
Bioenergetic and Proteomic Profiling of Immune Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients: An Exploratory Study

 , , ,
, , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Isolation of Human PBMCs Using a Vacutainer Cell Preparation Tube
2.3. Bioenergetics Profile
- (A)
- 1 μmol/L Oligomycin (inhibitor of complex V); 2 μmol/LM Rotenone/2 μM Antimycin (inhibitor of complex I and complex III, respectively);
- (B)
- 6 μmol/L FCCP (uncoupler); 2 μmol/L Rotenone/2 μM Antimycin;
- (C)
- 1 μmol/L Oligomycin; 50 mmol/L 2-Deoxy-D-glucose (2-DG) (inhibitor of glycolysis).
2.4. Protein Carbonyl Content
2.5. Protein Extraction and Sample Preparation
2.6. Tandem Mass Tag Labelling, Isoelectric Focusing Separation, and Purification of Peptides
2.7. Nano-Liquid Chromatography and Mass Spectrometry (MS) Analysis
2.8. Proteomic Database Search and Statistical Analysis
2.9. Targeted Metabolomics Profiling
2.10. Acylcarnitines and Organic Acids Profile
2.11. Steroid Analysis
2.12. Statistical Analysis
3. Results
3.1. Description of the Cohort
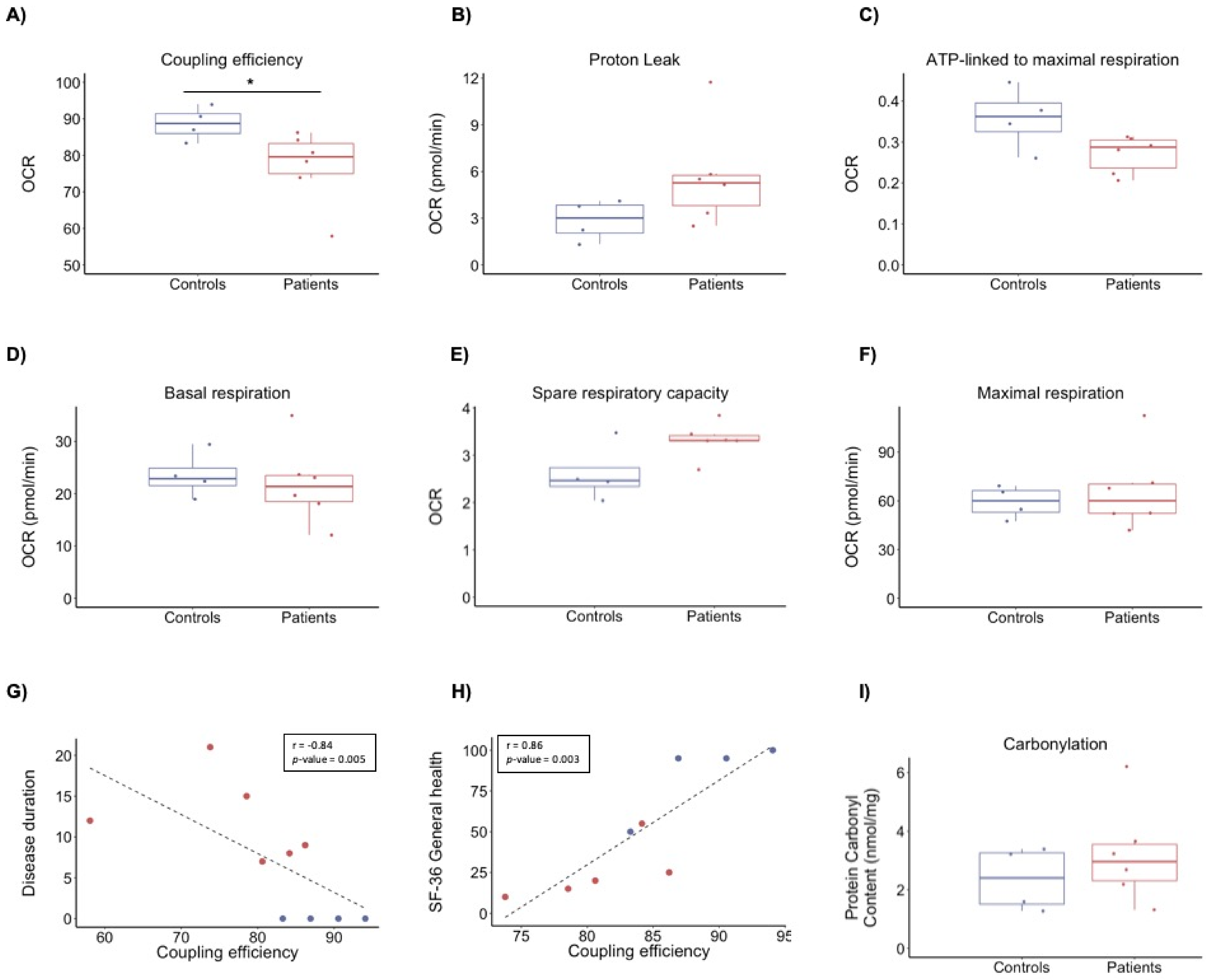
3.2. Bioenergetics Profile
3.3. Protein Carbonyl Content
3.4. Proteomics Profile
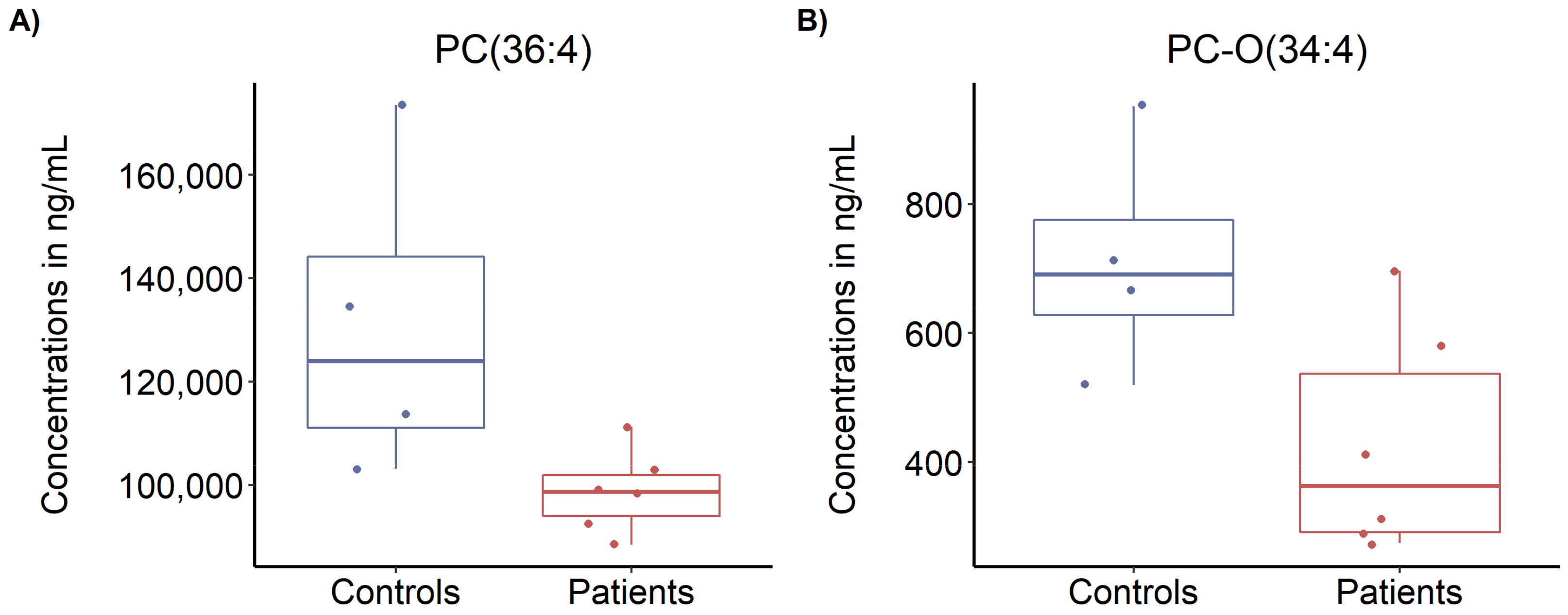
3.5. Targeted Metabolomics Profiling
3.6. Steroid Analysis
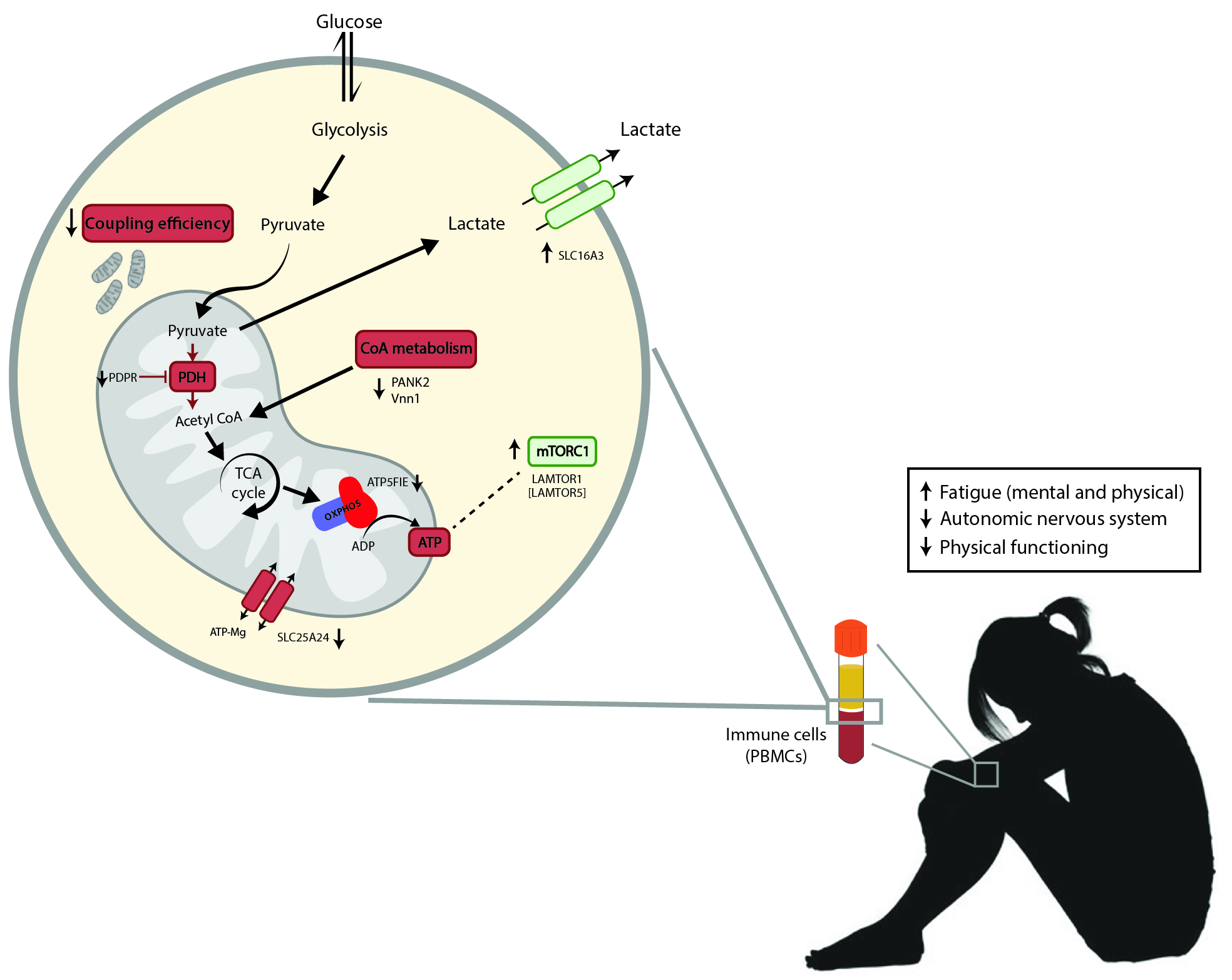
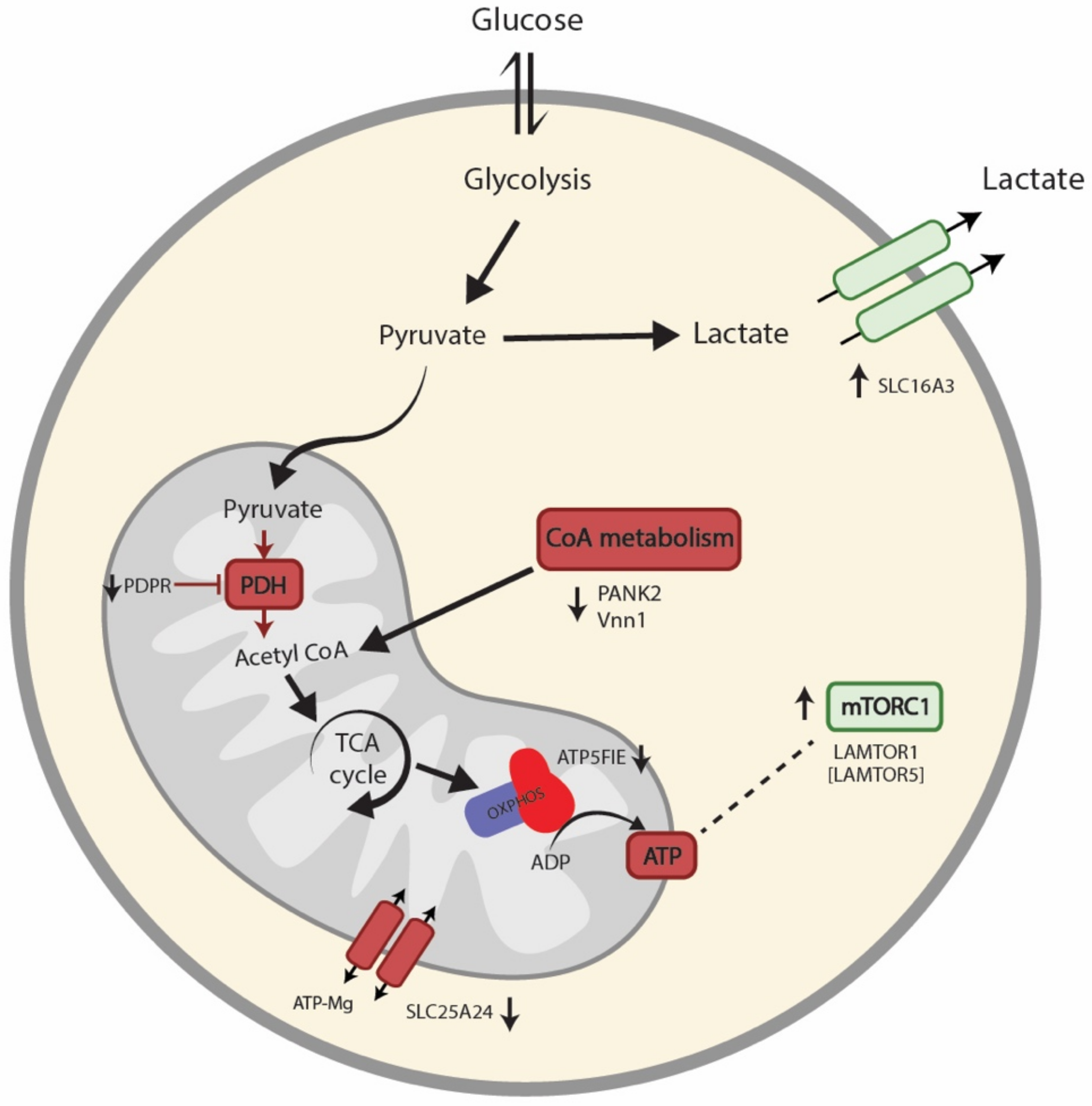
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aaron, L.A.; Ms, R.H.; Bs, S.A.; Belcourt, M.; Schmaling, K.; Goldberg, J.; Buchwald, D. Comorbid Clinical Conditions in Chronic Fatigue. A Co-Twin Control Study. J. Gen. Intern. Med. 2001, 16, 24–31. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reynolds, G.K.; Lewis, D.P.; Richardson, A.; Lidbury, B.A. Comorbidity of postural orthostatic tachycardia syndrome and chronic fatigue syndrome in an Australian cohort. J. Intern. Med. 2013, 275, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Mil. Med. 2015, 180, 721–723. [Google Scholar] [CrossRef]
- Carruthers, B.M. Definitions and aetiology of myalgic encephalomyelitis: How the Canadian consensus clinical definition of myalgic encephalomyelitis works. J. Clin. Pathol. 2006, 60, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The Chronic Fatigue Syndrome: A Comprehensive Approach to Its Definition and Study. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Chalder, T.; Cleare, A.; Hotopf, M.; Wessely, S. Chronic fatigue syndrome. BMJ 2000, 320, 292–296. [Google Scholar] [CrossRef]
- Nacul, L.C.; Lacerda, E.M.; Pheby, D.; Campion, P.; Molokhia, M.; Fayyaz, S.; Leite, J.C.; Poland, F.; Howe, A.; Drachler, M.L. Prevalence of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) in three regions of England: A repeated cross-sectional study in primary care. BMC Med. 2011, 9, 91. [Google Scholar] [CrossRef]
- Valdez, A.R.; Hancock, E.E.; Adebayo, S.; Kiernicki, D.J.; Proskauer, D.; Attewell, J.R.; Bateman, L.; DeMaria, A.J.; Lapp, C.W.; Rowe, P.C.; et al. Estimating Prevalence, Demographics, and Costs of ME/CFS Using Large Scale Medical Claims Data and Machine Learning. Front. Pediatr. 2019, 6, 412. [Google Scholar] [CrossRef]
- Hvidberg, M.F.; Brinth, L.; Olesen, A.V.; Petersen, K.D.; Ehlers, L. The Health-Related Quality of Life for Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS ONE 2015, 10, e0132421. [Google Scholar] [CrossRef]
- Anderson, J.S.; Ferrans, C.E. The Quality of Life of Persons with Chronic Fatigue Syndrome. J. Nerv. Ment. Dis. 1997, 185, 359–367. [Google Scholar] [CrossRef]
- Meeus, M.; Goubert, D.; De Backer, F.; Struyf, F.; Hermans, L.; Coppieters, I.; De Wandele, I.; Da Silva, H.; Calders, P. Heart rate variability in patients with fibromyalgia and patients with chronic fatigue syndrome: A systematic review. Semin. Arthritis Rheum. 2013, 43, 279–287. [Google Scholar] [CrossRef]
- Frith, J.; Zalewski, P.; Klawe, J.J.; Pairman, J.; Bitner, A.; Tafil-Klawe, M.; Newton, J.L. Impaired blood pressure variability in chronic fatigue syndrome—A potential biomarker. Qjm Int. J. Med. 2012, 105, 831–838. [Google Scholar] [CrossRef]
- Van Cauwenbergh, D.; Nijs, J.; Kos, D.; Van Weijnen, L.; Struyf, F.; Meeus, M. Malfunctioning of the autonomic nervous system in patients with chronic fatigue syndrome: A systematic literature review. Eur. J. Clin. Investig. 2014, 44, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.J.; Bahl, J.S.; Buckley, J.D.; Thomson, R.L.; Davison, K. Evidence of altered cardiac autonomic regulation in myalgic encephalomyelitis/chronic fatigue syndrome: A systematic review and meta-analysis. Medicine 2019, 98, e17600. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.L.; Pairman, J.; Hallsworth, K.; Trenell, M.; Moore, S.; Plotz, T. Physical activity intensity but not sedentary activity is reduced in chronic fatigue syndrome and is associated with autonomic regulation. Qjm Int. J. Med. 2011, 104, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Okonkwo, O.; Sutcliffe, K.; Seth, A.; Shin, J.; Jones, D. Symptoms of autonomic dysfunction in chronic fatigue syndrome. Qjm Int. J. Med. 2007, 100, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Escorihuela, R.M.; Capdevila, L.; Castro, J.R.; Zaragozà, M.C.; Maurel, S.; Alegre, J.; Castro-Marrero, J. Reduced heart rate variability predicts fatigue severity in individuals with chronic fatigue syndrome/myalgic encephalomyelitis. J. Transl. Med. 2020, 18, 4–12. [Google Scholar] [CrossRef]
- Kohr, D.; Singh, P.; Tschernatsch, M.; Kaps, M.; Pouokam, E.; Diener, M.; Kummer, W.; Birklein, F.; Vincent, A.; Goebel, A.; et al. Autoimmunity against the β2 adrenergic receptor and muscarinic-2 receptor in complex regional pain syndrome. Pain 2011, 152, 2690–2700. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.; Liles, C.; Khan, M.; Vanderlinde-Wood, M.; Galloway, A.; Zillner, C.; Benbrook, A.; Reim, S.; Collier, D.; et al. Autoimmune Basis for Postural Tachycardia Syndrome. J. Am. Heart Assoc. 2014, 3, e000755. [Google Scholar] [CrossRef]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.-D.; Fluge, O.; et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef]
- Bynke, A.; Julin, P.; Gottfries, C.-G.; Heidecke, H.; Scheibenbogen, C.; Bergquist, J. Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors in Myalgic Encephalomyelitis (ME) patients—A validation study in plasma and cerebrospinal fluid from two Swedish cohorts. Brain Behav. Immun. Health 2020, 7, 100107. [Google Scholar] [CrossRef]
- Hostrup, M.; Onslev, J.; Jacobson, G.A.; Wilson, R.; Bangsbo, J. Chronic β2 -adrenoceptor agonist treatment alters muscle proteome and functional adaptations induced by high intensity training in young men. J. Physiol. 2017, 596, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wu, Y.; Wang, L.; Bai, Y.; Du, Y.; Li, Y.; Cao, N.; Zhao, Y.; Zhang, Y.; Liu, H. Autoantibody against β1-adrenoceptor promotes the differentiation of natural regulatory T cells from activated CD4+ T cells by up-regulating AMPK-mediated fatty acid oxidation. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocr. 2018, 49, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol. BioSyst. 2017, 13, 371–379. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Barupal, D.K.; Lee, B.; Che, X.; Williams, B.L.; Kahn, E.J.R.; Ukaigwe, J.E.; Bateman, L.; Klimas, N.G.; Komaroff, A.L.; et al. Insights into myalgic encephalomyelitis/chronic fatigue syndrome phenotypes through comprehensive metabolomics. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Fluge, O.; Mella, O.; Bruland, O.; Risa, K.; Dyrstad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Røsland, G.V.; Fosså, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef]
- Soes, S.; Brinth, L.S.; Gregersen, N.; Olsen, R.K. CoQ10 as a modulator of mitochondrial dysfunction and maladaptive stress responses in chronic fatigue syndrome. In Coenzyme Q10: From Fact to Fiction; Nova Biomedical: Waltham, MA, USA, 2015. [Google Scholar]
- Missailidis, D.; Annesley, S.J.; Allan, C.Y.; Sanislav, O.; Lidbury, B.A.; Lewis, D.P.; Fisher, P.R. An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients. Int. J. Mol. Sci. 2020, 21, 1074. [Google Scholar] [CrossRef]
- Sweetman, E.; Kleffmann, T.; Edgar, C.; De Lange, M.; Vallings, R.; Tate, W. A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J. Transl. Med. 2020, 18, 1–18. [Google Scholar] [CrossRef]
- Mandarano, A.H.; Maya, J.; Giloteaux, L.; Peterson, D.L.; Maynard, M.; Gottschalk, C.G.; Hanson, M.R. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J. Clin. Investig. 2020, 130, 1491–1505. [Google Scholar] [CrossRef]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.-L.; Shirihai, O.S.; Holbrook, M.; Xu, G.; Kocherla, M.; Shah, A.; Fetterman, J.L.; Kluge, M.A.; Frame, A.A.; Hamburg, N.M.; et al. Relation of mitochondrial oxygen consumption in peripheral blood mononuclear cells to vascular function in type 2 diabetes mellitus. Vasc. Med. 2014, 19, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Bharadwaj, M.S.; Van Horn, C.G.; Marsh, A.P.; Nicklas, B.J.; Molina, A.J. Blood-cell bioenergetics are associated with physical function and inflammation in overweight/obese older adults. Exp. Gerontol. 2015, 70, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Czajka, A.; Ajaz, S.; Gnudi, L.; Parsade, C.K.; Jones, P.; Reid, F.; Malik, A.N. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients with Diabetic Nephropathy. EBioMedicine 2015, 2, 499–512. [Google Scholar] [CrossRef]
- Maynard, S.; Keijzers, G.; Gram, M.; Desler, C.; Bendix, L.; Budtz-Jørgensen, E.; Molbo, D.; Croteau, D.L.; Osler, M.; Stevnsner, T.; et al. Relationships between human vitality and mitochondrial respiratory parameters, reactive oxygen species production and dNTP levels in peripheral blood mononuclear cells. Aging 2013, 5, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Penner, I.K.; Raselli, C.; Stöcklin, M.; Opwis, K.; Kappos, L.; Calabrese, P. The Fatigue Scale for Motor and Cognitive Functions (FSMC): Validation of a new instrument to assess multiple sclerosis-related fatigue. Mult. Scler. 2009, 15, 1509–1517. [Google Scholar] [CrossRef]
- Ware, J.E. SF-36 Health Survey Update. Spine 2000, 25, 3130–3139. [Google Scholar] [CrossRef]
- Sletten, D.M.; Suarez, G.A.; Low, P.A.; Mandrekar, J.; Singer, W. COMPASS 31: A Refined and Abbreviated Composite Autonomic Symptom Score. Mayo Clin. Proc. 2012, 87, 1196–1201. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Skotte, L.; Courraud, J.; Geller, F.; Gørtz, S.; Wohlfahrt, J.; Melbye, M.; Cohen, A.S.; Feenstra, B. Integrating genetics with newborn metabolomics in infantile hypertrophic pyloric stenosis. Metabolomics 2021, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, N.J.W.; Junker, A.E.; Christensen, M.; Hædersdal, S.; Wibrand, F.; Lund, A.M.; Galsgaard, K.D.; Holst, J.J.; Knop, F.K.; Vilsbøll, T. Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G91–G96. [Google Scholar] [CrossRef]
- Vissing, C.R.; Duno, M.; Wibrand, F.; Christensen, M.; Vissing, J. Hydroxylated long-chain acylcarnitines are biomarkers of mitochondrial myopathy. J. Clin. Endocrinol. Metab. 2019, 104, 5968–5976. [Google Scholar] [CrossRef]
- R Studio. Team RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2020. [Google Scholar]
- Jason, L.A.; Evans, M.; Porter, N.; Brown, M.; Brown, A.; Hunnell, J.; Anderson, V.; Lerch, A.; De Meirleir, K.; Friedberg, F. The Development of a Revised Canadian Myalgic Encephalomyelitis Chronic Fatigue Syndrome Case Definition. Am. J. Biochem. Biotechnol. 2010, 6, 120–135. [Google Scholar] [CrossRef]
- Carruthers, B.M.; Van De Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.P.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef]
- Wang, J.; Tadeo, X.; Hou, H.; Andrews, S.; Moresco, J.J.; Yates, J.R.; Nagy, P.L.; Jia, S. Tls1 regulates splicing of shelterin components to control telomeric heterochromatin assembly and telomere length. Nucleic Acids Res. 2014, 42, 11419–11432. [Google Scholar] [CrossRef][Green Version]
- Mao, D.Y.; Neculai, D.; Downey, M.; Orlicky, S.; Haffani, Y.Z.; Ceccarelli, D.F.; Ho, J.S.; Szilard, R.K.; Zhang, W.; Ho, C.S.; et al. Atomic Structure of the KEOPS Complex: An Ancient Protein Kinase-Containing Molecular Machine. Mol. Cell 2008, 32, 259–275. [Google Scholar] [CrossRef]
- Downey, M.; Houlsworth, R.; Maringele, L.; Rollie, A.; Brehme, M.; Galicia, S.; Guillard, S.; Partington, M.; Zubko, M.K.; Krogan, N.J.; et al. A Genome-Wide Screen Identifies the Evolutionarily Conserved KEOPS Complex as a Telomere Regulator. Cell 2006, 124, 1155–1168. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; He, M.-H.; Liu, J.-C.; Lu, Y.-S.; Peng, J.; Zhou, J.-Q. Yeast KEOPS complex regulates telomere length independently of its t6A modification function. J. Genet. Genom. 2018, 45, 247–257. [Google Scholar] [CrossRef]
- Gorman, G.S.; Elson, J.L.; Newman, J.; Payne, B.; McFarland, R.; Newton, J.L.; Turnbull, D.M. Perceived fatigue is highly prevalent and debilitating in patients with mitochondrial disease. Neuromuscul. Disord. 2015, 25, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Hubacher, M.; Calabrese, P.; Bassetti, C.; Carota, A.; Stöcklin, M.; Penner, I.-K. Assessment of Post-Stroke Fatigue: The Fatigue Scale for Motor and Cognitive Functions. Eur. Neurol. 2012, 67, 377–384. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Fagioli, L.R.; Doolittle, T.H.; Gandek, B.; Gleit, M.A.; Guerriero, R.T.; Kornish, R.J.; Ware, N.C.; Ware, J.E.; Bates, D.W. Health status in patients with chronic fatigue syndrome and in general population and disease comparison groups. Am. J. Med. 1996, 101, 281–290. [Google Scholar] [CrossRef]
- Adler, B.; Russell, J.W.; Hummers, L.K.; Mcmahan, Z.H. Symptoms of Autonomic Dysfunction in Systemic Sclerosis Assessed by the COMPASS-31 Questionnaire. J. Rheumatol. 2018, 45, 1145–1152. [Google Scholar] [CrossRef]
- Andersson, D.; Fauconnier, J.; Yamada, T.; Lacampagne, A.; Zhang, S.-J.; Katz, A.; Westerblad, H. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J. Physiol. 2011, 589, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Mazurek, S.R.; De Tombe, P.P.; Zima, A.V. Increased Energy Demand during Adrenergic Receptor Stimulation Contributes to Ca 2+ Wave Generation. Biophys. J. 2015, 109, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Ruas, J.S.; Siqueira-Santos, E.S.; Amigo, I.; Rodrigues-Silva, E.; Kowaltowski, A.J.; Castilho, R.F. Underestimation of the Maximal Capacity of the Mitochondrial Electron Transport System in Oligomycin-Treated Cells. PLoS ONE 2016, 11, e0150967. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Burkhard, D.; Ririe, K.; Rasmussen, R.; Brown, J.; Wyse, B.W.; Hansen, R.G. Purification and properties of a pantetheine-hydrolyzing enzyme from pig kidney. J. Biol. Chem. 1983, 258, 9733–9738. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Bröer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350, 219–227. [Google Scholar] [CrossRef]
- Seenappa, V.; Joshi, M.; Satyamoorthy, K. Intricate Regulation of Phosphoenolpyruvate Carboxykinase (PEPCK) Isoforms in Normal Physiology and Disease. Curr. Mol. Med. 2019, 19, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Balsa-Martinez, E.; Puigserver, P. Cancer Cells Hijack Gluconeogenic Enzymes to Fuel Cell Growth. Mol. Cell 2015, 60, 509–511. [Google Scholar] [CrossRef]
- Germain, A.; Barupal, D.K.; Levine, S.M.; Hanson, M.R. Comprehensive Circulatory Metabolomics in ME/CFS Reveals Disrupted Metabolism of Acyl Lipids and Steroids. Metabolites 2020, 10, 34. [Google Scholar] [CrossRef]
- Tomas, C.; Elson, J.L.; Newton, J.L.; Walker, M. Substrate utilisation of cultured skeletal muscle cells in patients with CFS. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Mu, Z.; Wang, L.; Deng, W.; Wang, J.; Wu, G. Structural insight into the Ragulator complex which anchors mTORC1 to the lysosomal membrane. Cell Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.-P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Missailidis, D.; Sanislav, O.; Allan, C.; Smith, P.; Annesley, S.; Fisher, P. Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts. Int. J. Mol. Sci. 2021, 22, 2046. [Google Scholar] [CrossRef]
- Writzl, K.; Maver, A.; Kovačič, L.; Martinez-Valero, P.; Contreras, L.; Satrustegui, J.; Castori, M.; Faivre, L.; Lapunzina, P.; Van Kuilenburg, A.B.; et al. De Novo Mutations in SLC25A24 Cause a Disorder Characterized by Early Aging, Bone Dysplasia, Characteristic Face, and Early Demise. Am. J. Hum. Genet. 2017, 101, 844–855. [Google Scholar] [CrossRef]
- Van Der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Wurtman, R.J. How Anticholinergic Drugs Might Promote Alzheimer’s Disease: More Amyloid-β and Less Phosphatidylcholine. J. Alzheimer Dis. 2015, 46, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Comhaire, F. Why do some ME/CFS patients benefit from treatment with sodium dichloroacetate, but others do not? Med. Hypotheses 2018, 120, 65–67. [Google Scholar] [CrossRef]
- Comhaire, F. Treating patients suffering from myalgic encephalopathy/chronic fatigue syndrome (ME/CFS) with sodium dichloroacetate: An open-label, proof-of-principle pilot trial. Med. Hypotheses 2018, 114, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Elson, J.L.; Strassheim, V.; Newton, J.L.; Walker, M. The effect of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) severity on cellular bioenergetic function. PLoS ONE 2020, 15, e0231136. [Google Scholar] [CrossRef]
- Min, B.-K.; Park, S.; Kang, H.-J.; Kim, D.W.; Ham, H.J.; Ha, C.-M.; Choi, B.-J.; Lee, J.Y.; Oh, C.J.; Yoo, E.K.; et al. Pyruvate Dehydrogenase Kinase Is a Metabolic Checkpoint for Polarization of Macrophages to the M1 Phenotype. Front. Immunol. 2019, 10, 944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef]
- Lee, I.-K. The Role of Pyruvate Dehydrogenase Kinase in Diabetes and Obesity. Diabetes Metab. J. 2014, 38, 181–186. [Google Scholar] [CrossRef]
- Jha, M.K.; Song, G.J.; Lee, M.G.; Jeoung, N.H.; Go, Y.; Harris, R.A.; Park, D.H.; Kook, H.; Lee, I.-K.; Suk, K. Metabolic Connection of Inflammatory Pain: Pivotal Role of a Pyruvate Dehydrogenase Kinase-Pyruvate Dehydrogenase-Lactic Acid Axis. J. Neurosci. 2015, 35, 14353–14369. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Thoudam, T.; Choi, E.J.; Kim, M.; Harris, R.A.; Lee, I. Loss of metabolic flexibility as a result of overexpression of pyruvate dehydrogenase kinases in muscle, liver and the immune system: Therapeutic targets in metabolic diseases. J. Diabetes Investig. 2021, 12, 21–31. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Controls (n = 4) | ME/CFS Patients (n = 6) |
|---|---|---|
| Age (years) | 42 ± 11 | 39 ± 5 |
| Height (cm) | 173 ± 6.8 | 169 ± 4.7 |
| Weight (kg) | 65.5 ± 4 | 60.3 ± 4.5 |
| BMI (kg/m2) | 21.9 ± 1.6 | 21.2 ± 1.2 |
| Systolic blood pressure (mmHg) | 103 ± 12 | 110 ± 9 |
| Diastolic blood pressure (mmHg) | 69 ± 5 | 73 ± 9 |
| Heart Rate (bpm) | 66 ± 2 | 69 ± 6 |
| Disease duration (years) | NA | 12 ± 5.3 |
| Characteristics | Controlsprekidanje(n = 4) | ME/CFS Patientsprekidanje(n = 6) |
|---|---|---|
| FSCM | 22 ± 2.4 | 74.8 ± 15.5 *** |
| FSCM Mental fatigue | 11.8 ± 2.1 | 37 ± 8.9 ** |
| FSCM Physical fatigue | 10.3 ± 0.5 | 37.8 ± 7.2 *** |
| SF-36 Physical functioning | 100 ± 0 | 34 ± 18.5 *** |
| SF-36 Role limitations due to physical health | 100 ± 0 | 5 ± 11.2 *** |
| SF-36 Role limitations due to emotional problems | 100 ± 0 | 100 ± 0 |
| SF-36 Energy/fatigue | 75 ± 18 | 24 ± 30.1 * |
| SF-36 Emotional well-being | 93 ± 6 | 62 ± 26 |
| SF-36 Social functioning | 97 ± 6 | 35 ± 32.4 * |
| SF-36 Pain | 91 ± 12 | 45 ± 20.2 ** |
| SF-36 General health | 85 ± 23 | 25 ± 17.7 ** |
| SF-36 Health change | 50 ± 0 | 55 ± 20.9 |
| Compass-31 | 1.6 ± 1.3 | 35.5 ± 20.6 * |
| Accession Number | Description | Gene Name | Fold Change | p-Value |
|---|---|---|---|---|
| P56202 | Cathepsin W | CTSW | 1.53 | 0.005 |
| P13747 | HLA class I histocompatibility antigen, alpha chain E | HLA-E | 1.34 | 0.010 |
| Q6IAA8 | Ragulator complex protein LAMTOR1 | LAMTOR1 | 1.32 | 0.031 |
| Q9BYM8 | RanBP-type and C3HC4-type zinc finger-containing protein 1 | RBCK1 | 1.22 | 0.025 |
| Q9BSJ2 | Gamma-tubulin complex component 2 | TUBGCP2 | 1.20 | 0.022 |
| Q9NZ63 | Telomere length and silencing protein 1 homolog | C9orf78 | 1.19 | 0.010 |
| O75718 | Cartilage-associated protein | CRTAP | 1.17 | 0.045 |
| Q9NYM9 | BET1-like protein | BET1L | 1.17 | 0.018 |
| Q96DM3 | Regulator of MON1-CCZ1 complex | RMC1 | 1.15 | 0.024 |
| O95372 | Acyl-protein thioesterase 2 | LYPLA2 | 1.14 | 0.036 |
| O15427 | Monocarboxylate transporter 4 | SLC16A3 | 1.13 | 0.028 |
| Q8TCD5 | 5’(3’)-deoxyribonucleotidase, cytosolic type | NT5C | 1.12 | 0.043 |
| P49459 | Ubiquitin-conjugating enzyme E2 A | UBE2A | 1.12 | 0.016 |
| Q8NB16 | Mixed lineage kinase domain-like protein | MLKL | 0.89 | 0.049 |
| Q6NUK1 | Calcium-binding mitochondrial carrier protein SCaMC-1 | SLC25A24 | 0.88 | 0.026 |
| Q8NCN5 | Pyruvate dehydrogenase phosphatase regulatory subunit, mitochondrial | PDPR | 0.87 | 0.015 |
| P56381 | ATP synthase subunit epsilon, mitochondrial | ATP5F1E | 0.86 | 0.030 |
| Q9BZ23 | Pantothenate kinase 2, mitochondrial | PANK2 | 0.85 | 0.017 |
| Q14657 | EKC/KEOPS complex subunit LAGE3 | LAGE3 | 0.84 | 0.023 |
| O95497 | Pantetheinase | VNN1 | 0.75 | 0.028 |
| P13645 | Keratin, type I cytoskeletal 10 | KRT10 | 0.66 | 0.036 |
| Q7Z3D6 | D-glutamate cyclase, mitochondrial | DGLUCY | 0.60 | 0.030 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Guerra, P.; Gonzalez-Ebsen, A.C.; Boonen, S.E.; Courraud, J.; Gregersen, N.; Mehlsen, J.; Palmfeldt, J.; Olsen, R.K.J.; Brinth, L.S. Bioenergetic and Proteomic Profiling of Immune Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients: An Exploratory Study. Biomolecules 2021, 11, 961. https://doi.org/10.3390/biom11070961
Fernandez-Guerra P, Gonzalez-Ebsen AC, Boonen SE, Courraud J, Gregersen N, Mehlsen J, Palmfeldt J, Olsen RKJ, Brinth LS. Bioenergetic and Proteomic Profiling of Immune Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients: An Exploratory Study. Biomolecules. 2021; 11(7):961. https://doi.org/10.3390/biom11070961
Chicago/Turabian StyleFernandez-Guerra, Paula, Ana C. Gonzalez-Ebsen, Susanne E. Boonen, Julie Courraud, Niels Gregersen, Jesper Mehlsen, Johan Palmfeldt, Rikke K. J. Olsen, and Louise Schouborg Brinth. 2021. "Bioenergetic and Proteomic Profiling of Immune Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients: An Exploratory Study" Biomolecules 11, no. 7: 961. https://doi.org/10.3390/biom11070961
APA StyleFernandez-Guerra, P., Gonzalez-Ebsen, A. C., Boonen, S. E., Courraud, J., Gregersen, N., Mehlsen, J., Palmfeldt, J., Olsen, R. K. J., & Brinth, L. S. (2021). Bioenergetic and Proteomic Profiling of Immune Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients: An Exploratory Study. Biomolecules, 11(7), 961. https://doi.org/10.3390/biom11070961