtiukhov, A. V., Kolesanova, E. F., Boyko, A. I., Chashnikova, A. A., Gnedoy, S. N., Kaehne, T., Ivanova, D. A., Kolesnichenko, A. V., Aleshin, V. A., & Bunik, V. I. (2021). Preparation of Affinity Purified Antibodies against ε-Glutaryl-Lysine Residues in Proteins for Investigation of Glutarylated Proteins in Animal Tissues. Biomolecules, 11(8), 1168. https://doi.org/10.3390/biom11081168