
The Role of Cholesterol in Amyloidogenic Substrate Binding to the γ-Secretase Complex



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. APP Processing
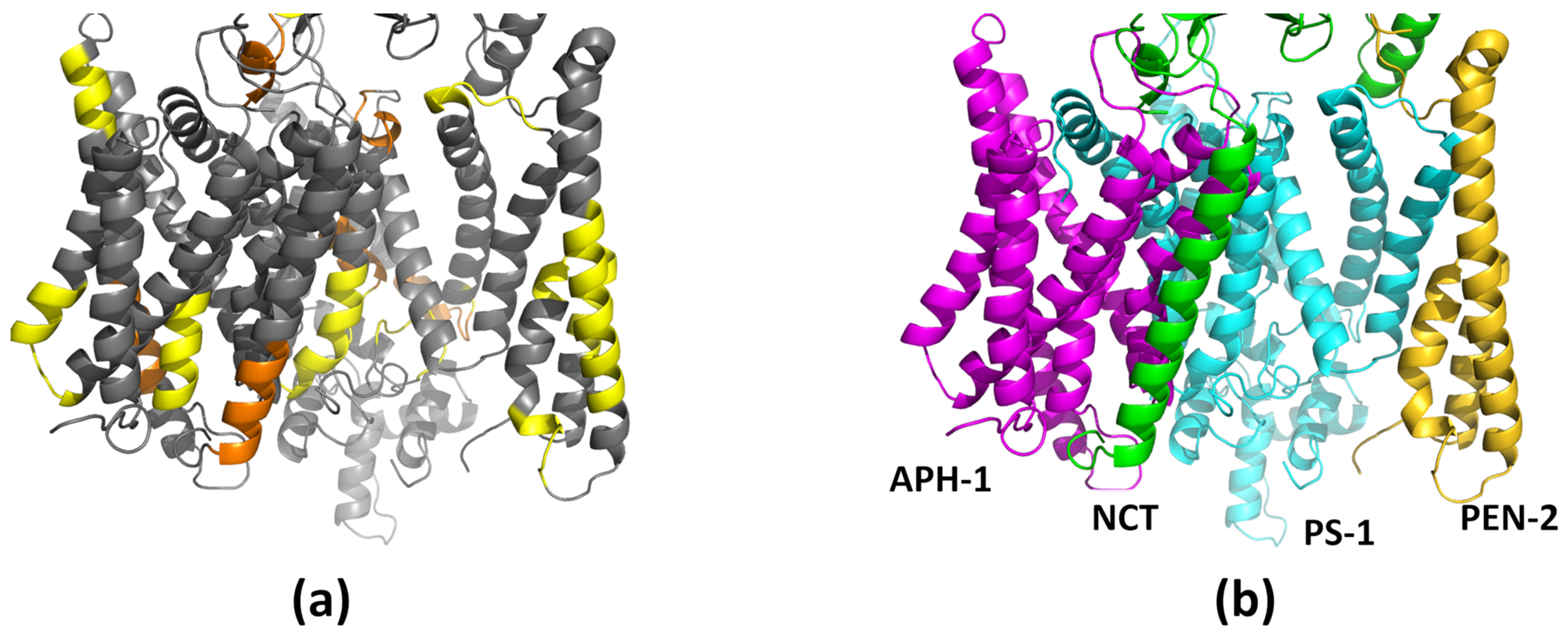
1.2. The γ-Secretase Structure
1.3. Recent Studies Involving MD Simulations of the γ-Secretase Complex
1.4. Our Investigations
2. Materials and Methods
2.1. Missing Fragments’ Modeling
2.2. Substrate Docking
2.3. Molecular Dynamics (MD) Simulations
2.4. Programs for Making Figures and Analyses
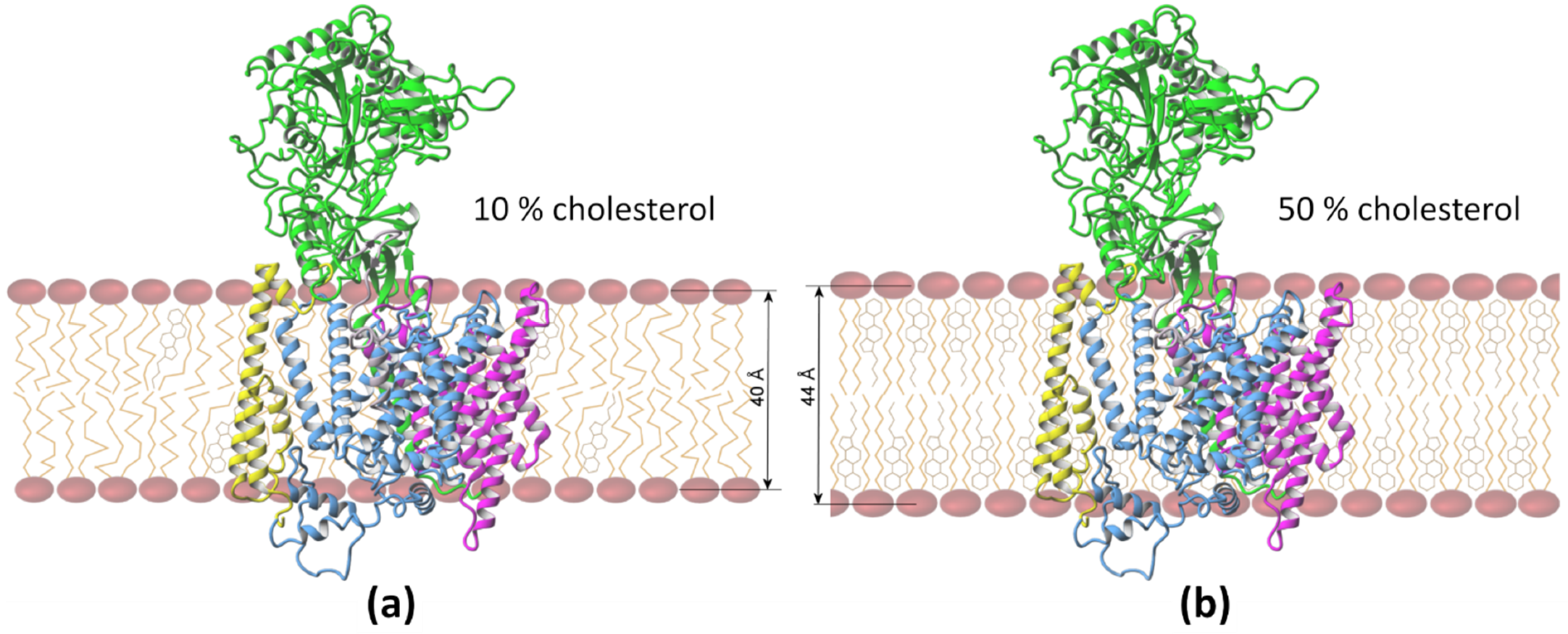
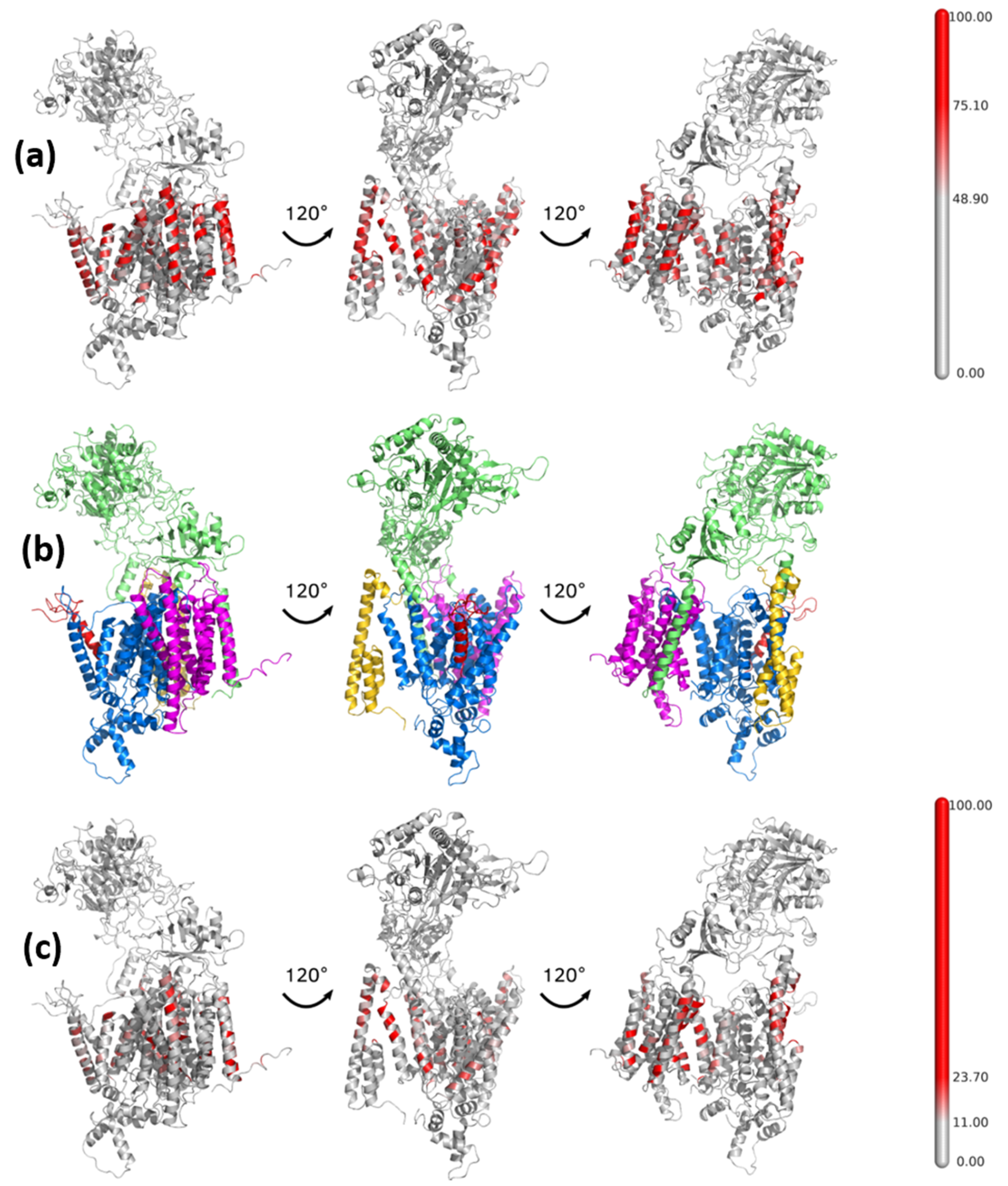
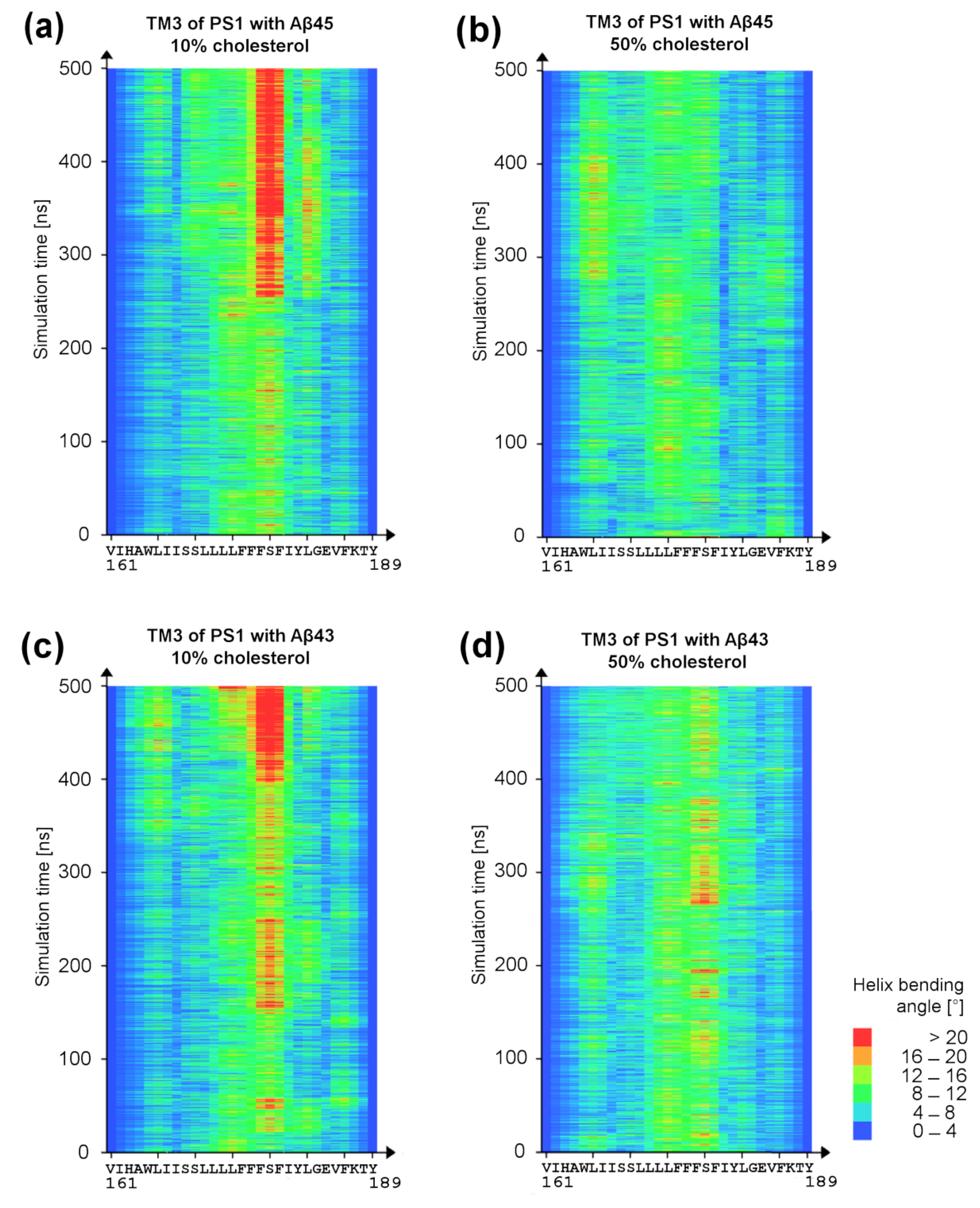
3. Results and Discussion
3.1. Mapping of the Cholesterol Binding Regions
3.2. Positioning of Substrate in the Binding Site
3.3. Bending of TM3 PS-1 at Low Cholesterol Level
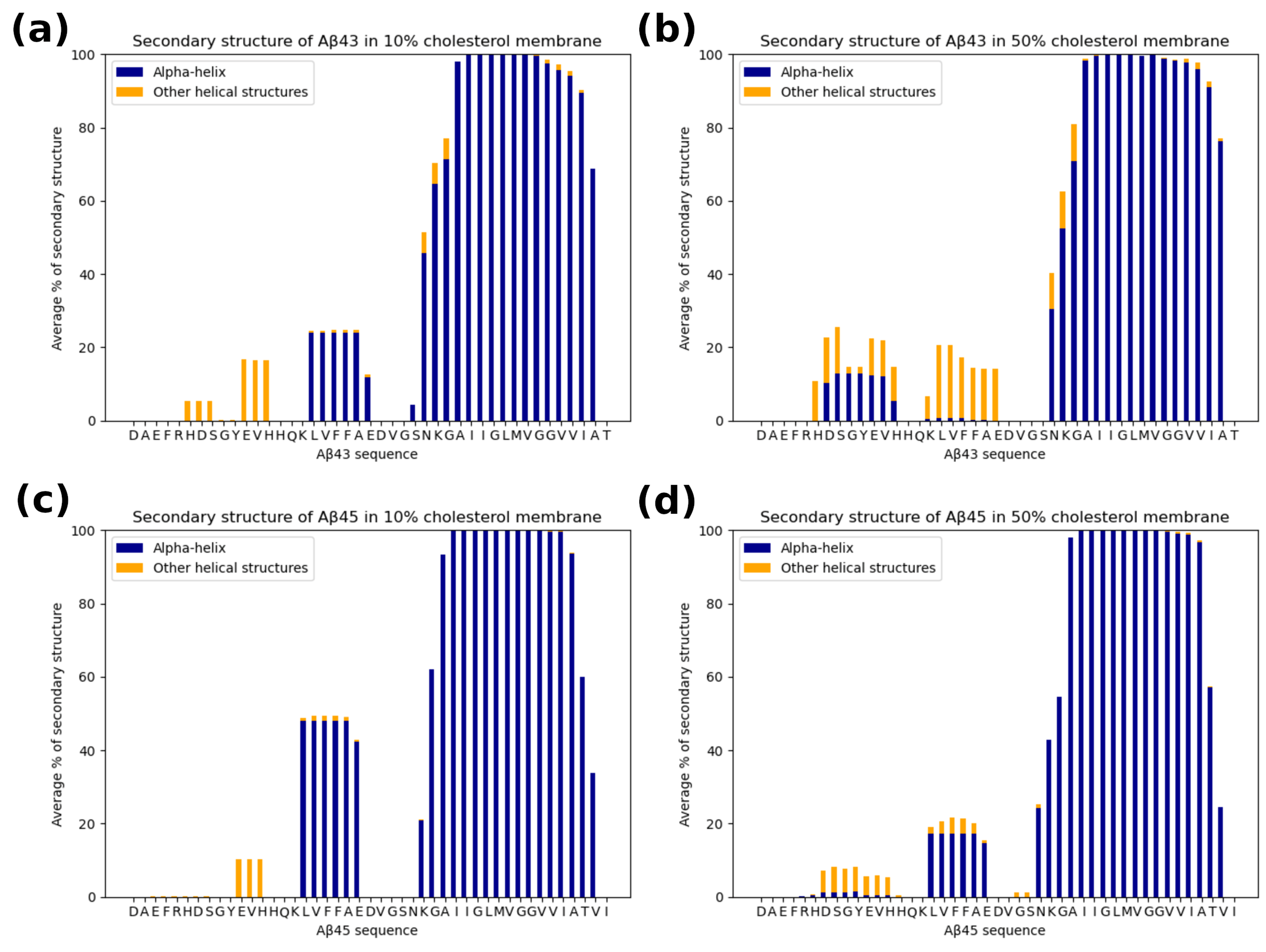
3.4. The Secondary Structure of the Substrate and Interactions with the Protease
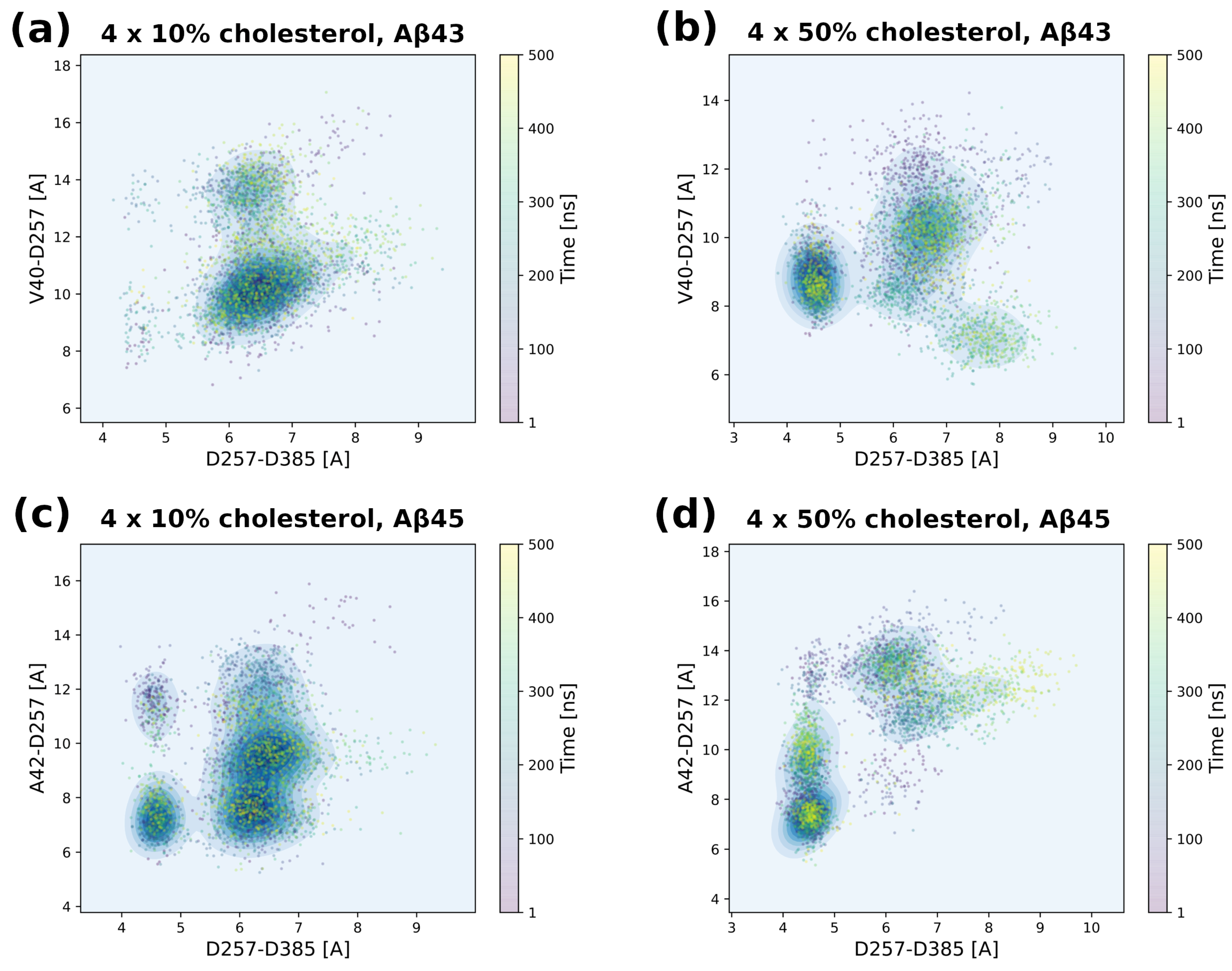
3.5. Tracing Structural Changes in the Active Site
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pensalfini, A.; Albay, R., III; Rasool, S.; Wu, J.W.; Hatami, A.; Arai, H.; Margol, L.; Milton, S.; Poon, W.W.; Corrada, M.M.; et al. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol. Dis. 2014, 71, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, H.; Fukumori, A.; Tagami, S.; Okochi, M. Making the final cut: Pathogenic amyloid-beta peptide generation by gamma-secretase. Cell Stress 2018, 2, 292–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, R.E.; Greig, N.H.; Giacobini, E.; Schneider, L.S.; Ferrucci, L. A new roadmap for drug development for Alzheimer’s disease. Nat. Rev. Drug Discov. 2014, 13, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, K.; Ji, N.; Zhang, X.; Qiao, W.; Tang, Z.; Gou, X. Drug development for Alzheimer’s disease: Review. J. Drug Target. 2019, 27, 164–173. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Haass, C.; Steiner, H. Regulated intramembrane proteolysis--lessons from amyloid precursor protein processing. J. Neurochem. 2011, 117, 779–796. [Google Scholar] [CrossRef]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef]
- Sanders, C.R. How gamma-secretase hits a moving target. ELife 2016, 5, e20043. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The amyloid-beta forming tripeptide cleavage mechanism of gamma-secretase. ELife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Pantelopulos, G.A.; Straub, J.E.; Thirumalai, D.; Sugita, Y. Structure of APP-C99 1-99 and implications for role of extra-membrane domains in function and oligomerization. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1698–1708. [Google Scholar] [CrossRef]
- Miyashita, N.; Straub, J.E.; Thirumalai, D. Structures of beta-amyloid peptide 1-40, 1-42, and 1-55-the 672-726 fragment of APP-in a membrane environment with implications for interactions with gamma-secretase. J. Am. Chem. Soc. 2009, 131, 17843–17852. [Google Scholar] [CrossRef] [Green Version]
- Gotz, A.; Scharnagl, C. Dissecting conformational changes in APP’s transmembrane domain linked to epsilon-efficiency in familial Alzheimer’s disease. PLoS ONE 2018, 13, e0200077. [Google Scholar] [CrossRef]
- Langosch, D.; Steiner, H. Substrate processing in intramembrane proteolysis by gamma-secretase-the role of protein dynamics. Biol. Chem. 2016, 398, 441–453. [Google Scholar] [CrossRef]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.; Shi, Y. An atomic structure of human gamma-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human gamma-secretase. ELife 2015, 4, e11182. [Google Scholar] [CrossRef]
- Elad, N.; De Strooper, B.; Lismont, S.; Hagen, W.; Veugelen, S.; Arimon, M.; Horre, K.; Berezovska, O.; Sachse, C.; Chavez-Gutierrez, L. The dynamic conformational landscape of gamma-secretase. J. Cell Sci. 2015, 128, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Matsumura, N.; Takami, M.; Okochi, M.; Wada-Kakuda, S.; Fujiwara, H.; Tagami, S.; Funamoto, S.; Ihara, Y.; Morishima-Kawashima, M. gamma-Secretase associated with lipid rafts: Multiple interactive pathways in the stepwise processing of beta-carboxyl-terminal fragment. J. Biol. Chem. 2014, 289, 5109–5121. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.Y.; Welander, H.; Behbahani, H.; Aoki, M.; Franberg, J.; Winblad, B.; Frykman, S.; Tjernberg, L.O. Active gamma-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 2008, 275, 1174–1187. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2010, 1801, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.H.; Yu, J.T.; Tan, L. The role of cholesterol metabolism in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 947–965. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Ye, W.; Wang, R.; Wolfe, M.S.; Selkoe, D.J. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 2008, 283, 22529–22540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langosch, D.; Scharnagl, C.; Steiner, H.; Lemberg, M.K. Understanding intramembrane proteolysis: From protein dynamics to reaction kinetics. Trends Biochem. Sci. 2015, 40, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Hitzenberger, M.; Zacharias, M. gamma-Secretase Studied by Atomistic Molecular Dynamics Simulations: Global Dynamics, Enzyme Activation, Water Distribution and Lipid Binding. Front. Chem. 2018, 6, 640. [Google Scholar] [CrossRef]
- Hitzenberger, M.; Zacharias, M. Structural Modeling of gamma-Secretase Abeta n Complex Formation and Substrate Processing. ACS Chem. Neurosci. 2019, 10, 1826–1840. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Dominguez, L. APH-1A Component of gamma-Secretase Forms an Internal Water and Ion-Containing Cavity. ACS Chem. Neurosci. 2019, 10, 2931–2938. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Straub, J.E.; Dominguez, L. Influence of membrane lipid composition on the structure and activity of gamma-secretase. Phys. Chem. Chem. Phys. 2018, 20, 27294–27304. [Google Scholar] [CrossRef]
- Bhattarai, A.; Devkota, S.; Bhattarai, S.; Wolfe, M.S.; Miao, Y. Mechanisms of gamma-Secretase Activation and Substrate Processing. ACS Cent. Sci. 2020, 6, 969–983. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human gamma-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Canutescu, A.A.; Dunbrack, R.L., Jr. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003, 12, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Im, W.; Feig, M.; Brooks, C.L. An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003, 85, 2900–2918. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- ICM-Pro; Version 3.8; Molsoft, L.L.C.: San Diego, CA, USA, 2020.
- Fernandez-Recio, J.; Totrov, M.; Abagyan, R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins 2003, 52, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Krautler, V.; Van Gunsteren, W.F.; Hunenberger, P.H. A fast SHAKE: Algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dahl, A.C.E.; Chavent, M.; Sansom, M.S.P. Bendix: Intuitive helix geometry analysis and abstraction. Bioinformatics 2012, 28, 2193–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orzeł, U.; Jakowiecki, J.; Młynarczyk, K.; Filipek, S. The Role of Cholesterol in Amyloidogenic Substrate Binding to the γ-Secretase Complex. Biomolecules 2021, 11, 935. https://doi.org/10.3390/biom11070935
Orzeł U, Jakowiecki J, Młynarczyk K, Filipek S. The Role of Cholesterol in Amyloidogenic Substrate Binding to the γ-Secretase Complex. Biomolecules. 2021; 11(7):935. https://doi.org/10.3390/biom11070935
Chicago/Turabian StyleOrzeł, Urszula, Jakub Jakowiecki, Krzysztof Młynarczyk, and Sławomir Filipek. 2021. "The Role of Cholesterol in Amyloidogenic Substrate Binding to the γ-Secretase Complex" Biomolecules 11, no. 7: 935. https://doi.org/10.3390/biom11070935
APA StyleOrzeł, U., Jakowiecki, J., Młynarczyk, K., & Filipek, S. (2021). The Role of Cholesterol in Amyloidogenic Substrate Binding to the γ-Secretase Complex. Biomolecules, 11(7), 935. https://doi.org/10.3390/biom11070935