Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing – Review


 , ,
, , 
{kind=link}
Abstract
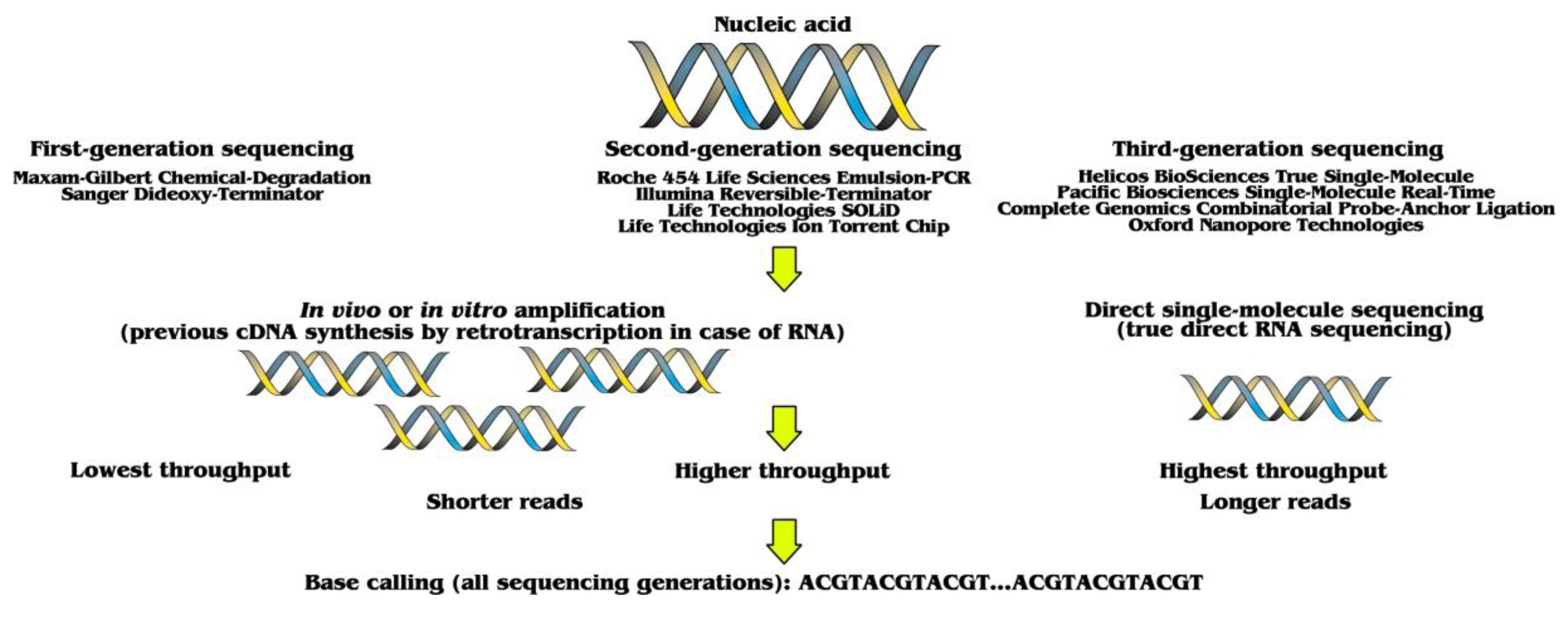
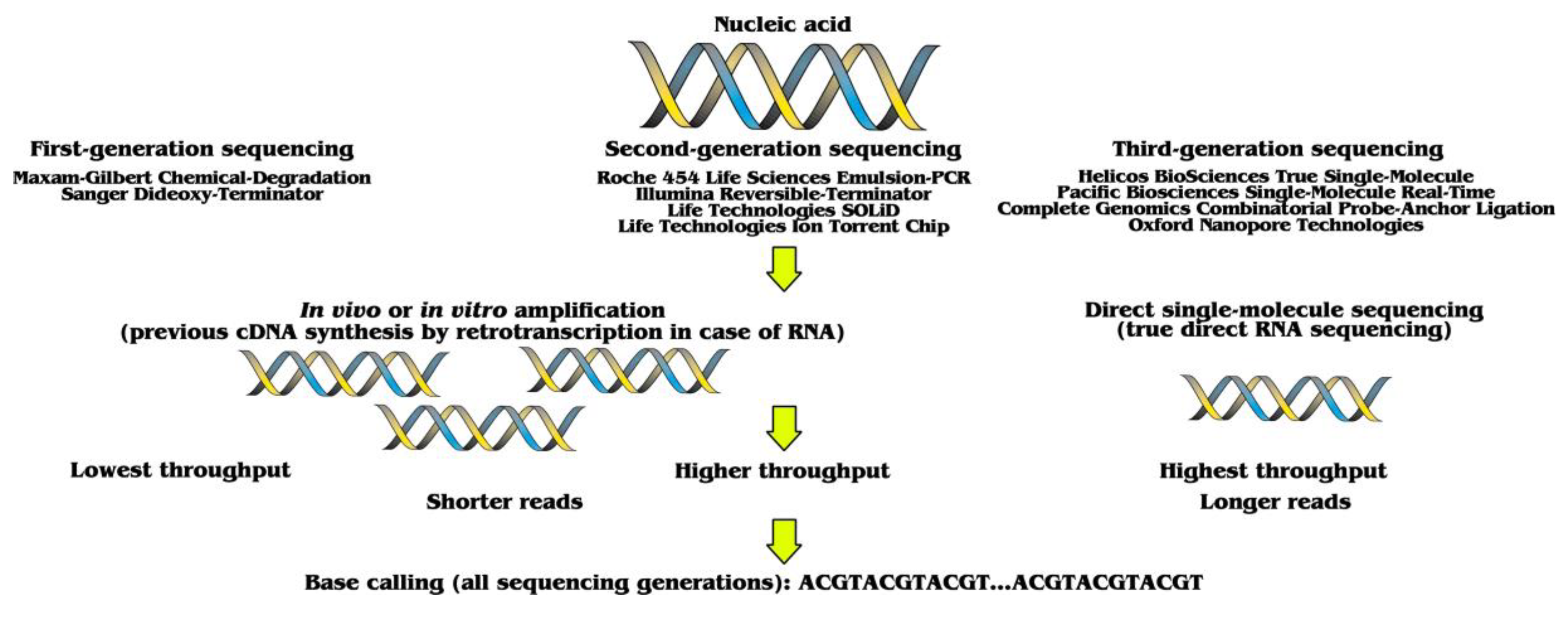
:1. Three Sequencing Generations
2. Applications of Nucleic-Acid Sequencing
2.1. Structural Genomics
2.2. Functional Genomics
2.3. Epigenomics
2.4. Metagenomics
3. Future Prospects and Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lario, A.; Gonzalez, A.; Dorado, G. Automated laser-induced fluorescence DNA sequencing: Equalizing signal-to-noise ratios significantly enhances overall performance. Anal. Biochem. 1997, 247, 30–33. [Google Scholar] [CrossRef]
- Ozsolak, F.; Platt, A.R.; Jones, D.R.; Reifenberger, J.G.; Sass, L.E.; McInerney, P.; Thompson, J.F.; Bowers, J.; Jarosz, M.; Milos, P.M. Direct RNA sequencing. Nature 2009, 461, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Heydari, M.; Miclotte, G.; Van de Peer, Y.; Fostier, J. Illumina error correction near highly repetitive DNA regions improves de novo genome assembly. BMC Bioinform. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Bleidorn, C. Third generation sequencing: Technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers. 2016, 14, 1–8. [Google Scholar] [CrossRef]
- Blom, M.P.K. Opportunities and challenges for high-quality biodiversity tissue archives in the age of long-read sequencing. Mol. Ecol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Broseus, L.; Thomas, A.; Oldfield, A.J.; Severac, D.; Dubois, E.; Ritchie, W. TALC: Transcript-level Aware Long-read Correction. Bioinformatics 2020, 36, 5000–5006. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Shang, J.Y.; Sun, Y.N. Improving protein domain classification for third-generation sequencing reads using deep learning. Bmc Genom. 2021, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hestand, M.S.; Ameur, A. The Versatility of SMRT Sequencing. Genes 2019, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, S.L.; Su, S.; Dong, X.Y.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.T.; Qu, L.; Yang, L.S.; Wang, Y.Y.; Zhu, H.Q. NanoReviser: An Error-Correction Tool for Nanopore Sequencing Based on a Deep Learning Algorithm. Front. Genet. 2020, 11, 900. [Google Scholar] [CrossRef]
- Feng, Z.X.; Clemente, J.C.; Wong, B.; Schadt, E.E. Detecting and phasing minor single-nucleotide variants from long-read sequencing data. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Bai, X.; Li, Y.X.; Zeng, X.M.; Zhao, Q.; Zhang, Z.W. Single-cell sequencing technology in tumor research. Clin. Chim. Acta 2021, 518, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Chappell, K.; Washam, C.L.; Gies, A.; Bird, J.; Robeson, M.S.; Byrum, S.D. Multi-omics data integration considerations and study design for biological systems and disease. Mol. Omics. 2021, 17, 170–185. [Google Scholar] [CrossRef]
- Reiter, T.; Brooks, P.T.; Irber, L.; Joslin, S.E.K.; Reid, C.M.; Scott, C.; Brown, C.T.; Pierce-Ward, N.T. Streamlining data-intensive biology with workflow systems. Gigascience 2021, 10, giaa140. [Google Scholar] [CrossRef]
- Li, Y.; Ma, A.J.; Mathe, E.A.; Li, L.; Liu, B.Q.; Ma, Q. Elucidation of Biological Networks across Complex Diseases Using Single-Cell Omics. Trends Genet. 2020, 36, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Khella, C.A.; Mehta, G.A.; Mehta, R.N.; Gatza, M.L. Recent Advances in Integrative Multi-Omics Research in Breast and Ovarian Cancer. J. Pers. Med. 2021, 11, 149. [Google Scholar] [CrossRef]
- Zhu, C.X.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Philpott, M.; Cribbs, A.P.; Brown, T.; Brown, T.; Oppermann, U. Advances and challenges in epigenomic single-cell sequencing applications. Curr. Opin. Chem. Biol. 2020, 57, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Jovcevska, I. Next Generation Sequencing and Machine Learning Technologies Are Painting the Epigenetic Portrait of Glioblastoma. Front. Oncol. 2020, 10, 798. [Google Scholar] [CrossRef]
- Chachar, S.; Liu, J.R.; Zhang, P.X.; Riaz, A.; Guan, C.F.; Liu, S.Y. Harnessing Current Knowledge of DNA N6-Methyladenosine From Model Plants for Non-model Crops. Front. Genet. 2021, 12, 668317. [Google Scholar] [CrossRef] [PubMed]
- Schultzhaus, Z.; Wang, Z.; Stenger, D. CRISPR-based enrichment strategies for targeted sequencing. Biotechnol. Adv. 2021, 46, 107672. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Castro-Wallace, S.L.; Chiu, C.Y.; John, K.K.; Stahl, S.E.; Rubins, K.H.; McIntyre, A.B.R.; Dworkin, J.P.; Lupisella, M.L.; Smith, D.J.; Botkin, D.J.; et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, K.K.; Botkin, D.S.; Burton, A.S.; Castro-Wallace, S.L.; Chaput, J.D.; Dworkin, J.P.; Lehman, N.; Lupisella, M.L.; Mason, C.E.; Smith, D.J.; et al. The Biomolecule Sequencer Project: Nanopore sequencing as a dual-use tool for crew health and astrobiology investigations. In Proceedings of the 47th Lunar and Planetary Science Conference, The Woodlands, TX, USA, 21–25 March 2016. [Google Scholar]
- Wong, S. Diagnostics in space: Will zero gravity add weight to new advances? Expert Rev. Mol. Diagn. 2020, 20, 1–4. [Google Scholar] [CrossRef]
- Stahl-Rommel, S.; Jain, M.; Nguyen, H.N.; Arnold, R.R.; Aunon-Chancellor, S.M.; Sharp, G.M.; Castro, C.L.; John, K.K.; Juul, S.; Turner, D.J.; et al. Real-Time Culture-Independent Microbial Profiling Onboard the International Space Station Using Nanopore Sequencing. Genes 2021, 12, 106. [Google Scholar] [CrossRef]
- Caspar, S.M.; Schneider, T.; Stoll, P.; Meienberg, J.; Matyas, G. Potential of whole-genome sequencing-based pharmacogenetic profiling. Pharmacogenomics 2021, 22, 177–190. [Google Scholar] [CrossRef]
- Gorcenco, S.; Ilinca, A.; Almasoudi, W.; Kafantari, E.; Lindgren, A.G.; Puschmann, A. New generation genetic testing entering the clinic. Parkinsonism Relat. D 2020, 73, 72–84. [Google Scholar] [CrossRef]
- Duan, Q.K.; Tang, C.; Ma, Z.; Chen, C.G.; Shang, X.B.; Yue, J.; Jiang, H.J.; Gao, Y.; Xu, B. Genomic Heterogeneity and Clonal Evolution in Gastroesophageal Junction Cancer Revealed by Single Cell DNA Sequencing. Front. Oncol. 2021, 11, 1574. [Google Scholar] [CrossRef] [PubMed]
- Barp, A.; Mosca, L.; Sansone, V.A. Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders. Diagnostics 2021, 11, 701. [Google Scholar] [CrossRef]
- Begum, G.; Albanna, A.; Bankapur, A.; Nassir, N.; Tambi, R.; Berdiev, B.K.; Akter, H.; Karuvantevida, N.; Kellam, B.; Alhashmi, D.; et al. Long-Read Sequencing Improves the Detection of Structural Variations Impacting Complex Non-Coding Elements of the Genome. Int. J. Mol. Sci. 2021, 22, 2060. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.S.; Jones, N.S. Discovering Cellular Mitochondrial Heteroplasmy Heterogeneity with Single Cell RNA and ATAC Sequencing. Biology 2021, 10, 503. [Google Scholar] [CrossRef]
- Macken, W.L.; Vandrovcova, J.; Hanna, M.G.; Pitceathly, R.D.S. Applying genomic and transcriptomic advances to mitochondrial medicine. Nat. Rev. Neurol. 2021, 17, 215–230. [Google Scholar] [CrossRef]
- Poole, O.V.; Pizzamiglio, C.; Murphy, D.; Falabella, M.; Macken, W.L.; Bugiardini, E.; Woodward, C.E.; Labrum, R.; Efthymiou, S.; Salpietro, V.; et al. Mitochondrial DNA Analysis from Exome Sequencing Data Improves Diagnostic Yield in Neurological Diseases. Ann. Neurol. 2021, 89, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Murphy, D.; Bugiardini, E.; Salem, R.; Jager, J.; Futema, M.; Akhtar, M.M.; Savvatis, K.; Woodward, C.; Pittman, A.M.; et al. Iterative Reanalysis of Hypertrophic Cardiomyopathy Exome Data Reveals Causative Pathogenic Mitochondrial DNA Variants. Circ-Genom. Precis. Me. 2021, 14, 379–382. [Google Scholar] [CrossRef]
- Gusic, M.; Prokisch, H. Genetic basis of mitochondrial diseases. Febs. Lett. 2021, 595, 1132–1158. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Stenton, S.L.; Hudson, G.; Prokisch, H.; Taylor, R.W. The genetics of mitochondrial disease: Dissecting mitochondrial pathology using multi-omic pipelines. J. Pathol. 2021, 254, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Anaya, L.Z.; Felix-Sastre, A.J.; Lares-Villa, F.; Lares-Jimenez, L.F.; Gonzalez-Galaviz, J.R. Application of the omics sciences to the study of Naegleria fowleri, Acanthamoeba spp., and Balamuthia mandrillaris: Current status and future projections. Parasite 2021, 28, 36. [Google Scholar] [CrossRef]
- Montarry, J.; Mimee, B.; Danchin, E.G.J.; Koutsovoulos, G.D.; Ste-Croix, D.T.; Grenier, E. Recent Advances in Population Genomics of Plant-Parasitic Nematodes. Phytopathology 2021, 111, 40–48. [Google Scholar] [CrossRef]
- Stam, R.; Gladieux, P.; Vinatzer, B.A.; Goss, E.M.; Potnis, N.; Candresse, T.; Brewer, M.T. Population Genomic- and Phylogenomic-Enabled Advances to Increase Insight Into Pathogen Biology and Epidemiology Introduction. Phytopathology 2021, 111, 8–11. [Google Scholar] [CrossRef]
- Ste-Croix, D.T.; St-Marseille, A.F.G.; Lord, E.; Belanger, R.R.; Brodeur, J.; Mimee, B. Genomic Profiling of Virulence in the Soybean Cyst Nematode Using Single-Nematode Sequencing. Phytopathology 2021, 111, 137–148. [Google Scholar] [CrossRef]
- Brito, I.L. Examining horizontal gene transfer in microbial communities. Nat. Rev. Microbiol. 2021, 19, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Lyu, R.; Tsui, V.; McCarthy, D.J.; Crismani, W. Personalized genome structure via single gamete sequencing. Genome Biol. 2021, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Campoy, J.A.; Sun, H.Q.; Goel, M.; Jiao, W.B.; Folz-Donahue, K.; Wang, N.; Rubio, M.; Liu, C.; Kukat, C.; Ruiz, D.; et al. Gamete binning: Chromosome-level and haplotype-resolved genome assembly enabled by high-throughput single-cell sequencing of gamete genomes. Genome Biol. 2020, 21, 1–20. [Google Scholar] [CrossRef]
- Wang, C.R.; Liu, H.F.; Wang, H.Y.; Tao, J.J.; Yang, T.W.; Chen, H.; An, R.; Wang, J.; Huang, N.; Gong, X.Y.; et al. Robust Storage of Chinese Language in a Pool of Small Single-Stranded DNA Rings and Its Facile Reading-Out. B Chem. Soc. Jpn. 2021, 94, 53–59. [Google Scholar] [CrossRef]
- Jha, A.B.; Gali, K.K.; Alam, Z.; Lachagari, V.B.R.; Warkentin, T.D. Potential Application of Genomic Technologies in Breeding for Fungal and Oomycete Disease Resistance in Pea. Agronomy 2021, 11, 1260. [Google Scholar] [CrossRef]
- van Dijk, A.D.J.; Kootstra, G.; Kruijer, W.; de Ridder, D. Machine learning in plant science and plant breeding. Iscience 2021, 24, 101890. [Google Scholar] [CrossRef]
- Awika, H.O.; Mishra, A.K.; Gill, H.; DiPiazza, J.; Avila, C.A.; Joshi, V. Selection of nitrogen responsive root architectural traits in spinach using machine learning and genetic correlations. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Varshney, R.K.; Bohra, A.; Yu, J.M.; Graner, A.; Zhang, Q.F.; Sorrells, M.E. Feature Review Designing Future Crops: Genomics-Assisted Breeding Comes of Age. Trends Plant Sci. 2021, 26, 631–649. [Google Scholar] [CrossRef]
- Anwar, K.; Joshi, R.; Dhankher, O.P.; Singla-Pareek, S.L.; Pareek, A. Elucidating the Response of Crop Plants towards Individual, Combined and Sequentially Occurring Abiotic Stresses. Int. J. Mol. Sci. 2021, 22, 6119. [Google Scholar] [CrossRef] [PubMed]
- Pazhamala, L.T.; Kudapa, H.; Weckwerth, W.; Millar, A.H.; Varshney, R.K. Systems biology for crop improvement. Plant Genome 2021, 1–23. [Google Scholar] [CrossRef]
- Saad, N.S.M.; Severn-Ellis, A.A.; Pradhan, A.; Edwards, D.; Batley, J. Genomics Armed With Diversity Leads the Way in Brassica Improvement in a Changing Global Environment. Front. Genet. 2021, 12, 600789. [Google Scholar] [CrossRef]
- Hu, D.D.; Jing, J.J.; Snowdon, R.J.; Mason, A.S.; Shen, J.X.; Meng, J.L.; Zou, J. Exploring the gene pool of Brassica napus by genomics-based approaches. Plant Biotechnol. J. 2021. [Google Scholar] [CrossRef]
- Witzel, K.; Kurina, A.B.; Artemyeva, A.M. Opening the Treasure Chest: The Current Status of Research on Brassica oleracea and B. rapa Vegetables From ex situ Germplasm Collections. Front. Plant Sci. 2021, 12, 925. [Google Scholar] [CrossRef] [PubMed]
- Scossa, F.; Alseekh, S.; Fernie, A.R. Integrating multi-omics data for crop improvement. J. Plant Physiol. 2021, 257, 153352. [Google Scholar] [CrossRef]
- Sinha, P.; Singh, V.K.; Bohra, A.; Kumar, A.; Reif, J.C.; Varshney, R.K. Genomics and breeding innovations for enhancing genetic gain for climate resilience and nutrition traits. Theor. Appl. Genet. 2021, 134, 1829–1843. [Google Scholar] [CrossRef]
- Kuo, R.I.; Cheng, Y.Y.; Zhang, R.X.; Brown, J.W.S.; Smith, J.; Archibald, A.L.; Burt, D.W. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genom. 2020, 21, 1–22. [Google Scholar] [CrossRef]
- Sahlin, K.; Medvedev, P. Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis (vol 12, 2, 2021). Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Rabadan, R. Computing the Role of Alternative Splicing in Cancer. Trends Cancer 2021, 7, 347–358. [Google Scholar] [CrossRef]
- Mertes, C.; Scheller, I.F.; Yepez, V.A.; Celik, M.H.; Liang, Y.J.Q.; Kremer, L.S.; Gusic, M.; Prokisch, H.; Gagneur, J. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Hu, Y.; Fang, L.; Chen, X.L.; Zhong, J.F.; Li, M.Y.; Wang, K. LIQA: Long-read isoform quantification and analysis. Genome Biol. 2021, 22, 1–21. [Google Scholar] [CrossRef]
- Riepe, T.V.; Khan, M.; Roosing, S.; Cremers, F.P.M.; ’t Hoen, P.A.C. Benchmarking deep learning splice prediction tools using functional splice assays. Hum. Mutat. 2021, 42, 799–810. [Google Scholar] [CrossRef]
- Pan, Y.; Kadash-Edmondson, K.E.; Wang, R.; Phillips, J.; Liu, S.; Ribas, A.; Aplenc, R.; Witte, O.N.; Xing, Y. RNA Dysregulation: An Expanding Source of Cancer Immunotherapy Targets. Trends Pharmacol. Sci. 2021, 42, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Gephart, M.G.H.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [Green Version]
- Kaster, A.K.; Sobol, M.S. Microbial single-cell omics: The crux of the matter. Appl. Microbiol. Biot. 2020, 104, 8209–8220. [Google Scholar] [CrossRef] [PubMed]
- Adil, A.; Kumar, V.; Jan, A.T.; Asger, M. Single-Cell Transcriptomics: Current Methods and Challenges in Data Acquisition and Analysis. Front. Neurosci-Switz 2021, 15, 398. [Google Scholar] [CrossRef]
- Song, Y.L.; Xu, X.; Wang, W.; Tian, T.; Zhu, Z.; Yang, C.Y. Single cell transcriptomics: Moving towards multi-omics. Analyst 2019, 144, 3172–3189. [Google Scholar] [CrossRef]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Arauzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [Green Version]
- Hahn, O.; Fehlmann, T.; Zhang, H.; Munson, C.N.; Vest, R.T.; Borcherding, A.; Liu, S.; Villarosa, C.; Drmanac, S.; Drmanac, R.; et al. CooIMPS for robust sequencing of single-nuclear RNAs captured by droplet-based method. Nucleic Acids Res. 2021, 49, e11. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Ma, J.Y.; Meng, T.G.; Wang, Z.B.; Yue, W.; Zhou, Q.; Li, S.; Feng, X.; Hou, Y.; Schatten, H.; et al. Single-cell RNA sequencing reveals the landscape of early female germ cell development. Faseb J. 2020, 34, 12634–12645. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F.C. Human Germline Cell Development: From the Perspective of Single-Cell Sequencing. Mol. Cell 2019, 76, 320–328. [Google Scholar] [CrossRef]
- Brandt, L.; Cristinelli, S.; Ciuffi, A. Single-Cell Analysis Reveals Heterogeneity of Virus Infection, Pathogenicity, and Host Responses: HIV as a Pioneering Example. Annu. Rev. Virol. 2020, 7, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, F.; Lupieri, A.; Aikawa, M.; Aikawa, E. Harnessing Single-Cell RNA Sequencing to Better Understand How Diseased Cells Behave the Way They Do in Cardiovascular Disease. Arterioscl. Throm. Vas. Biol. 2021, 41, 585–600. [Google Scholar] [CrossRef]
- Yu, S.G.; Li, C.H.; Lin, H.; Ou, M.L.; Tang, D.G.; Dai, Y.; Yan, Q. Application of single-cell RNA sequencing in embryonic development. Genomics 2020, 112, 4547–4551. [Google Scholar] [CrossRef]
- Yasen, A.; Aini, A.; Wang, H.; Li, W.D.; Zhang, C.S.; Ran, B.; Tuxun, T.; Maimaitinijiati, Y.; Shao, Y.M.; Aji, T.; et al. Progress and applications of single-cell sequencing techniques. Infect. Genet. Evol. 2020, 80, 104198. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef]
- Zang, J.Y.; Ye, K.Y.; Fei, Y.; Zhang, R.Y.; Chen, H.G.; Zhuang, G.L. Immunotherapy in the Treatment of Urothelial Bladder Cancer: Insights From Single-Cell Analysis. Front. Oncol. 2021, 11, 2020. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Y.Y.; Qing, T.; Xing, K.C.; Li, J.; Zhen, T.M.; Zhu, S.B.; Zhan, X.B. Comprehensive analysis of metastatic gastric cancer tumour cells using single-cell RNA-seq. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Mercatelli, D.; Balboni, N.; Palma, A.; Aleo, E.; Sanna, P.P.; Perini, G.; Giorgi, F.M. Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines. Biomolecules 2021, 11, 177. [Google Scholar] [CrossRef]
- Ysebaert, L.; Quillet-Mary, A.; Tosolini, M.; Pont, F.; Laurent, C.; Fournie, J.J. Lymphoma Heterogeneity Unraveled by Single-Cell Transcriptomics. Front. Immunol. 2021, 12, 202. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.L.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.J.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 1–26. [Google Scholar] [CrossRef]
- Liu, J.; Xu, T.M.; Jin, Y.M.; Huang, B.Y.; Zhang, Y. Progress and Clinical Application of Single-Cell Transcriptional Sequencing Technology in Cancer Research. Front. Oncol. 2021, 10, 3367. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Meng, D.; Guo, H.Y.; Sun, C.L.; Chen, P.X.; Jiang, M.L.; Xu, Y.; Yu, J.; Fang, Q.Y.; Zhu, J.; et al. Single-Cell Sequencing, an Advanced Technology in Lung Cancer Research. Oncotargets Ther. 2021, 14, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.T.; Li, W.M.; Cai, X.Y. Applications of Single-Cell Omics to Dissect Tumor Microenvironment. Front. Genet. 2020, 11, 548719. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, S.F.; Min, M.; Ni, Y.L.; Lu, Z.; Sun, X.T.; Wu, J.Q.; Liu, B.; Ying, X.M.; Liu, Y. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut 2021, 70, 464–475. [Google Scholar] [CrossRef]
- Cheng, S.J.; Li, Z.Y.; Gao, R.R.; Xing, B.C.; Gao, Y.N.; Yang, Y.; Qin, S.S.; Zhang, L.; Ouyang, H.Q.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809. [Google Scholar] [CrossRef]
- Sinjab, A.; Han, G.C.; Wang, L.H.; Kadara, H. Field Carcinogenesis in Cancer Evolution: What the Cell Is Going On? Cancer Res. 2020, 80, 4888–4891. [Google Scholar] [CrossRef]
- Cildir, G.; Yip, K.H.; Pant, H.; Tergaonkar, V.; Lopez, A.F.; Tumes, D.J. Understanding mast cell heterogeneity at single cell resolution. Trends Immunol. 2021, 42, 523–535. [Google Scholar] [CrossRef]
- de Jong, E.; Bosco, A. Unlocking immune-mediated disease mechanisms with transcriptomics. Biochem. Soc. Trans. 2021, 49, 705–714. [Google Scholar] [CrossRef]
- Derakhshani, A.; Rostami, Z.; Safarpour, H.; Shadbad, M.A.; Nourbakhsh, N.S.; Argentiero, A.; Taefehshokr, S.; Tabrizi, N.J.; Kooshkaki, O.; Astamal, R.V.; et al. From Oncogenic Signaling Pathways to Single-Cell Sequencing of Immune Cells: Changing the Landscape of Cancer Immunotherapy. Molecules 2021, 26, 2278. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Li, Y.R.; Cai, B.L.; He, Q.Z.; Chen, G.F.; Wang, M.F.; Wang, K.; Wan, X.P.; Yan, Q. Phenotyping of immune and endometrial epithelial cells in endometrial carcinomas revealed by single-cell RNA sequencing. Aging 2021, 13, 6565–6591. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Li, X.H. Dive into Single, Seek Out Multiple: Probing Cancer Metastases via Single-Cell Sequencing and Imaging Techniques. Cancers 2021, 13, 1067. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, M.C.; Yan, J.L.; Guo, L.; Zhang, B.; Liu, S.; Lei, J.; Zhang, W.T.; Zhou, B.H.; Gao, J.; et al. PNOC Expressed by B Cells in Cholangiocarcinoma Was Survival Related and LAIR2 Could Be a T Cell Exhaustion Biomarker in Tumor Microenvironment: Characterization of Immune Microenvironment Combining Single-Cell and Bulk Sequencing Technology. Front. Immunol. 2021, 12, 828. [Google Scholar] [CrossRef]
- Ren, X.W.; Zhang, L.; Zhang, Y.Y.; Li, Z.Y.; Siemers, N.; Zhang, Z.M. Insights Gained from Single-Cell Analysis of Immune Cells in the Tumor Microenvironment. Annu. Rev. Immunol. 2021, 39, 583–609. [Google Scholar] [CrossRef] [PubMed]
- Kashima, Y.; Togashi, Y.; Fukuoka, S.; Kamada, T.; Irie, T.; Suzuki, A.; Nakamura, Y.; Shitara, K.; Minamide, T.; Yoshida, T.; et al. Potentiality of multiple modalities for single-cell analyses to evaluate the tumor microenvironment in clinical specimens. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, J.X.; Huang, Y.X.; Shangguan, D.G.; Zhang, P. Single-Cell Profiling to Explore Immunological Heterogeneity of Tumor Microenvironment in Breast Cancer. Front. Immunol. 2021, 12, 471. [Google Scholar] [CrossRef]
- Feng, B.H.; Hess, J. Immune-Related Mutational Landscape and Gene Signatures: Prognostic Value and Therapeutic Impact for Head and Neck Cancer. Cancers 2021, 13, 1162. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, P.; Lee, Y.G.; Kim, K.H.; Ruella, M. The current landscape of single-cell transcriptomics for cancer immunotherapy. J. Exp. Med. 2021, 218, e20201574. [Google Scholar] [CrossRef]
- Dai, Z.; Gu, X.Y.; Xiang, S.Y.; Gong, D.D.; Man, C.F.; Fan, Y. Research and application of single-cell sequencing in tumor heterogeneity and drug resistance of circulating tumor cells. Biomark. Res. 2020, 8, 1–8. [Google Scholar] [CrossRef]
- Armand, E.J.; Li, J.H.; Xie, F.M.; Luo, C.Y.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef]
- Iqbal, M.M.; Hurgobin, B.; Holme, A.L.; Appels, R.; Kaur, P. Status and Potential of Single-Cell Transcriptomics for Understanding Plant Development and Functional Biology. Cytom. Part A 2020, 97, 997–1006. [Google Scholar] [CrossRef]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Ma, S.X.; Lim, S.B. Single-Cell RNA Sequencing in Parkinson’s Disease. Biomedicines 2021, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Ma, A.J.; Chang, Y.Z.; Gong, J.T.; Jiang, Y.X.; Qi, R.; Wang, C.K.; Fu, H.J.; Ma, Q.; Xu, D. scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Almeida, N.; Chung, M.W.H.; Drudi, E.M.; Engquist, E.N.; Hamrud, E.; Isaacson, A.; Tsang, V.S.K.; Watt, F.M.; Spagnoli, F.M. Employing core regulatory circuits to define cell identity. EMBO J. 2021, 40, e106785. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 6, 1–18. [Google Scholar] [CrossRef]
- Waylen, L.N.; Nim, H.T.; Martelotto, L.G.; Ramialison, M. From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Savulescu, A.F.; Jacobs, C.; Negishi, Y.; Davignon, L.; Mhlanga, M.M. Pinpointing Cell Identity in Time and Space. Front. Mol. Biosci. 2020, 7, 209. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Lu, X.Y.; Shao, X.; Zhu, L.; Fan, X.H. Uncovering an Organ’s Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics. Trends Biotechnol. 2021, 39, 43–58. [Google Scholar] [CrossRef]
- Chen, Y.W.; Song, J.; Ruan, Q.Y.; Zeng, X.; Wu, L.L.; Cai, L.F.; Wang, X.Q.; Yang, C.Y. Single-Cell Sequencing Methodologies: From Transcriptome to Multi-Dimensional Measurement. Small Methods 2021, 5, 2100111. [Google Scholar] [CrossRef]
- Troskie, R.L.; Jafrani, Y.; Mercer, T.R.; Ewing, A.D.; Faulkner, G.J.; Cheetham, S.W. Long-read cDNA sequencing identifies functional pseudogenes in the human transcriptome. Genome Biol. 2021, 22, 1–15. [Google Scholar] [CrossRef]
- Jin, S.Q.; Guerrero-Juarez, C.F.; Zhang, L.H.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Turei, D.; Valdeolivas, A.; Gul, L.; Palacio-Escat, N.; Klein, M.; Ivanova, O.; Olbei, M.; Gabor, A.; Theis, F.; Korcsmaros, T.; et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 2021, 17, e9923. [Google Scholar] [CrossRef] [PubMed]
- Ghoshdastider, U.; Rohatgi, N.; Naeini, M.M.; Baruah, P.; Revkov, E.; Guo, Y.A.; Rizzetto, S.; Wong, A.M.L.; Solai, S.; Nguyen, T.T.; et al. Pan-Cancer Analysis of Ligand-Receptor Cross-talk in the Tumor Microenvironment. Cancer Res. 2021, 81, 1802–1812. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Reyes-Robles, T.; Fadeyi, O.O.; Oslund, R.C. Strategies for monitoring cell-cell interactions. Nat. Chem. Biol. 2021, 17, 641–652. [Google Scholar] [CrossRef]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Shao, X.; Lu, X.Y.; Liao, J.; Chen, H.J.; Fan, X.H. New avenues for systematically inferring cell-cell communication: Through single-cell transcriptomics data. Protein Cell 2020, 11, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. Single-Cell Transcriptomics Supports a Role of CHD8 in Autism. Int. J. Mol. Sci. 2021, 22, 3261. [Google Scholar] [CrossRef] [PubMed]
- Xin, R.J.; Gao, Y.; Gao, Y.; Wang, R.; Kadash-Edmondson, K.E.; Liu, B.; Wang, Y.D.; Lin, L.; Xing, Y. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ilgisonis, E.; Vavilov, N.; Ponomarenko, E.; Lisitsa, A.; Poverennaya, E.; Zgoda, V.; Radko, S.; Archakov, A. Genome of the Single Human Chromosome 18 as a “Gold Standard” for Its Transcriptome. Front. Genet. 2021, 12, 958. [Google Scholar] [CrossRef]
- Bobrovskikh, A.; Doroshkov, A.; Mazzoleni, S.; Carteni, F.; Giannino, F.; Zubairova, U. A Sight on Single-Cell Transcriptomics in Plants Through the Prism of Cell-Based Computational Modeling Approaches: Benefits and Challenges for Data Analysis. Front. Genet. 2021, 12, 771. [Google Scholar] [CrossRef]
- Liu, J.J.; Fan, Z.W.; Zhao, W.L.; Zhou, X.B. Machine Intelligence in Single-Cell Data Analysis: Advances and New Challenges. Front. Genet. 2021, 12, 807. [Google Scholar] [CrossRef]
- Xiang, R.Z.; Wang, W.C.; Yang, L.; Wang, S.Y.; Xu, C.H.; Chen, X.W. A Comparison for Dimensionality Reduction Methods of Single-Cell RNA-seq Data. Front. Genet. 2021, 12, 646936. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.F.; Zhang, W.W.; Sun, X.Q.; Nan, S.W.; Wei, N.N.; Wu, H.J.; Zheng, X.Q. Deconvolution of heterogeneous tumor samples using partial reference signals. Plos Comput. Biol. 2020, 16, e1008452. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Ma, Y.L.; Huang, Y.K.; Zhang, Y.; Jiang, Q.; Zhou, M.; Su, J.Z. Benchmarking algorithms for pathway activity transformation of single-cell RNA-seq data. Comput. Struct. Biotec. 2020, 18, 2953–2961. [Google Scholar] [CrossRef]
- Schlieben, L.D.; Prokisch, H.; Yepez, V.A. How Machine Learning and Statistical Models Advance Molecular Diagnostics of Rare Disorders Via Analysis of RNA Sequencing Data. Front. Mol. Biosci. 2021, 8, 647277. [Google Scholar] [CrossRef]
- Seong, H.J.; Han, S.W.; Sul, W.J. Prokaryotic DNA methylation and its functional roles. J. Microbiol. 2021, 59, 242–248. [Google Scholar] [CrossRef]
- Cao, B.; Wu, X.L.; Zhou, J.L.; Wu, H.; Liu, L.L.; Zhang, Q.H.; DeMott, M.S.; Gu, C.; Wang, L.R.; You, D.L.; et al. Nick-seq for single-nucleotide resolution genomic maps of DNA modifications and damage. Nucleic Acids Res. 2020, 48, 6715–6725. [Google Scholar] [CrossRef]
- Wei, Y.; Huang, Q.Q.; Tian, X.H.; Zhang, M.M.; He, J.K.; Chen, X.X.; Chen, C.; Deng, Z.X.; Li, Z.Q.; Chen, S.; et al. Single-molecule optical mapping of the distribution of DNA phosphorothioate epigenetics. Nucleic Acids Res. 2021, 49, 3672–3680. [Google Scholar] [CrossRef]
- Tourancheau, A.; Mead, E.A.; Zhang, X.S.; Fang, G. Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat. Methods 2021, 18, 491–498. [Google Scholar] [CrossRef]
- Beaulaurier, J.; Schadt, E.E.; Fang, G. Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet. 2019, 20, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Mannweiler, O.; Pinto-Carbo, M.; Lardi, M.; Agnoli, K.; Eberl, L. Investigation of Burkholderia cepacia Complex Methylomes via Single-Molecule, Real-Time Sequencing and Mutant Analysis. J. Bacteriol. 2021, 203, e00683-20. [Google Scholar] [CrossRef]
- Payelleville, A.; Brillard, J. Novel Identification of Bacterial Epigenetic Regulations Would Benefit From a Better Exploitation of Methylomic Data. Front. Microbiol. 2021, 12, 1205. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Fang, G. Conserved DNA Methyltransferases: A Window into Fundamental Mechanisms of Epigenetic Regulation in Bacteria. Trends Microbiol. 2021, 29, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Spadar, A.; Perdigao, J.; Phelan, J.; Charleston, J.; Modesto, A.; Elias, R.; de Sessions, P.F.; Hibberd, M.L.; Campino, S.; Duarte, A.; et al. Methylation analysis of Klebsiella pneumoniae from Portuguese hospitals. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Morovic, W.; Budinoff, C.R. Epigenetics: A New Frontier in Probiotic Research. Trends Microbiol. 2021, 29, 117–126. [Google Scholar] [CrossRef]
- Carter, M.Q.; Pham, A.; Huynh, S.; Parker, C.T.; Miller, A.; He, X.H.; Hu, B.; Chain, P.S.G. DNA adenine methylase, not the PstI restriction-modification system, regulates virulence gene expression in Shiga toxin-producing Escherichia coli. Food Microbiol. 2021, 96, 103722. [Google Scholar] [CrossRef] [PubMed]
- Modlin, S.J.; Conkle-Gutierrez, D.; Kim, C.; Mitchell, S.N.; Morrissey, C.; Weinrick, B.C.; Jacobs, W.R.; Ramirez-Busby, S.M.; Hoffner, S.E.; Valafar, F. Drivers and sites of diversity in the DNA adenine methylomes of 93 Mycobacterium tuberculosis complex clinical isolates. Elife 2020, 9, e58542. [Google Scholar] [CrossRef]
- Gaultney, R.A.; Vincent, A.T.; Lorioux, C.; Coppee, J.Y.; Sismeiro, O.; Varet, H.; Legendre, R.; Cockram, C.A.; Veyrier, F.J.; Picardeau, M. 4-Methylcytosine DNA modification is critical for global epigenetic regulation and virulence in the human pathogen Leptospira interrogans. Nucleic Acids Res. 2020, 48, 12102–12115. [Google Scholar] [CrossRef] [PubMed]
- Allue-Guardia, A.; Garcia, J.I.; Torrelles, J.B. Evolution of Drug-Resistant Mycobacterium tuberculosis Strains and Their Adaptation to the Human Lung Environment. Front. Microbiol. 2021, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.R.; Xiao, R.; Hamilton-Brehm, S.D. Hybrid genome de novo assembly with methylome analysis of the anaerobic thermophilic subsurface bacterium Thermanaerosceptrum fracticalcis strain DRI-13(T). Bmc Genom. 2021, 22, 1–16. [Google Scholar] [CrossRef]
- Choi, W.L.; Mok, Y.G.; Huh, J.H. Application of 5-Methylcytosine DNA Glycosylase to the Quantitative Analysis of DNA Methylation. Int. J. Mol. Sci. 2021, 22, 1072. [Google Scholar] [CrossRef] [PubMed]
- Usai, G.; Vangelisti, A.; Simoni, S.; Giordani, T.; Natali, L.; Cavallini, A.; Mascagni, F. DNA Modification Patterns within the Transposable Elements of the Fig (Ficus carica L.) Genome. Plants 2021, 10, 451. [Google Scholar] [CrossRef]
- Liu, J.Z.; He, Z.H. Small DNA Methylation, Big Player in Plant Abiotic Stress Responses and Memory. Front. Plant Sci. 2020, 11, 1977. [Google Scholar] [CrossRef]
- Rojas-Rojas, F.U.; Vega-Arreguin, J.C. Epigenetic insight into regulatory role of chromatin covalent modifications in lifecycle and virulence of Phytophthora. Env. Microbiol. Rep. 2021, 13, 445–457. [Google Scholar] [CrossRef]
- Reva, O.N.; Larisa, S.A.; Mwakilili, A.D.; Tibuhwa, D.D.; Lyantagaye, S.; Chan, W.Y.; Lutz, S.; Ahrens, C.H.; Vater, J.; Borriss, R. Complete genome sequence and epigenetic profile of Bacillus velezensis UCMB5140 used for plant and crop protection in comparison with other plant-associated Bacillus strains. Appl. Microbiol. Biot. 2020, 104, 7643–7656. [Google Scholar] [CrossRef]
- Ashe, A.; Colot, V.; Oldroyd, B.P. How does epigenetics influence the course of evolution? Philos. T R Soc. B 2021, 376, 20200111. [Google Scholar] [CrossRef]
- Loughland, I.; Little, A.; Seebacher, F. DNA methyltransferase 3a mediates developmental thermal plasticity. Bmc Biol. 2021, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.; Ben Maamar, M.; Skinner, M.K. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics 2021, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paun, O.; Verhoeven, K.J.F.; Richards, C.L. Opportunities and limitations of reduced representation bisulfite sequencing in plant ecological epigenomics. New Phytol. 2019, 221, 738–742. [Google Scholar] [CrossRef] [Green Version]
- Horemans, N.; Spurgeon, D.J.; Lecomte-Pradines, C.; Saenen, E.; Bradshaw, C.; Oughton, D.; Rasnaca, I.; Kamstra, J.H.; Adam-Guillermin, C. Current evidence for a role of epigenetic mechanisms in response to ionizing radiation in an ecotoxicological context. Environ. Pollut. 2019, 251, 469–483. [Google Scholar] [CrossRef]
- Belli, M.; Tabocchini, M.A. Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. Int. J. Mol. Sci. 2020, 21, 5993. [Google Scholar] [CrossRef]
- Cabrera-Licona, A.; Perez-Anorve, X.I.; Flores-Fortis, M.; del Moral-Hernandez, O.; Gonzalez-de la Rosa, C.H.; Suarez-Sanchez, R.; Chavez-Saldana, M.; Arechaga-Ocampo, E. Deciphering the epigenetic network in cancer radioresistance. Radiother Oncol. 2021, 159, 48–59. [Google Scholar] [CrossRef]
- Schang, A.L.; Saberan-Djoneidi, D.; Mezger, V. The impact of epigenomic next-generation sequencing approaches on our understanding of neuropsychiatric disorders. Clin. Genet. 2018, 93, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Wagh, K.; Ishikawa, M.; Garcia, D.A.; Stavreva, D.A.; Upadhyaya, A.; Hager, G.L. Mechanical Regulation of Transcription: Recent Advances. Trends Cell Biol. 2021, 31, 457–472. [Google Scholar] [CrossRef]
- Gorini, F.; Scala, G.; Cooke, M.S.; Majello, B.; Amente, S. Towards a comprehensive view of 8-oxo-7,8-dihydro-2′-deoxyguanosine: Highlighting the intertwined roles of DNA damage and epigenetics in genomic instability. DNA Repair 2021, 97, 103027. [Google Scholar] [CrossRef]
- Scott, C.A.; Duryea, J.D.; MacKay, H.; Baker, M.S.; Laritsky, E.; Gunasekara, C.J.; Coarfa, C.; Waterland, R.A. Identification of cell type-specific methylation signals in bulk whole genome bisulfite sequencing data. Genome Biol. 2020, 21, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Men, S.; Yu, Y.Y. Prospects for Use of Single-Cell Sequencing to Assess DNA Methylation in Asthma. Med. Sci. Monitor 2020, 26, e925514-1. [Google Scholar] [CrossRef]
- Kamies, R.; Martinez-Jimenez, C.P. Advances of single-cell genomics and epigenomics in human disease: Where are we now? Mamm. Genome 2020, 31, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Jonaitis, P.; Kupcinskas, L.; Kupcinskas, J. Molecular Alterations in Gastric Intestinal Metaplasia. Int. J. Mol. Sci. 2021, 22, 5758. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Cheng, J.; Zhu, Z.A.; Catalfamo, M.; Goerlitz, D.; Lawless, O.J.; Tallon, L.; Sadzewicz, L.; Calderone, R.; Bellanti, J.A. Treg-inducing capacity of genomic DNA of Bifidobacterium longum subsp. infantis. Allergy Asthma Proc. 2020, 41, 372–385. [Google Scholar] [CrossRef]
- Bellanti, J.A.; Li, D.M. T Regulatory Cells in Human Health and Diseases. In Advances in Experimental Medicine and Biology; Zheng, S.G., Ed.; Springer: Singapore, 2021; Volume 1278, pp. 1–302. [Google Scholar]
- Trevino, A.E.; Sinnott-Armstrong, N.; Andersen, J.; Yoon, S.J.; Huber, N.; Pritchard, J.K.; Chang, H.Y.; Greenleaf, W.J.; Pasca, S.P. Chromatin accessibility dynamics in a model of human forebrain development. Science 2020, 367, eaay1645. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Childs, D.; Guzman-Karlsson, M.C.; Kibe, M.; Moulden, J.; Song, E.; Tahir, A.; Sweatt, J.D. DNA methylation regulates associative reward learning. Nat. Neurosci. 2013, 16, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- MacBean, L.F.; Smith, A.R.; Lunnon, K. Exploring Beyond the DNA Sequence: A Review of Epigenomic Studies of DNA and Histone Modifications in Dementia. Curr. Genet. Med. Rep. 2020, 8, 79–92. [Google Scholar] [CrossRef]
- Perkovic, M.N.; Paska, A.V.; Konjevod, M.; Kouter, K.; Strac, D.S.; Erjavec, G.N.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef]
- Zeng, S.Y.; Hua, Y.W.; Zhang, Y.; Liu, G.F.; Zhao, C.C. GLEANER: A web server for GermLine cycle Expression ANalysis and Epigenetic Roadmap visualization. Bmc Bioinform. 2021, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Khaneghah, A.M.; Fakhri, Y.; Nematollahi, A.; Seilani, F.; Vasseghian, Y. The Concentration of Acrylamide in Different Food Products: A Global Systematic Review, Meta-Analysis, and Meta-Regression. Food Rev. Int. 2020, 7, 1–19. [Google Scholar] [CrossRef]
- Seal, C.J.; de Mul, A.; Eisenbrand, G.; Haverkort, A.J.; Franke, K.; Lalljie, S.P.D.; Mykkaenen, H.; Reimerdes, E.; Scholz, G.; Somoza, V.; et al. Risk-benefit considerations of mitigation measures on acrylamide content of foods—A case study on potatoes, cereals and coffee. Brit. J. Nutr. 2008, 99, S1–S46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Non, A.L. Social epigenomics: Are we at an impasse? Epigenomics 2021. [Google Scholar] [CrossRef]
- Brasil, S.; Neves, C.J.; Rijoff, T.; Falcao, M.; Valadao, G.; Videira, P.A.; Ferreira, V.D. Artificial Intelligence in Epigenetic Studies: Shedding Light on Rare Diseases. Front. Mol. Biosci. 2021, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.C. Omics-based microbiome analysis in microbial ecology: From sequences to information. J. Microbiol. 2021, 59, 229–232. [Google Scholar] [CrossRef]
- Davey, L.; Valdivia, R.H. Bacterial genetics and molecular pathogenesis in the age of high throughput DNA sequencing. Curr. Opin. Microbiol. 2020, 54, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, J.M.; Singleton, C.; Clegg, L.A.; Petriglieri, F.; Nielsen, P.H. High Diversity and Functional Potential of Undescribed “Acidobacteriota” in Danish Wastewater Treatment Plants. Front. Microbiol. 2021, 12, 906. [Google Scholar] [CrossRef]
- Singleton, C.M.; Petriglieri, F.; Kristensen, J.M.; Kirkegaard, R.H.; Michaelsen, T.Y.; Andersen, M.H.; Kondrotaite, Z.; Karst, S.M.; Dueholm, M.S.; Nielsen, P.H.; et al. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Infante-Villamil, S.; Huerlimann, R.; Jerry, D.R. Microbiome diversity and dysbiosis in aquaculture. Rev. Aquacult. 2021, 13, 1077–1096. [Google Scholar] [CrossRef]
- Seeneevassen, L.; Bessede, E.; Megraud, F.; Lehours, P.; Dubus, P.; Varon, C. Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 3418. [Google Scholar] [CrossRef]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying biases and their potential solutions in human microbiome studies. Microbiome 2021, 9, 1–22. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Ziemski, M.; Robeson, M.S.; Kaehler, B.D. Measuring the microbiome: Best practices for developing and benchmarking microbiomics methods. Comput. Struct. Biotec. 2020, 18, 4048–4062. [Google Scholar] [CrossRef]
- Cusco, A.; Perez, D.; Vines, J.; Fabregas, N.; Francino, O. Long-read metagenomics retrieves complete single-contig bacterial genomes from canine feces. Bmc Genom. 2021, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dang, N.; Ren, D.Y.; Zhao, F.Y.; Lv, R.R.; Ma, T.; Bao, Q.H.; Menghe, B.; Liu, W.J. Comparison of Bacterial Microbiota in Raw ’Using PacBio Single Molecule Real-Time Sequencing Technology. Front. Microbiol. 2020, 11, 2708. [Google Scholar] [CrossRef]
- Marco, M.L. Defining how microorganisms benefit human health. Microb. Biotechnol. 2021, 14, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Su, L.K.; Wang, Y.; Deng, S.L. Improved High-Throughput Sequencing of the Human Oral Microbiome: From Illumina to PacBio. Can. J. Infect. Dis. Med. 2020, 2020, 13. [Google Scholar] [CrossRef]
- Xu, F.; Ge, C.T.; Li, S.T.; Tang, S.L.; Wu, X.W.; Luo, H.; Deng, X.Y.; Zhang, G.T.; Stevenson, A.; Baker, R.C. Evaluation of nanopore sequencing technology to identify Salmonella enterica Choleraesuis var. Kunzendorf and Orion var. 15(+), 34(+). Int. J. Food Microbiol. 2021, 346, 109167. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, K.; Bessarab, I.; Haryono, M.A.S.; Liu, X.H.; Zuniga-Montanez, R.E.; Roy, S.; Qiu, G.L.; Drautz-Moses, D.I.; Law, Y.Y.; Wuertz, S.; et al. Recovery of complete genomes and non-chromosomal replicons from activated sludge enrichment microbial communities with long read metagenome sequencing. Npj Biofilms Microbi. 2021, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, I.N.; Muller, C.P.; He, F.Q. Applying next-generation sequencing to unravel the mutational landscape in viral quasispecies. Virus Res. 2020, 283, 197963. [Google Scholar] [CrossRef] [PubMed]
- He, W.Q.; Gao, Y.H.; Wen, Y.Q.; Ke, X.M.; Ou, Z.J.; Li, Y.Z.; He, H.; Chen, Q. Detection of Virus-Related Sequences Associated With Potential Etiologies of Hepatitis in Liver Tissue Samples From Rats, Mice, Shrews, and Bats. Front. Microbiol. 2021, 12, 1409. [Google Scholar] [CrossRef]
- Ono, Y.; Asai, K.; Hamada, M. PBSIM2: A simulator for long-read sequencers with a novel generative model of quality scores. Bioinformatics 2021, 37, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Zablocki, O.; Michelsen, M.; Burris, M.; Solonenko, N.; Warwick-Dugdale, J.; Ghosh, R.; Pett-Ridge, J.; Sullivan, M.B.; Temperton, B. VirION2: A short- and long-read sequencing and informatics workflow to study the genomic diversity of viruses in nature. Peerj 2021, 9, e11088. [Google Scholar] [CrossRef]
- Arbas, S.M.; Busi, S.B.; Queiros, P.; de Nies, L.; Herold, M.; May, P.; Wilmes, P.; Muller, E.E.L.; Narayanasamy, S. Challenges, Strategies, and Perspectives for Reference-Independent Longitudinal Multi-Omic Microbiome Studies. Front. Genet. 2021, 12, 858. [Google Scholar] [CrossRef]
- Barry, G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol. Psychiatr. 2014, 19, 410–416. [Google Scholar] [CrossRef]
- Guennewig, B.; Cooper, A.A. The Central Role of Noncoding RNA in the Brain. Int. Rev. Neurobiol. 2014, 116, 153–194. [Google Scholar] [CrossRef] [PubMed]
- Muir, P.; Li, S.T.; Lou, S.K.; Wang, D.F.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorado, G.; Gálvez, S.; Rosales, T.E.; Vásquez, V.F.; Hernández, P. Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing – Review. Biomolecules 2021, 11, 1111. https://doi.org/10.3390/biom11081111
Dorado G, Gálvez S, Rosales TE, Vásquez VF, Hernández P. Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing – Review. Biomolecules. 2021; 11(8):1111. https://doi.org/10.3390/biom11081111
Chicago/Turabian StyleDorado, Gabriel, Sergio Gálvez, Teresa E. Rosales, Víctor F. Vásquez, and Pilar Hernández. 2021. "Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing – Review" Biomolecules 11, no. 8: 1111. https://doi.org/10.3390/biom11081111
APA StyleDorado, G., Gálvez, S., Rosales, T. E., Vásquez, V. F., & Hernández, P. (2021). Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing – Review. Biomolecules, 11(8), 1111. https://doi.org/10.3390/biom11081111