
New Hybrid Compounds Combining Fragments of Usnic Acid and Monoterpenoids for Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibition

 , , , ,
, , , ,  ,
,  ,
, 
Abstract
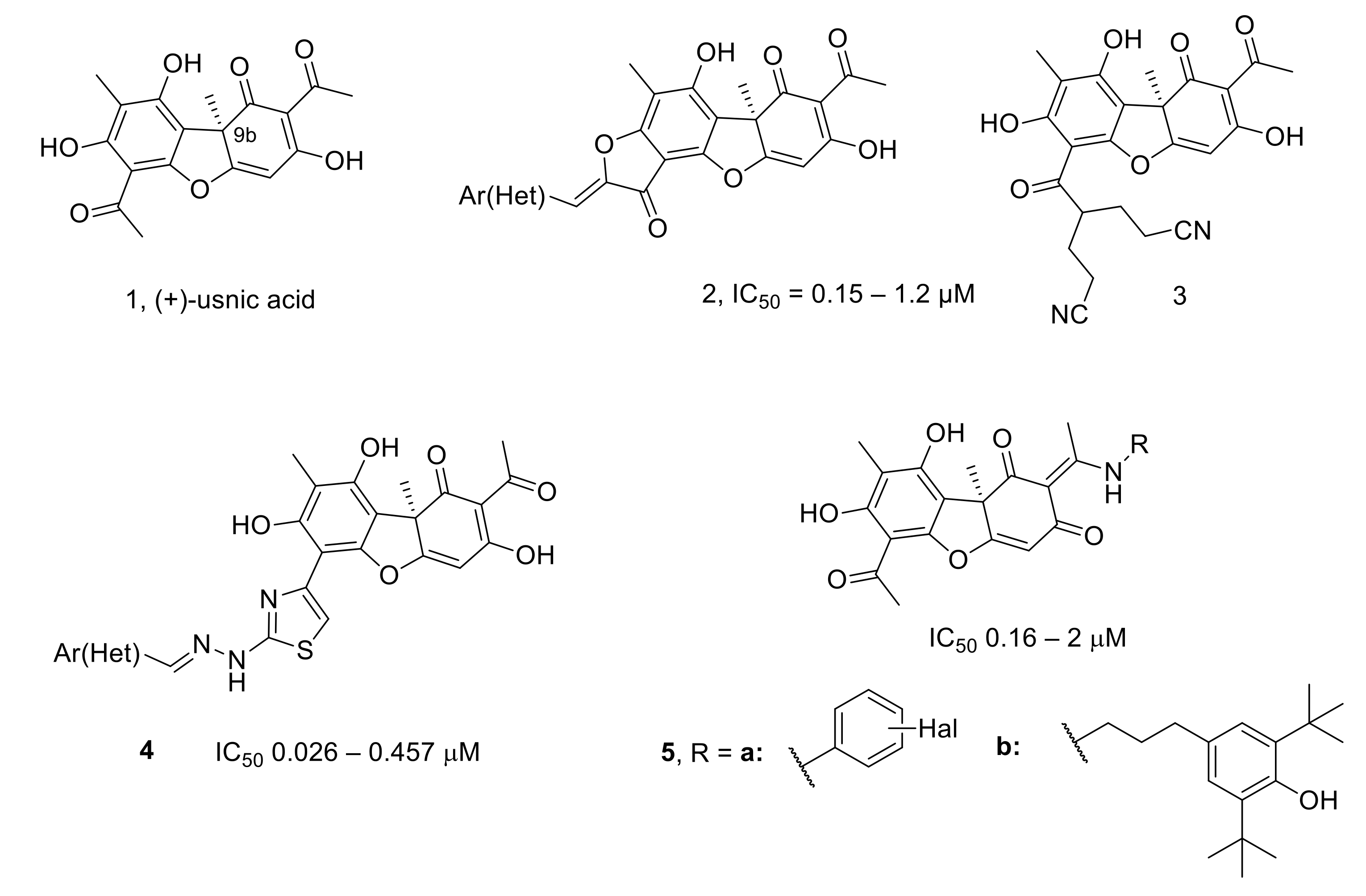
:1. Introduction
2. Materials and Methods
2.1. General Information
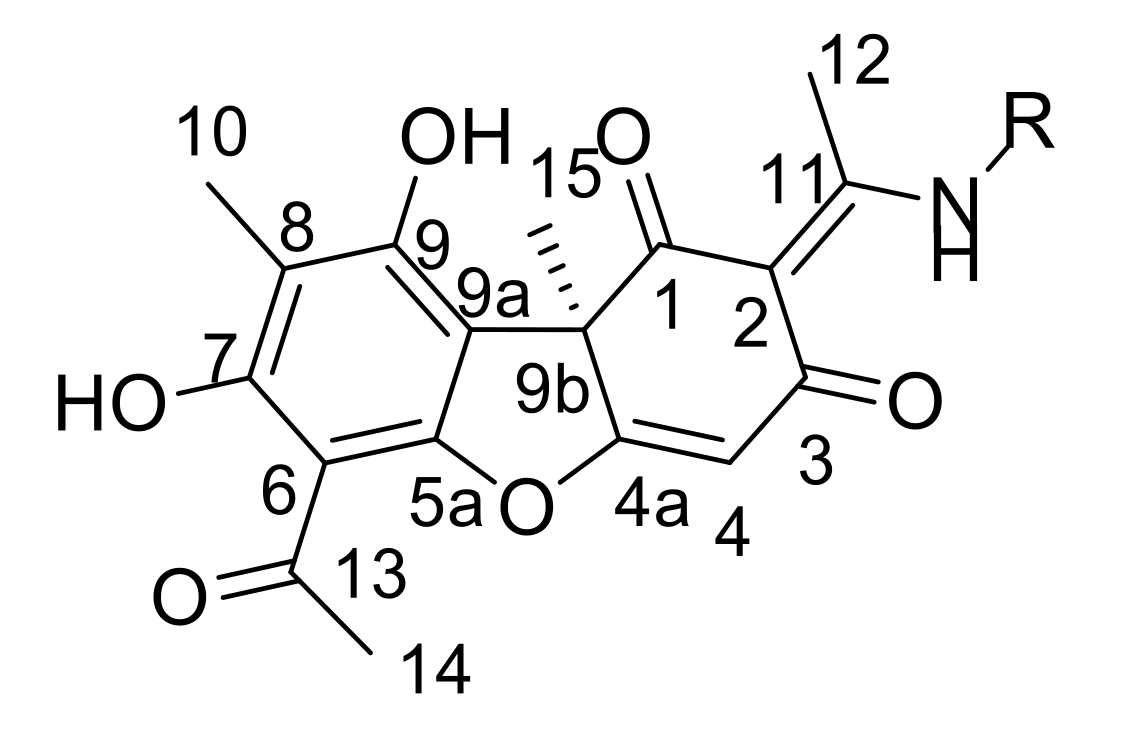
2.2. Chemistry
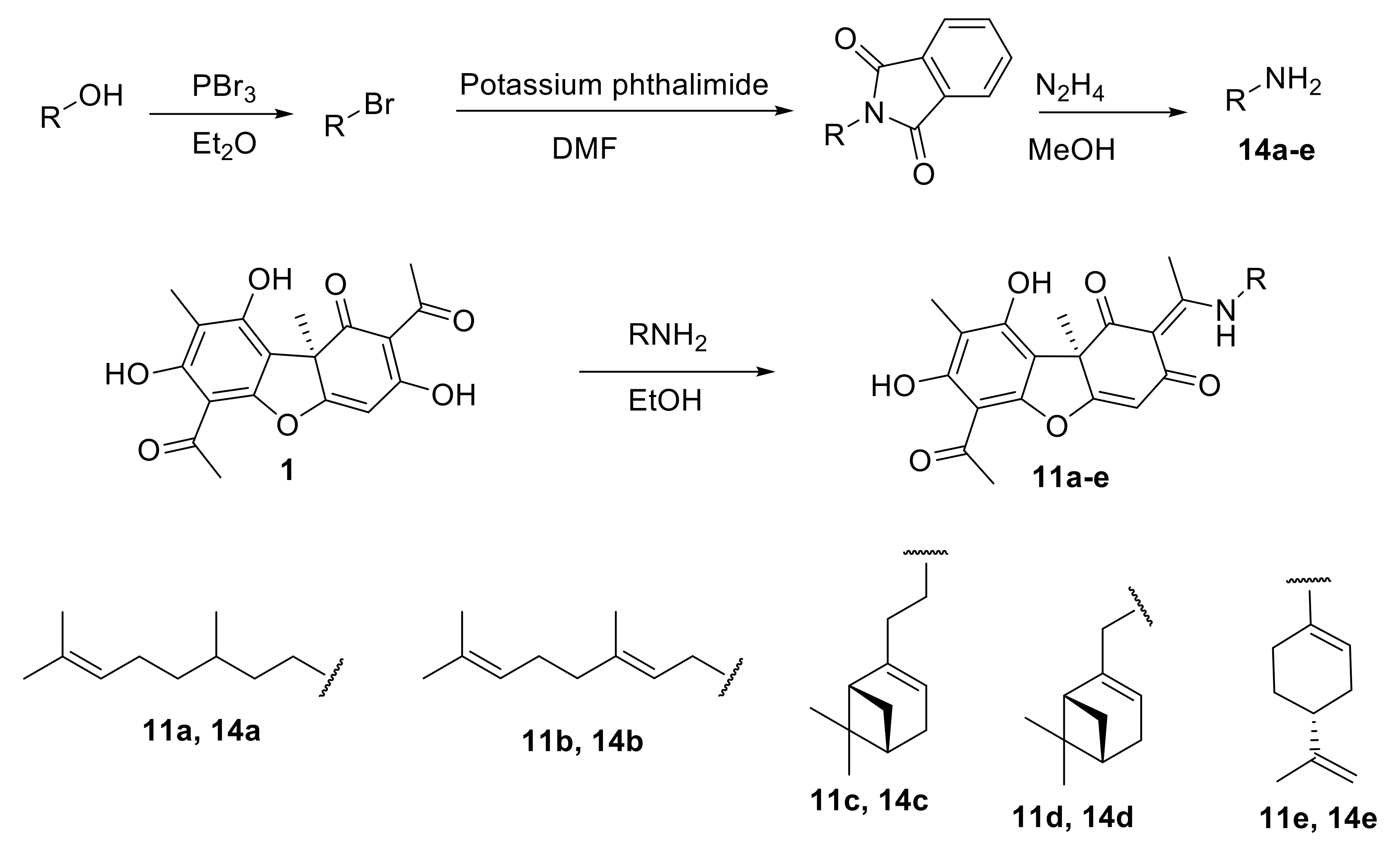
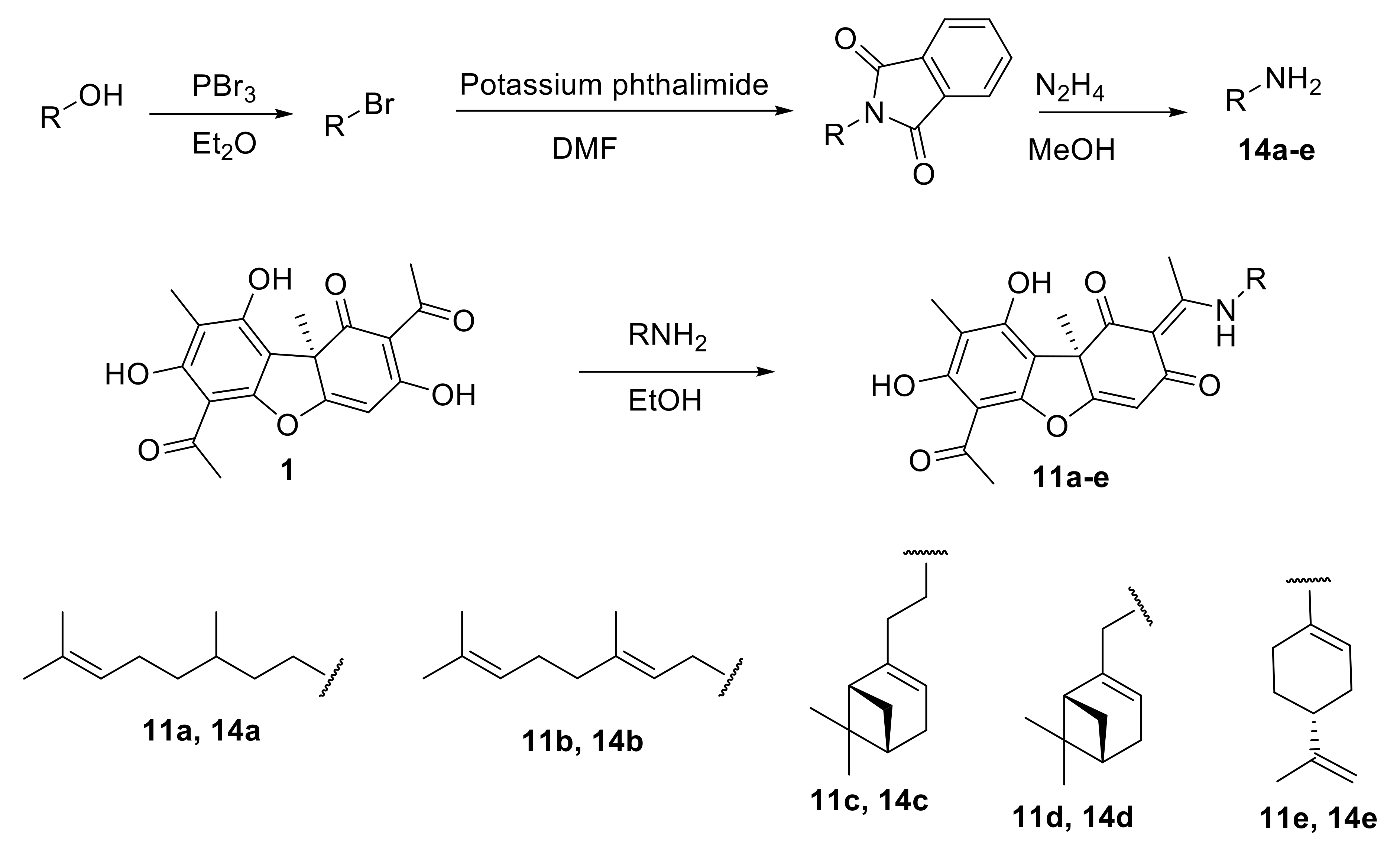
2.2.1. General Procedure for the Synthesis of Enamine Compounds 11a–e
2.2.2. NMR Spectra of Compounds 11a–e
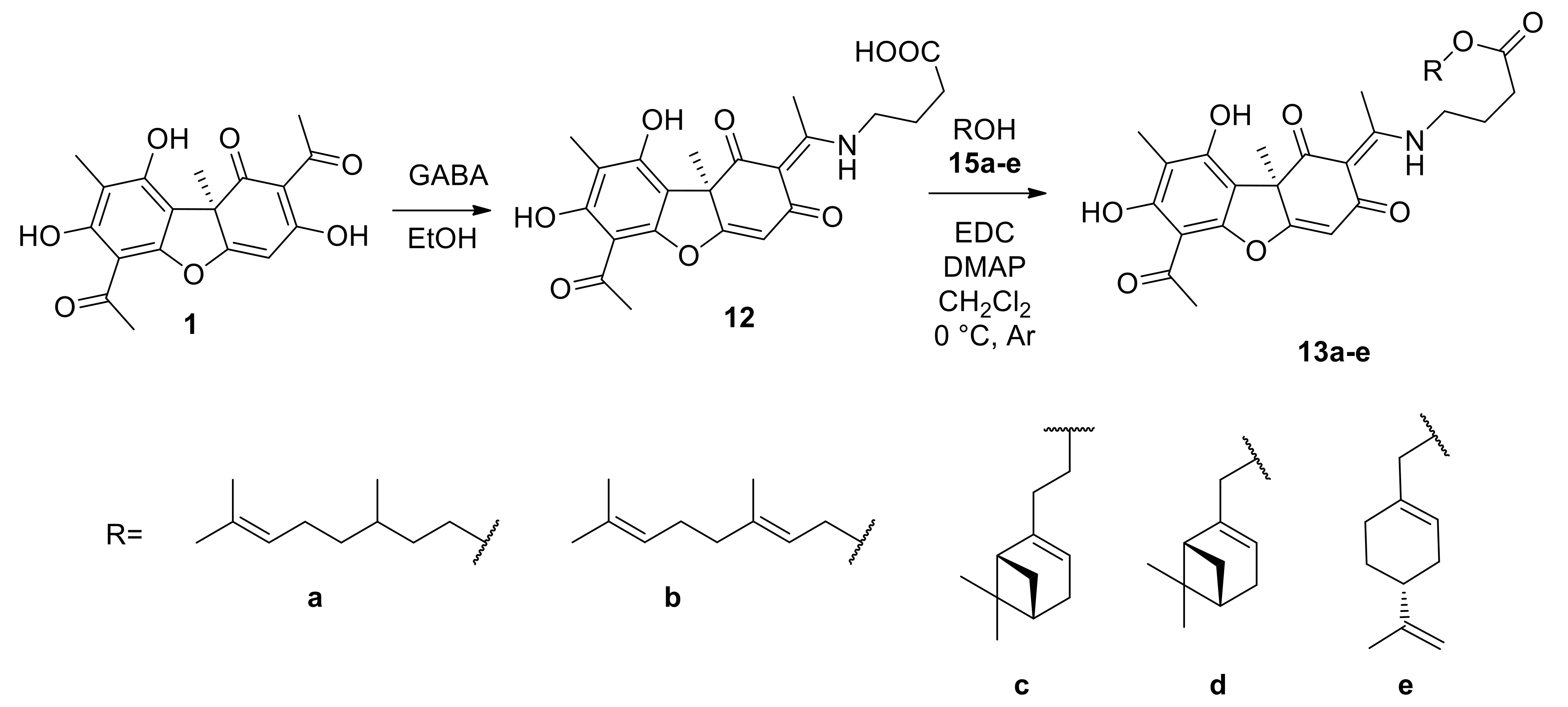
2.2.3. General Procedure for the Synthesis of Enamine Compounds 13a–e
2.2.4. NMR Spectra of Compounds 13a–e
2.3. Biology
2.3.1. Real-Time Detection of TDP1 Activity
2.3.2. TDP1 Activity by gel-Based Assay
2.3.3. TDP1 Knockout HEK293A Clones
2.3.4. Cell Culture Cytotoxicity Assay
2.4. Modelling and Screening
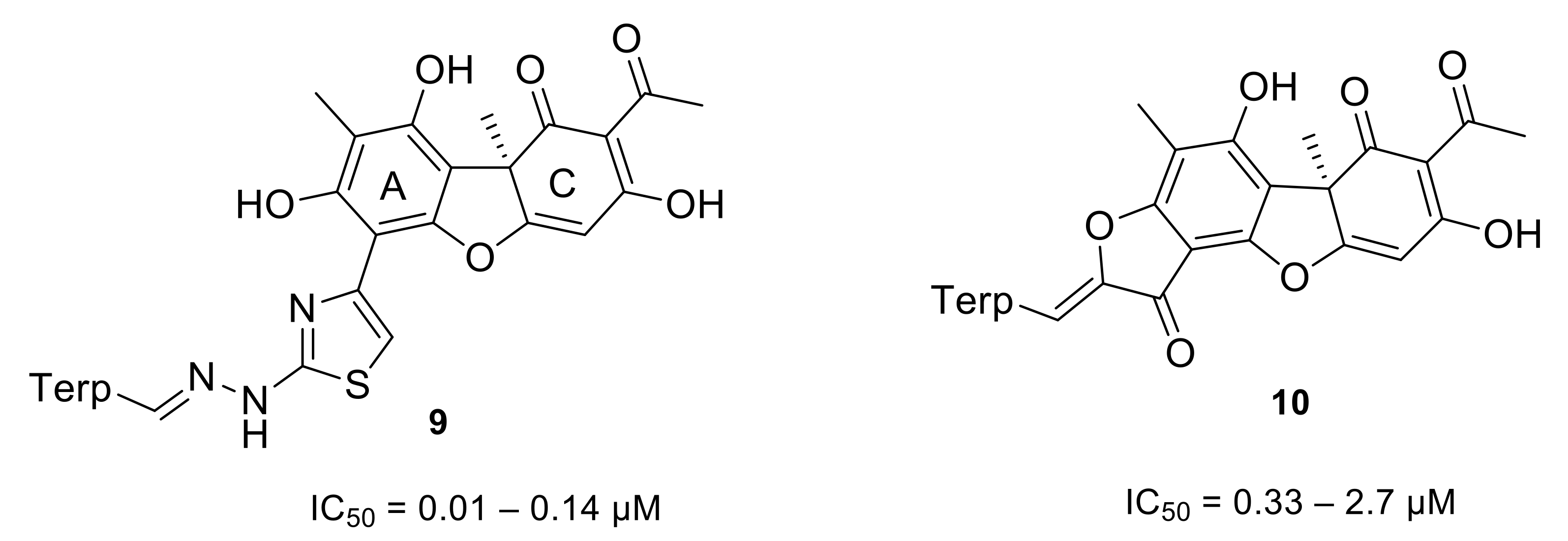
3. Results
3.1. Chemistry
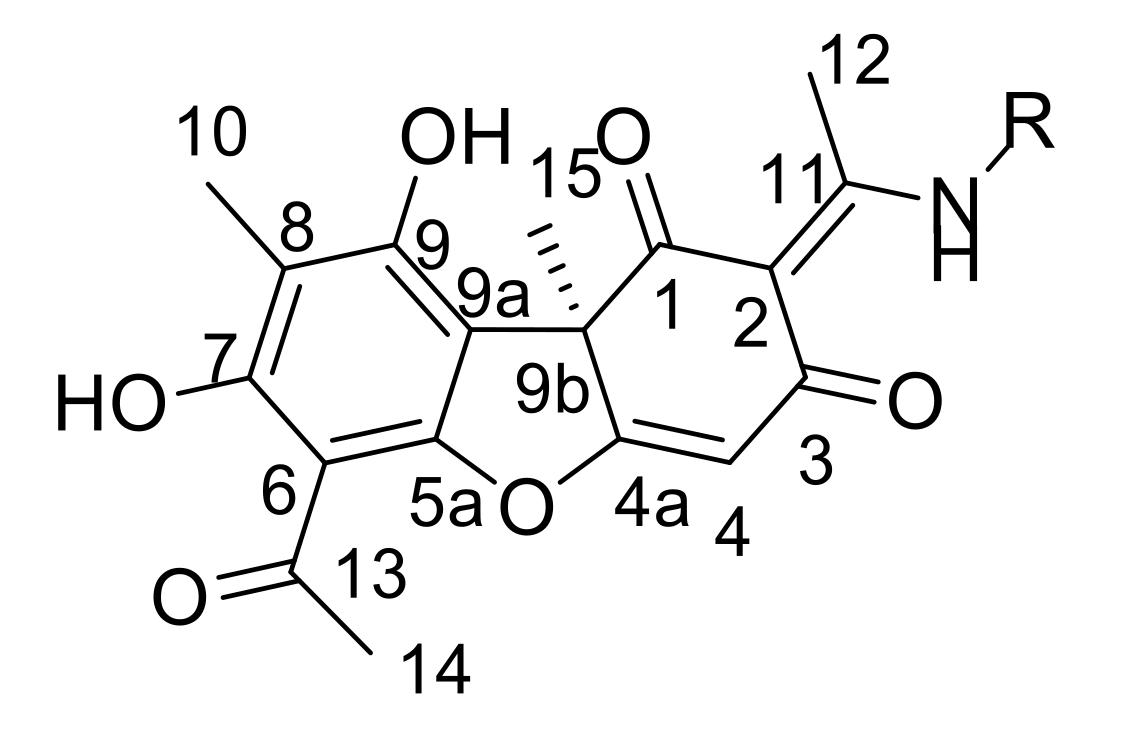
3.1.1. Synthesis of Usnic Acid Enamine Derivatives 11a–e
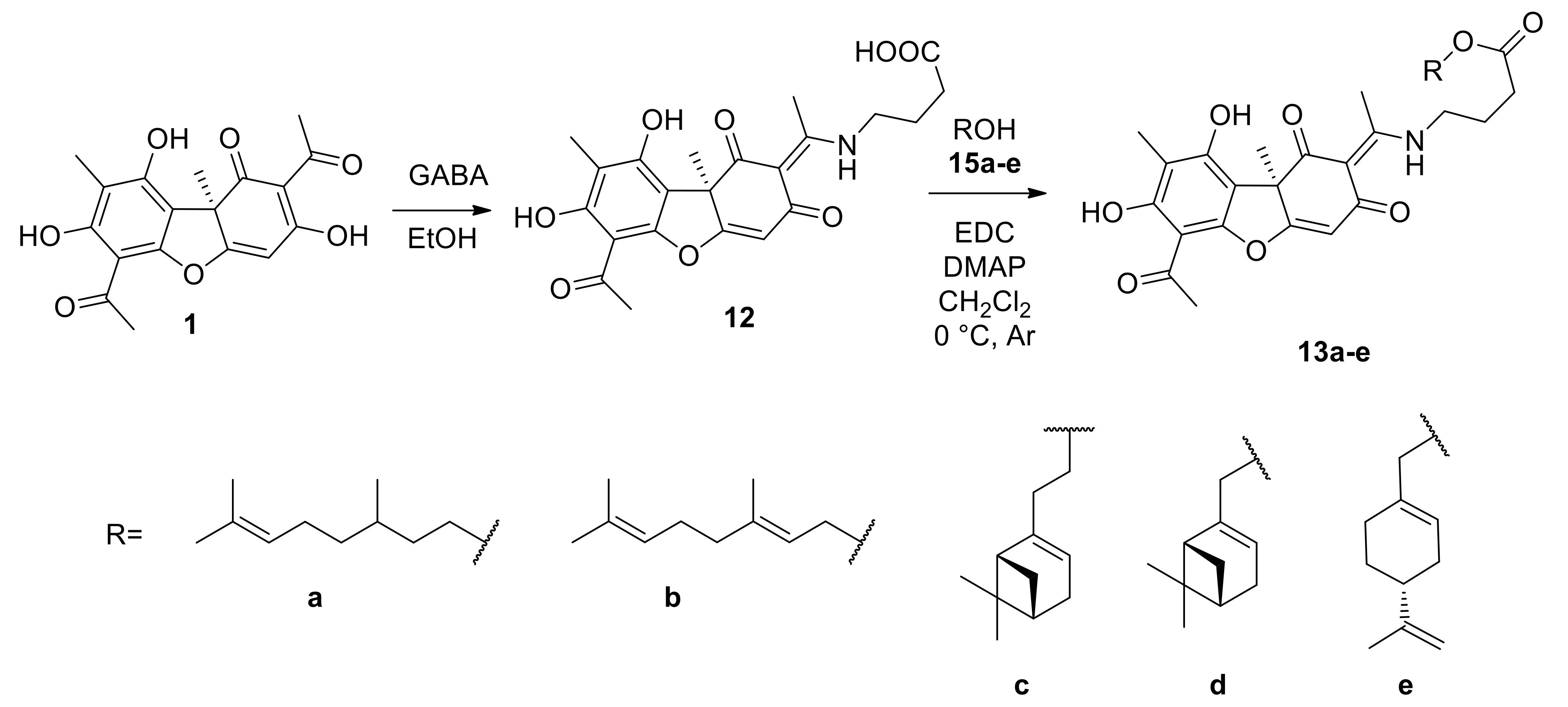
3.1.2. Synthesis of Usnic Acid Enamine Derivatives with an Ether Bond 13a–e
3.2. Biology
3.2.1. Real-Time Detection of TDP1 Activity
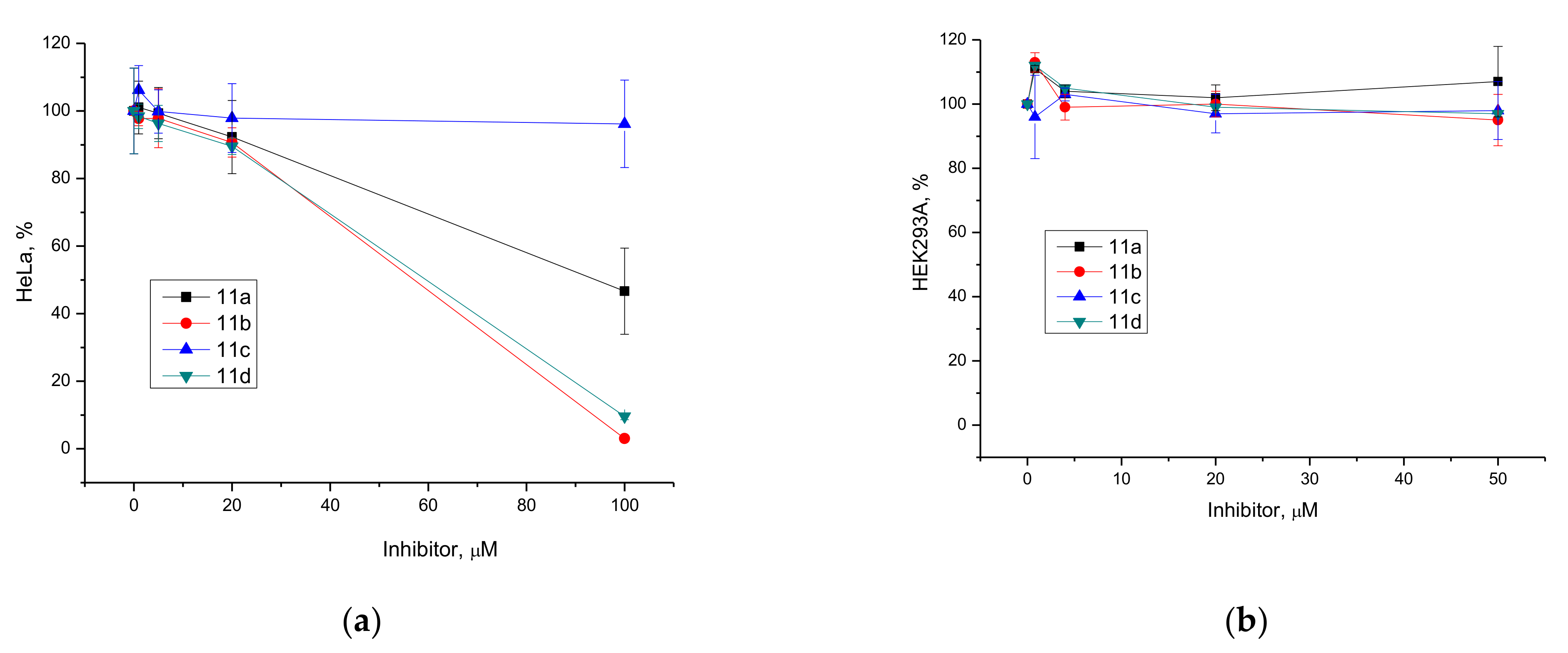
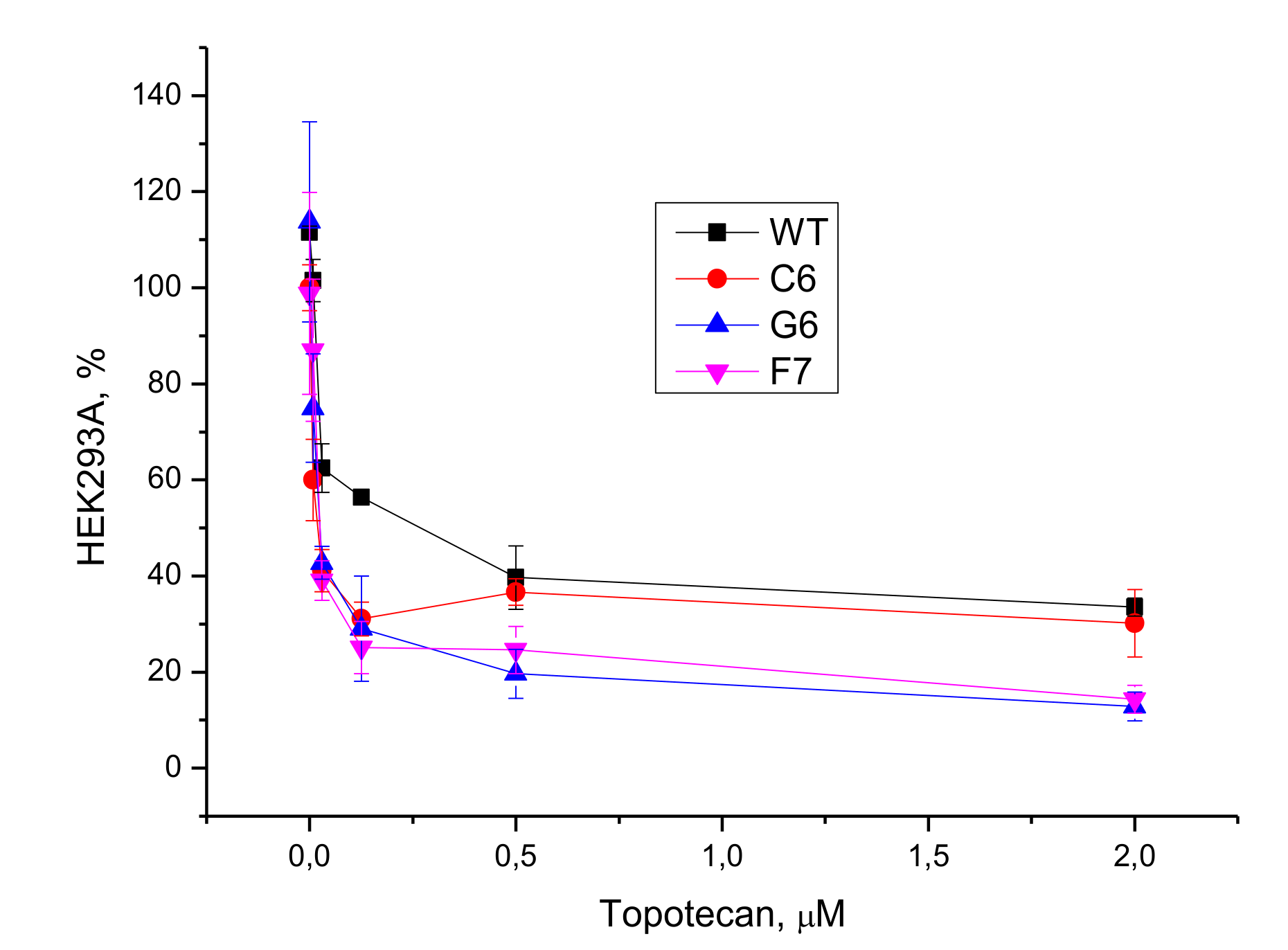
3.2.2. Cell Culture Cytotoxicity Assay
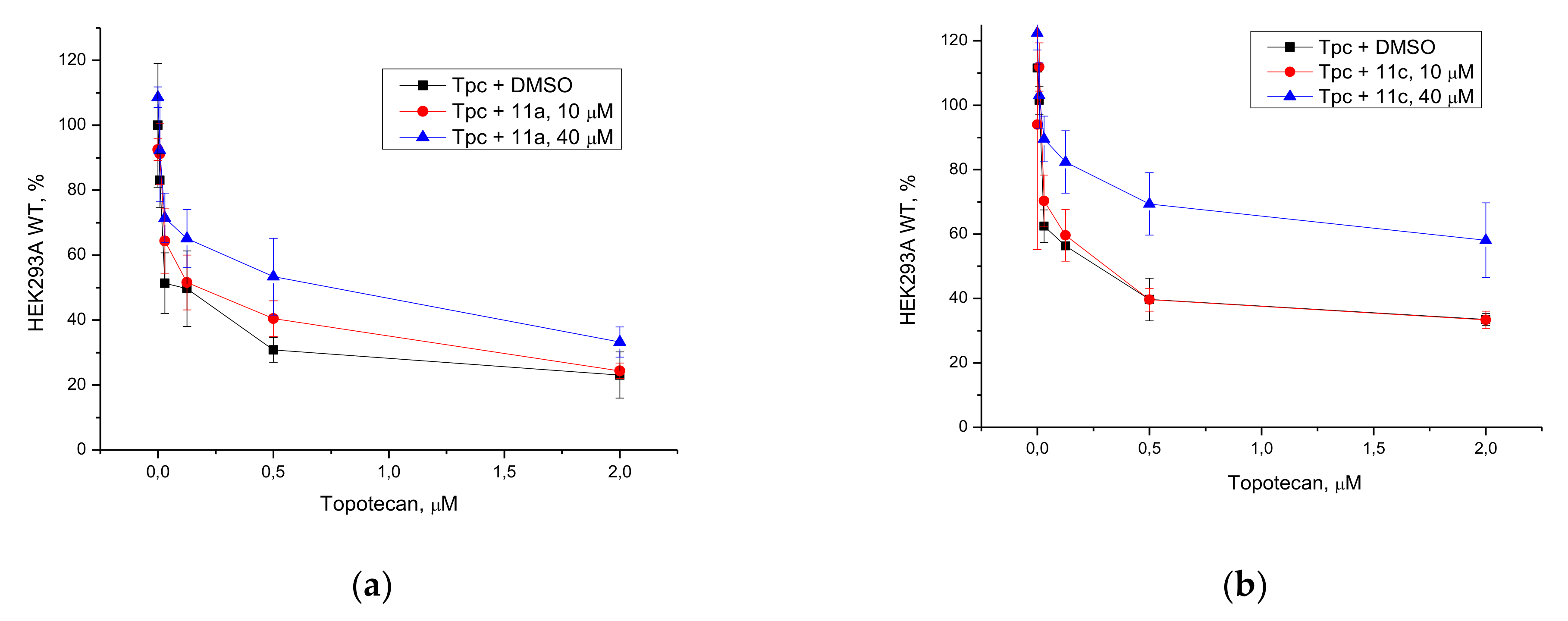
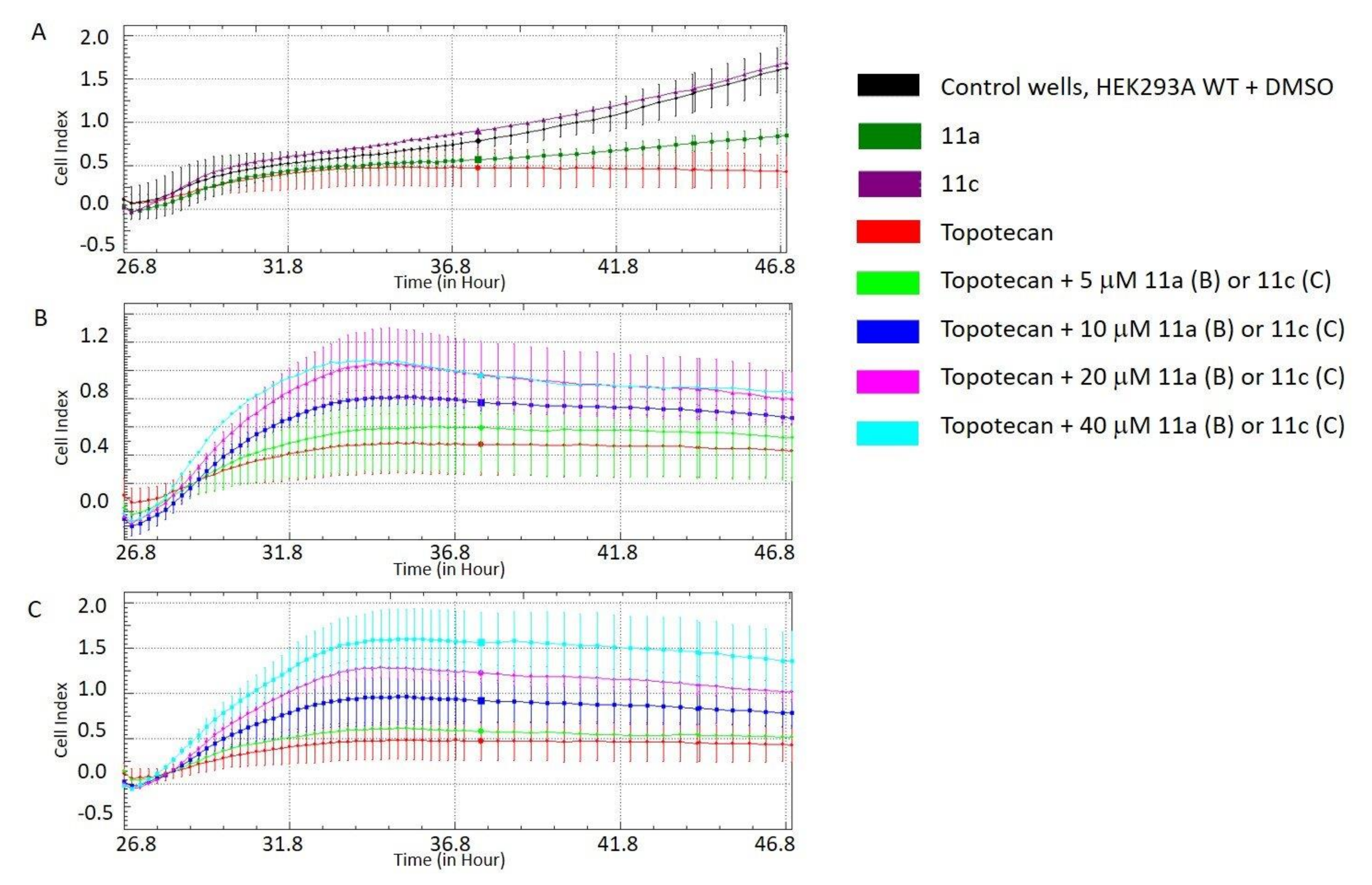
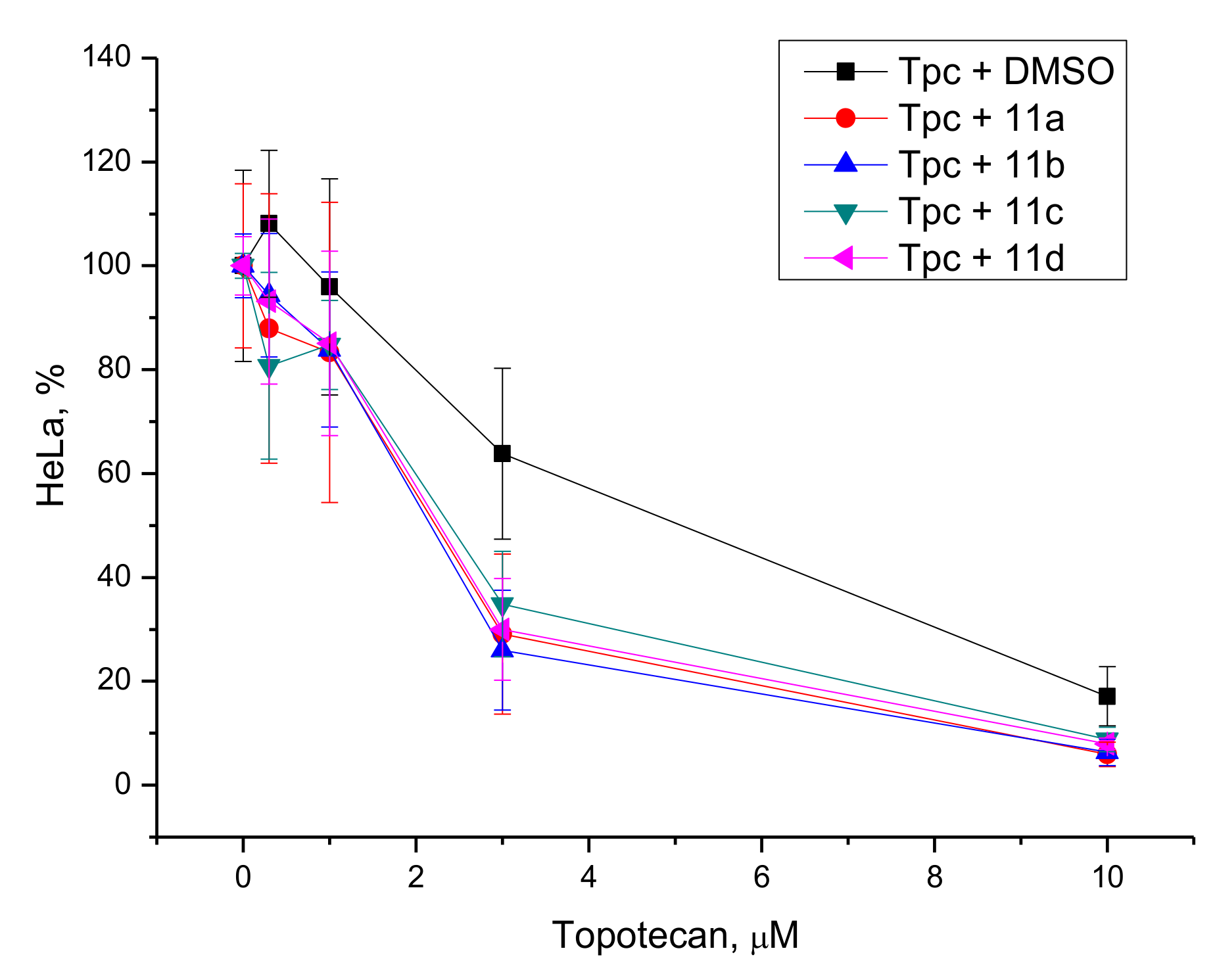
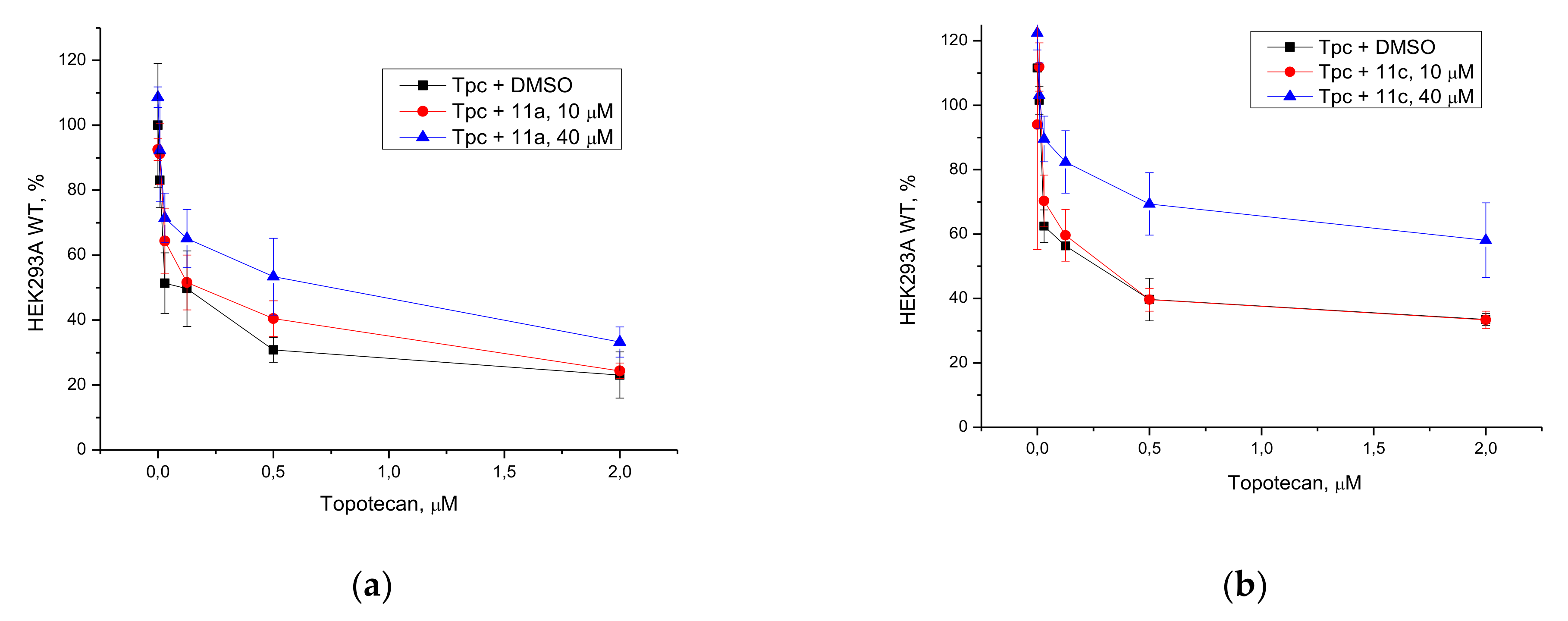
3.2.3. Potentiation with Topotecan
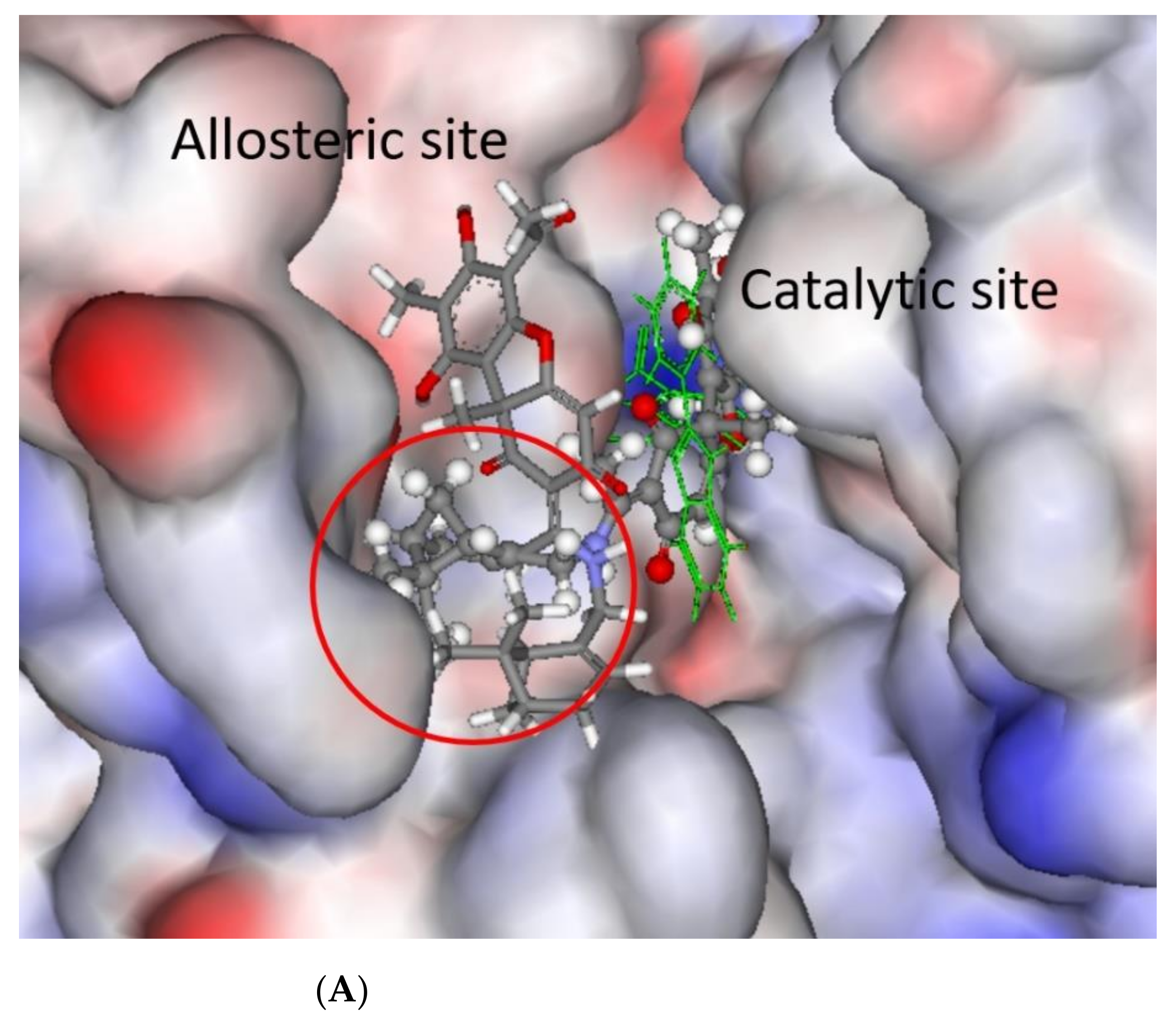
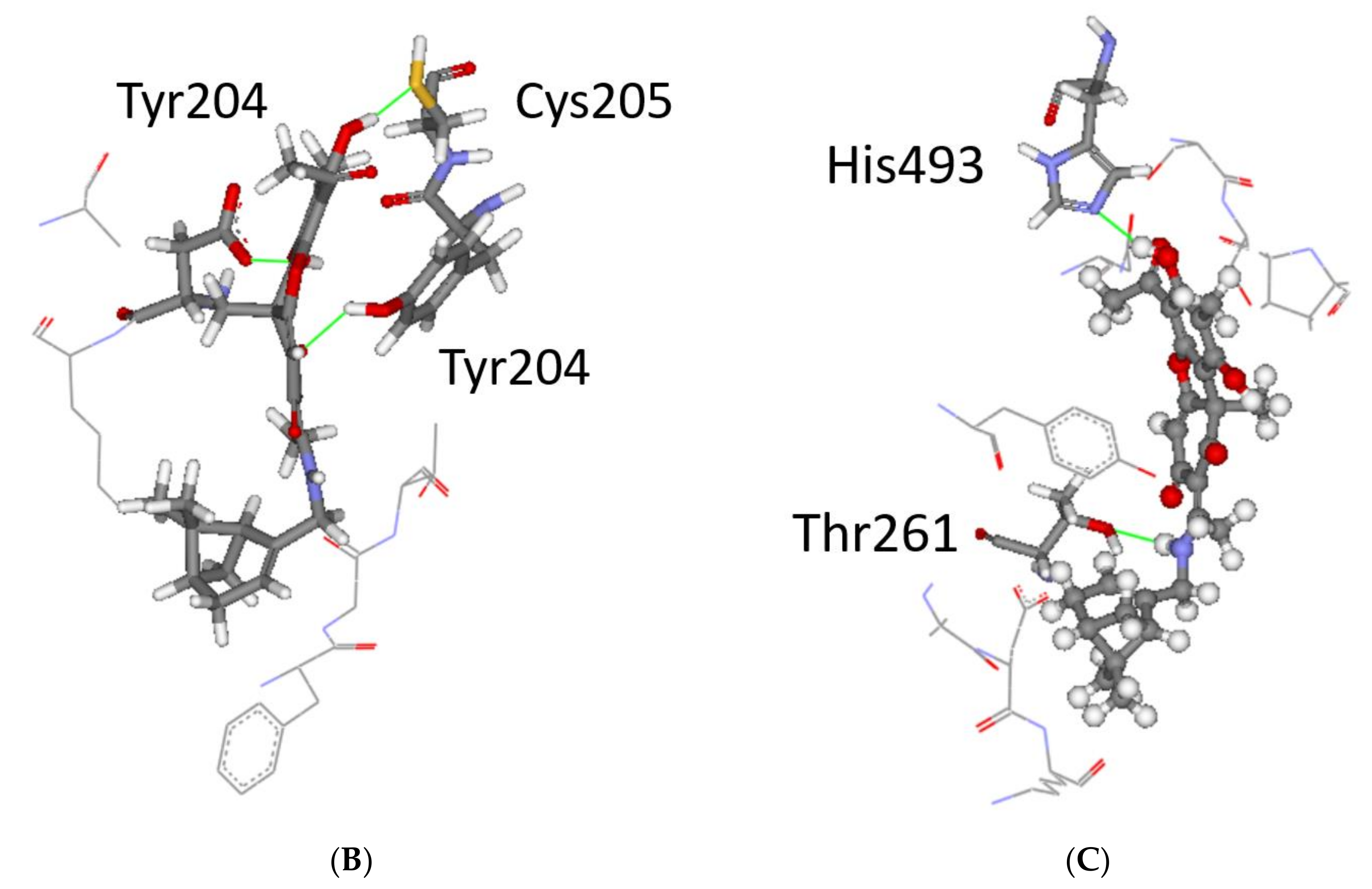
3.3. Modelling and Screening
3.4. Chemical Space
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TDP1 | tyrosyl-DNA phosphodiesterase 1: |
| TG7 | 4-[(2-phenylimidazo[1,2-a]pyridin-3-yl)amino]benzene-1,2-dicarboxylic acid; |
| TOP1 | DNA-topoisomerase 1; |
| Tpc | topotecan; |
| UA | usnic acid; |
| GABA | γ-aminobutyricacid; |
| EDC | 1-(3-dimethylamino-propyl)-3-ethylcarbodiimide hydrochloride; |
| DMAP | dimethylaminopyridine. |
References
- Ingolfsdottir, K. Usnic acid. Phytochemistry 2002, 61, 729–736. [Google Scholar] [CrossRef]
- Luzina, O.A.; Salakhutdinov, N.F. Usnic acid and its derivatives for pharmaceutical use: A patent review (2000–2017). Expert Opin. Ther. Pat. 2018, 28, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Neff, G.W.; Reddy, K.R.; Durazo, F.A.; Meyer, D.; Marrero, R.; Kaplowitz, N. Severe hepatotoxicity associated with the use of weight loss diet supplements containing ma huang or usnic acid. J. Hepatol. 2004, 41, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, V.N.; Strigina, L.I. (+)-Usnic acid in Far Eastern lichens. Chem. Nat. Compd. 1976, 12, 75. [Google Scholar] [CrossRef] [Green Version]
- Galanty, A.; Pasko, P.; Podolak, I. Enantioselective activity of usnic acid: A comprehensive review and future perspectives. Phytochem. Rev. 2019, 18, 527–548. [Google Scholar] [CrossRef] [Green Version]
- Bekker, O.B.; Sokolov, D.N.; Luzina, O.A.; Komarova, N.I.; Gatilov, Y.V.; Andreevskaya, S.N.; Smirnova, T.G.; Maslov, D.A.; Chernousova, L.N.; Salakhutdinov, N.F.; et al. Synthesis and activity of (+)-usnic acid and (-)-usnic acid derivatives containing 1,3-thiazole cycle against Mycobacterium tuberculosis. Med. Chem. Res. 2015, 24, 2926–2938. [Google Scholar] [CrossRef]
- Shtro, A.A.; Zarubaev, V.V.; Luzina, O.A.; Sokolov, D.N.; Salakhutdinov, N.F. Derivatives of usnic acid inhibit broad range of influenza viruses and protect mice from lethal influenza infection. Antivir. Chem. Chemother. 2015, 24, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Shtro, A.A.; Zarubaev, V.V.; Luzina, O.A.; Sokolov, D.N.; Kiselev, O.I.; Salakhutdinov, N.F. Novel derivatives of usnic acid effectively inhibiting reproduction of influenza A virus. Bioorg. Med. Chem. 2014, 22, 6826–6836. [Google Scholar] [CrossRef]
- Krukov, V.Y.; Tomilova, O.G.; Luzina, O.A.; Yaroslavtseva, O.N.; Akhanaev, Y.B.; Tyurin, M.V.; Duisembekov, B.A.; Salakhutdinov, N.F.; Glupov, V.V. Effects of fluorine-containing usnic acid and fungus Beauveria bassiana on the survival and immune-physiological reactions of Colorado potato beetle larvae. Pest Manag. Sci. 2018, 74, 598–606. [Google Scholar] [CrossRef]
- Luzina, O.A.; Salakhutdinov, N.F. Biological activity of usnic acid and its derivatives: Part 1. Activity against unicellular organisms. Russ. J. Bioorg. Chem. 2016, 42, 115–132. [Google Scholar] [CrossRef]
- Luzina, O.A.; Salakhutdinov, N.F. Biological activity of usnic acid and its derivatives: Part 2. effects on higher organisms. Molecular and physicochemical aspects. Russ. J. Bioorg. Chem. 2016, 42, 249–268. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Patel, J.; Zakharova, O.D.; Chepanova, A.A.; Zafar, A.; et al. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem. 2019, 161, 581–593. [Google Scholar] [CrossRef]
- Zakharova, O.; Luzina, O.; Zakharenko, A.; Sokolov, D.; Filimonov, A.; Dyrkheeva, N.; Chepanova, A.; Ilina, E.; Ilyina, A.; Klabenkova, K.; et al. Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors. Bioorg. Med. Chem. 2018, 26, 4470–4480. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Zakharova, O.D.; Rakhmanova, M.E.; Chepanova, A.A.; Dyrkheeva, N.S.; Lavrik, O.I.; Salakhutdinov, N.F. Usnic Acid Derivatives Are Effective Inhibitors of Tyrosyl-DNA Phosphodiesterase 1. Russ. J. Bioorg. Chem. 2017, 43, 84–90. [Google Scholar] [CrossRef]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef]
- Comeaux, E.Q.; van Waardenburg, R.C. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab. Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef]
- Kawale, A.S.; Povirk, L.F. Tyrosyl-DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 2018, 46, 520–537. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Camptothecins and topoisomerase I: A foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs: Importance of DNA replication, repair and cell cycle checkpoints. Curr. Med. Chem. Anticancer Agents 2004, 4, 429–434. [Google Scholar] [CrossRef]
- Beretta, G.L.; Cossa, G.; Gatti, L.; Zunino, F.; Perego, P. Tyrosyl-DNA phosphodiesterase 1 targeting for modulation of camptothecin-based treatment. Curr. Med. Chem. 2010, 17, 1500–1508. [Google Scholar] [CrossRef]
- Alagoz, M.; Gilbert, D.C.; El-Khamisy, S.; Chalmers, A.J. DNA repair and resistance to topoisomerase I inhibitors: Mechanisms, biomarkers and therapeutic targets. Curr. Med. Chem. 2012, 19, 3874–3885. [Google Scholar] [CrossRef]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential. Bioorg. Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Filimonov, A.S.; Chepanova, A.A.; Luzina, O.A.; Zakharenko, A.L.; Zakharova, O.D.; Ilina, E.S.; Dyrkheeva, N.S.; Kuprushkin, M.S.; Kolotaev, A.V.; Khachatryan, D.S.; et al. New Hydrazinothiazole Derivatives of Usnic Acid as Potent Tdp1 Inhibitors. Molecules 2019, 24, 3711. [Google Scholar] [CrossRef] [Green Version]
- Koldysheva, E.V.; Men’shchikova, A.P.; Lushnikova, E.L.; Popova, N.A.; Kaledin, V.I.; Nikolin, V.P.; Zakharenko, A.L.; Luzina, O.A.; Salakhutdinov, N.F.; Lavrik, O.I. Antimetastatic Activity of Combined Topotecan and Tyrosyl-DNA Phosphodiesterase-1 Inhibitor on Modeled Lewis Lung Carcinoma. Bull. Exp. Biol. Med. 2019, 166, 661–666. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Zakharenko, A.L.; Novoselova, E.S.; Chepanova, A.A.; Popova, N.A.; Nikolin, V.P.; Luzina, O.A.; Salakhutdinov, N.F.; Ryabchikova, E.I.; Lavrik, O.I. Antitumor Activity of the Combination of Topotecan and Tyrosyl-DNA-Phosphodiesterase 1 Inhibitor on Model Krebs-2 Mouse Ascite Carcinoma. Mol. Biol. 2021, 55, 312–317. [Google Scholar] [CrossRef]
- Salakhutdinov, N.F.; Volcho, K.P.; Yarovaya, O.I. Monoterpenes as a renewable source of biologically active compounds. Pure Appl. Chem. 2017, 89, 1105–1117. [Google Scholar] [CrossRef]
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef]
- Munkuev, A.A.; Mozhaitsev, E.S.; Chepanova, A.A.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Ilina, E.S.; Dyrkheeva, N.S.; Zakharenko, A.L.; Reynisson, J.; et al. Novel Tdp1 inhibitors based on adamantane connected with monoterpene moieties via heterocyclic fragments. Molecules 2021, 26, 3128. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising New Inhibitors of Tyrosyl-DNA Phosphodiesterase I (Tdp 1) Combining 4-Arylcoumarin and Monoterpenoid Moieties as Components of Complex Antitumor Therapy. Int. J. Mol. Sci. 2019, 21, 126. [Google Scholar] [CrossRef] [Green Version]
- Dyrkheeva, N.; Luzina, O.; Filimonov, A.; Zakharova, O.; Ilina, E.; Zakharenko, A.; Kuprushkin, M.; Nilov, D.; Gushchina, I.; Švedas, V.; et al. Inhibitory Effect of New Semisynthetic Usnic Acid Derivatives on Human Tyrosyl-DNA Phosphodiesterase 1. Planta Med. 2019, 85, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzina, O.; Filimonov, A.; Zakharenko, A.; Chepanova, A.; Zakharova, O.; Ilina, E.; Dyrkheeva, N.; Likhatskaya, G.; Salakhutdinov, N.; Lavrik, O. Usnic Acid Conjugates with Monoterpenoids as Potent Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. J. Nat. Prod. 2020, 83, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Polovinka, M.P.; Salakhutdinov, N.F.; Panchenko, M.Y. Method for obtaining usnic acid. Patent RU2317076, 20 February 2008. Application granted date 17 Apr 2006. [Google Scholar]
- Luzina, O.A.; Polovinka, M.P.; Salakhutdinov, N.F.; Tolstikov, G.A. Chemical modification of usnic acid 2. Reactions of (+)-usnic acid with amino acids. Russ. Chem. Bull. 2007, 56, 1249–1251. [Google Scholar] [CrossRef]
- Suslov, E.V.; Mozhaytsev, E.S.; Korchagina, D.V.; Bormotov, N.I.; Yarovaya, O.I.; Volcho, K.P.; Salakhutdinov, N.F. New chemical agents based on adamantane–monoterpene conjugates against orthopoxvirus infections. RSC Med. Chem. 2020, 11. [Google Scholar] [CrossRef]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef]
- Dyrkheeva, N.; Anarbaev, R.; Lebedeva, N.; Kuprushkin, M.; Kuznetsova, A.; Kuznetsov, N.; Rechkunova, N.; Lavrik, O. Human Tyrosyl-DNA Phosphodiesterase 1 Possesses Transphosphooligonucleotidation Activity with Primary Alcohols. Front. Cell Dev. Biol. 2020, 8, 604732. [Google Scholar] [CrossRef]
- Interthal, H.; Chen, H.J.; Champoux, J.J. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J. Biol. Chem. 2005, 280, 36518–36528. [Google Scholar] [CrossRef] [Green Version]
- Il’ina, I.V.; Dyrkheeva, N.S.; Zakharenko, A.L.; Sidorenko, A.Y.; Li-Zhulanov, N.S.; Korchagina, D.V.; Chand, R.; Ayine-Tora, D.M.; Chepanova, A.A.; Zakharova, O.D.; et al. Design, Synthesis, and Biological Investigation of Novel Classes of 3-Carene-Derived Potent Inhibitors of TDP1. Molecules 2020, 25, 3496. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Kiselev, E.; Lountos, G.T.; Wang, W.; Tropea, J.E.; Needle, D.; Hilimire, T.A.; Schneekloth, J.S.; Waugh, D.S.; Pommier, Y.; et al. Small Molecule Microarray Identifies Inhibitors of Tyrosyl-DNA Phosphodiesterase 1 that Simultaneously Access the Catalytic Pocket and Two Substrate Binding Sites. Chem. Sci. 2021, 12, 3876–3884. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nuc. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Scigress Ultra, V.F. J 2.6. (EU 3.1.7); Fujitsu Limited: Tokyo, Japan, 2008–2016. [Google Scholar]
- Allinger, N.L. Conformational Analysis. 130. MM2. A Hydrocarbon Force Field Utilizing V1 and V2 Torsional Terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Gotō, H.; Ōsawa, E. An Efficient Algorithm for Searching Low-Energy Conformers of Cyclic and Acyclic Molecules. J. Chem. Soc., Perkin Trans. 1993, 2, 187–198. [Google Scholar] [CrossRef]
- Jones, G.; Willet, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, M.D.; Murray, C.; Auton, T.R.; Paolini, G.V.; Mee, P.M. Empirical Scoring Functions: I. the Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J. Comp. Aid. Mol. Design 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Mooij, W.T.M.; Verdonk, M.L. General and Targeted Statistical Potentials for Protein–ligand Interactions. Proteins 2005, 61, 272–287. [Google Scholar] [CrossRef]
- QikProp, Version 6.2; Schrödinger: New York, NY, USA, 2021.
- Ioakimidis, L.; Thoukydidis, L.; Naeem, S.; Mirza, A.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Eurtivong, C.; Reynisson, J. The Development of a Weighted Index to Optimise Compound Libraries for High Throughput Screening. Mol. Inf. 2018, 37, 1800068. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Bolhofer, V.A. An Improved Procedure for the Condensation of Potassium Phthalimide with Organic Halides. J. Am. Chem. Soc. 1950, 72, 2786. [Google Scholar] [CrossRef]
- Zhu, F.; Logan, G.; Reynisson, J. Wine Compounds as a Source for HTS Screening Collections. A Feasibility Study. Mol. Inf. 2012, 31, 847–855. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Popadyuk, I.I.; Zakharenko, A.L.; Zakharova, O.D.; Chepanova, A.A.; Dyrkheeva, N.S.; Komarova, N.I.; Reynisson, J.; Anarbaev, R.O.; Salakhutdinov, N.F.; et al. Deoxycholic Acid as a Molecular Scaffold for Tyrosyl-DNA Phosphodiesterase 1 Inhibition: A Synthesis, Structure–activity Relationship and Molecular Modeling Study. Steroids 2021, 165, 108771. [Google Scholar] [CrossRef]
- Gladkova, E.D.; Chepanova, A.A.; Ilina, E.S.; Zakharenko, A.L.; Reynisson, J.; Luzina, O.A.; Volcho, K.P.; Lavrik, O.I.; Salakhutdinov, N.F. Discovery of Novel Sultone Fused Berberine Derivatives as Promising TDP1 Inhibitors. Molecules 2021, 26, 1945. [Google Scholar] [CrossRef]
- Chepanova, A.A.; Mozhaitsev, E.S.; Munkuev, A.A.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Zakharenko, A.L.; Patel, J.; Ayine-Tora, D.M.; Reynisson, J.; et al. The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges. Appl. Sci. 2019, 9, 2767. [Google Scholar] [CrossRef] [Green Version]
- Mozhaitsev, E.; Suslov, E.; Demidova, Y.; Korchagina, D.; Volcho, K.; Zakharenko, A.; Vasi’eva, I.; Kupryushkin, M.; Chepanova, A.; Ayine-Tora, D.M.; et al. The Development of Tyrosyl-DNA Phosphodyesterase 1 (TDP1) Inhibitors Based on the Amines Combining Aromatic/Heteroaromatic and Monoterpenoid Moieties. Lett. Drug Des. Discov. 2019, 6, 597–605. [Google Scholar] [CrossRef]
- Mozhaitsev, E.S.; Zakharenko, A.L.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Vasil’eva, I.A.; Chepanova, A.A.; Black, E.; Patel, J.; Chand, R.; et al. Novel Inhibitors of DNA Repair Enzyme TDP1 Combining Monoterpenoid and Adamantane Fragments. Anticancer Agents Med. Chem. 2019, 19, 463–472. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | a | b | c | d | e |
|---|---|---|---|---|---|
| Structure |  |  | 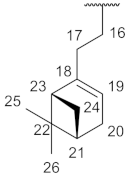 | 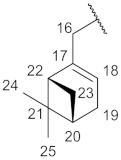 |  |
| H-16 | 3.45, m | 4.0, m | 3.44, m | 3.95, m | 3.94, m |
| H-17 | 1.15–1.52, m | 5.28, m | 2.18–2.38, m | --- | --- |
| H-18 | 1.73, m | --- | --- | 5.47, ss | 5.67, ss |
| H-19 | 1.15–1.52, m | 2.08, s | 5.44, s | 2.30, m | 1.94–2.20, m |
| H-20 | 1.99, m | 2.09, s | 2.18–2.38, m | 2.44, m | 1.94–2.20, m |
| H-21 | 5.06, m | 5.05, m | 2.18–2.38, m | --- | 1.50, m and 1.86, m |
| H-22 | --- | --- | --- | 2.0, m | 1.94–2.20, m |
| H-23 | 1.59, s | 1.58, s | 2.0, m | 1.2 d (8.9) and 2.1, m | --- |
| H-24 | 0.94, d (6.25) | 1.65, s | 1.1, d (8.6) and 2.1, m | 0.84, s | 4.7, d (13.5) |
| H-25 | 1.68, s | 1.77, s | 0.78, s | 1.29, s | 1.71, s |
| H-26 | --- | --- | 1.25, s | --- | --- |
| № | a | b | c | d | e |
|---|---|---|---|---|---|
| Structure | | | | 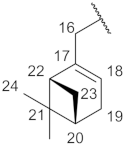 | |
| C-16 | 42.00 | 41.8 | 45.2 | 48.4 | 49.5 |
| C-17 | 36.6 | 117.0 | 35.7 | 120.2 | 148.9 |
| C-18 | 31.9 | 142.15 | 120.4 | 141.5 | 125.0 |
| C-19 | 35.8 | 39.2 | 143.0 | 31.4 | 30.2 |
| C-20 | 25.2 | 26.0 | 31.2 | 40.5 | 40.5 |
| C-21 | 123.9 | 123.2 | 40.5 | 38.1 | 26.9 |
| C-22 | 131.7 | 132.0 | 38.0 | 44.0 | 27.0 |
| C-23 | 17.6 | 25.6 | 42.0 | 31.9 | 131.1 |
| C-24 | 25.6 | 16.5 | 32.0 | 18.8 | 108.9 |
| C-25 | 18.2 | 17.6 | 18.4 | 25.9 | 18.1 |
| C-26 | --- | --- | 26.1 | --- | --- |
| № | a | b | c | d | e |
|---|---|---|---|---|---|
| Structure | | | | | |
| H-16 | 4.10, m | 4.61, m | 4.01, m | 4.45, m | 4.45, m |
| H-17 | 1.15−1.52, m | 5.31, m | 2.10−2.30, m | --- | --- |
| H-18 | 1.68, m | --- | --- | 5.55, m | 5.73, s |
| H-19 | 1.15−1.52, m | 2.09, s | 5.27, s | 2.25, m | 1.86−2.17, m |
| H-20 | 1.95, m | 2.09, s | 2.10−2.30, m | 2.38, m | 1.86−2.17, m |
| H-21 | 5.06, m | 5.06, m | 2.40, m | --- | 1.44, m and 1.80, m |
| H-22 | --- | --- | --- | 2.0, m | 1.86−2.17, m |
| H-23 | 1.57, s | 1.57, s | 2.0, m | 1.1 d (8.9)and 2.1, m | --- |
| H-24 | 0.96, d (6.5) | 1.65, s | 1.1, d (8.6)and 2.1, m | 0.78, s | 4.68, m |
| H-25 | 1.65, s | 1.68, s | 0.78, s | 1.28, s | 1.69, s |
| H-26 | --- | 1.23, s | --- | --- |
| № | a | b | c | d | e |
|---|---|---|---|---|---|
| Structure | | | | | |
| C-16 | 63.3 | 61.7 | 63.0 | 67.3 | 68.65 |
| C-17 | 36.8 | 117.7 | 35.7 | 121.8 | 149.1 |
| C-18 | 31.7 | 142.7 | 118.8 | 142.5 | 126.1 |
| C-19 | 35.2 | 39.4 | 143.7 | 31.1 | 30.2 |
| C-20 | 25.2 | 26.1 | 31.2 | 42.9 | 40.5 |
| C-21 | 124.3 | 123.5 | 40.5 | 37.9 | 26.2 |
| C-22 | 131.3 | 132.8 | 37.8 | 43.4 | 27.0 |
| C-23 | 17.5 | 25.6 | 45.4 | 31.8 | 132.1 |
| C-24 | 25.6 | 16.4 | 32.0 | 18.5 | 108.9 |
| C-25 | 18.1 | 17.6 | 18.2 | 25.9 | 18.0 |
| C-26 | --- | --- | 26.1 | --- | --- |
| Terpene Fragment Structure | Code | IC50, µM | Code | IC50, µM |
|---|---|---|---|---|
| 11a–e | 13a–e | |||
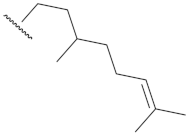 | 11a | 0.40 ± 0.09 | 13a | >10 |
 | 11b | 0.32 ± 0.13 | 13b | >10 |
 | 11c | 0.32 ± 0.10 | 13c | >10 |
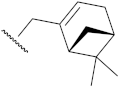 | 11d | 0.23 ± 0.10 | 13d | >10 |
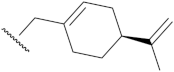 | 11e | >10 | 13e | >10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyrkheeva, N.S.; Filimonov, A.S.; Luzina, O.A.; Zakharenko, A.L.; Ilina, E.S.; Malakhova, A.A.; Medvedev, S.P.; Reynisson, J.; Volcho, K.P.; Zakian, S.M.; et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Monoterpenoids for Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibition. Biomolecules 2021, 11, 973. https://doi.org/10.3390/biom11070973
Dyrkheeva NS, Filimonov AS, Luzina OA, Zakharenko AL, Ilina ES, Malakhova AA, Medvedev SP, Reynisson J, Volcho KP, Zakian SM, et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Monoterpenoids for Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibition. Biomolecules. 2021; 11(7):973. https://doi.org/10.3390/biom11070973
Chicago/Turabian StyleDyrkheeva, Nadezhda S., Aleksandr S. Filimonov, Olga A. Luzina, Alexandra L. Zakharenko, Ekaterina S. Ilina, Anastasia A. Malakhova, Sergey P. Medvedev, Jóhannes Reynisson, Konstantin P. Volcho, Suren M. Zakian, and et al. 2021. "New Hybrid Compounds Combining Fragments of Usnic Acid and Monoterpenoids for Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibition" Biomolecules 11, no. 7: 973. https://doi.org/10.3390/biom11070973
APA StyleDyrkheeva, N. S., Filimonov, A. S., Luzina, O. A., Zakharenko, A. L., Ilina, E. S., Malakhova, A. A., Medvedev, S. P., Reynisson, J., Volcho, K. P., Zakian, S. M., Salakhutdinov, N. F., & Lavrik, O. I. (2021). New Hybrid Compounds Combining Fragments of Usnic Acid and Monoterpenoids for Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibition. Biomolecules, 11(7), 973. https://doi.org/10.3390/biom11070973