
Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement

 ,
,  , , ,
, , ,  , , , and
, , , and 
Abstract
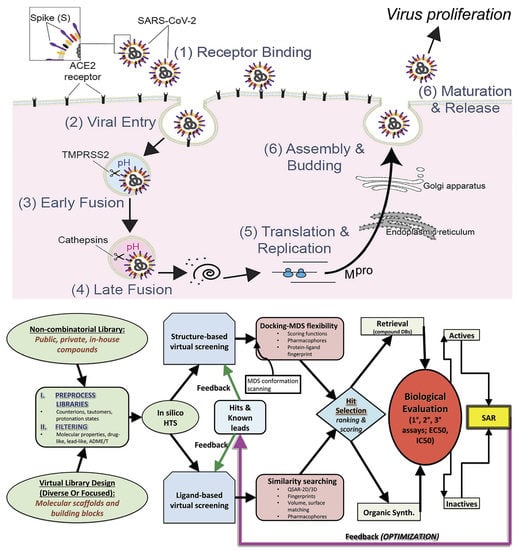
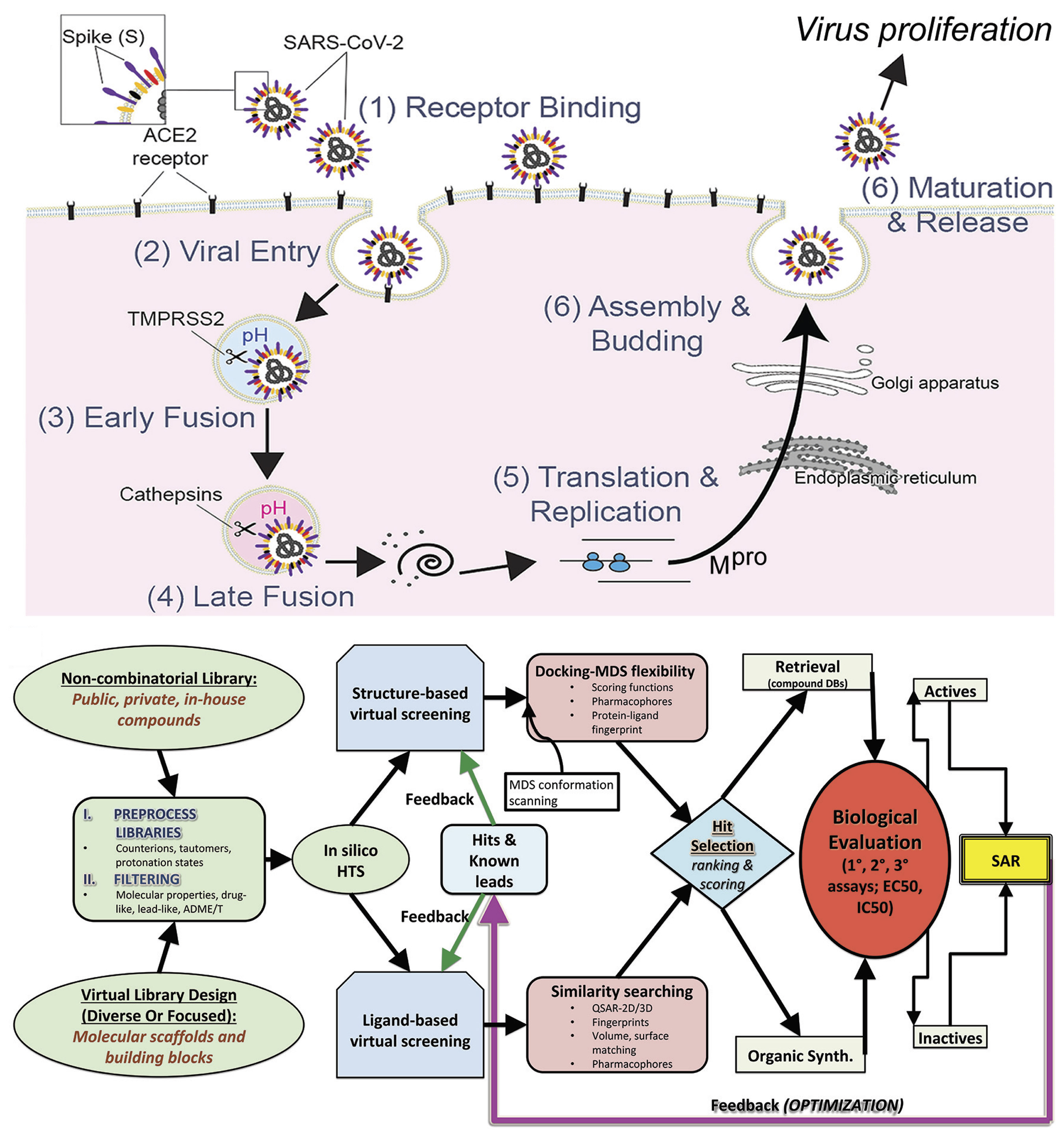
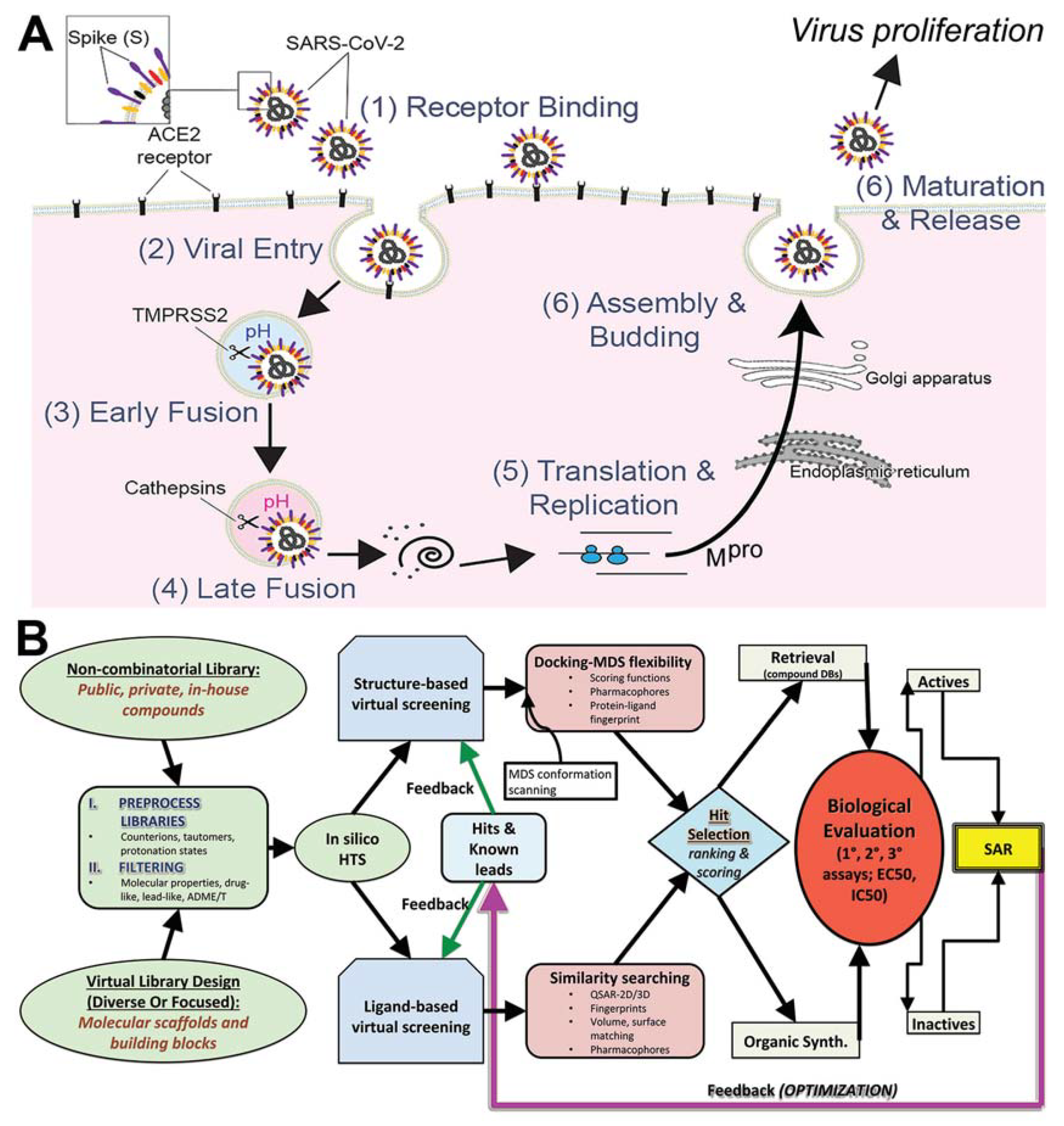
1. Introduction
2. Materials and Methods
2.1. Structural Modeling of ACE2–S protein
2.2. Structural Modeling of TMPRSS2
2.3. Structural Modeling of Mpro
2.4. Molecular Simulation Refinement of Protein Models
2.5. Production (Analytical) MDS Protocol
2.6. Docking Methods
2.6.1. Site Mapping and Preparation of Proteins
2.6.2. Libraries Used
2.6.3. Docking Parameters
2.7. In Vitro Experimentation
2.7.1. In Vitro Materials
2.7.2. Synthesis of Gelatin Methacryloyl (GelMA)
2.7.3. Cells
2.7.4. 3D Bioprinting of the Human Hepatic Microtissue Model
2.7.5. Characterizations of the Bioprinted 3D Hepatic Microtissues
2.7.6. Evaluation of Toxicity of the Compounds
2.7.7. Live Virus Screening
3. Results
3.1. Modeling and Simulations for Improved Docking Outcome
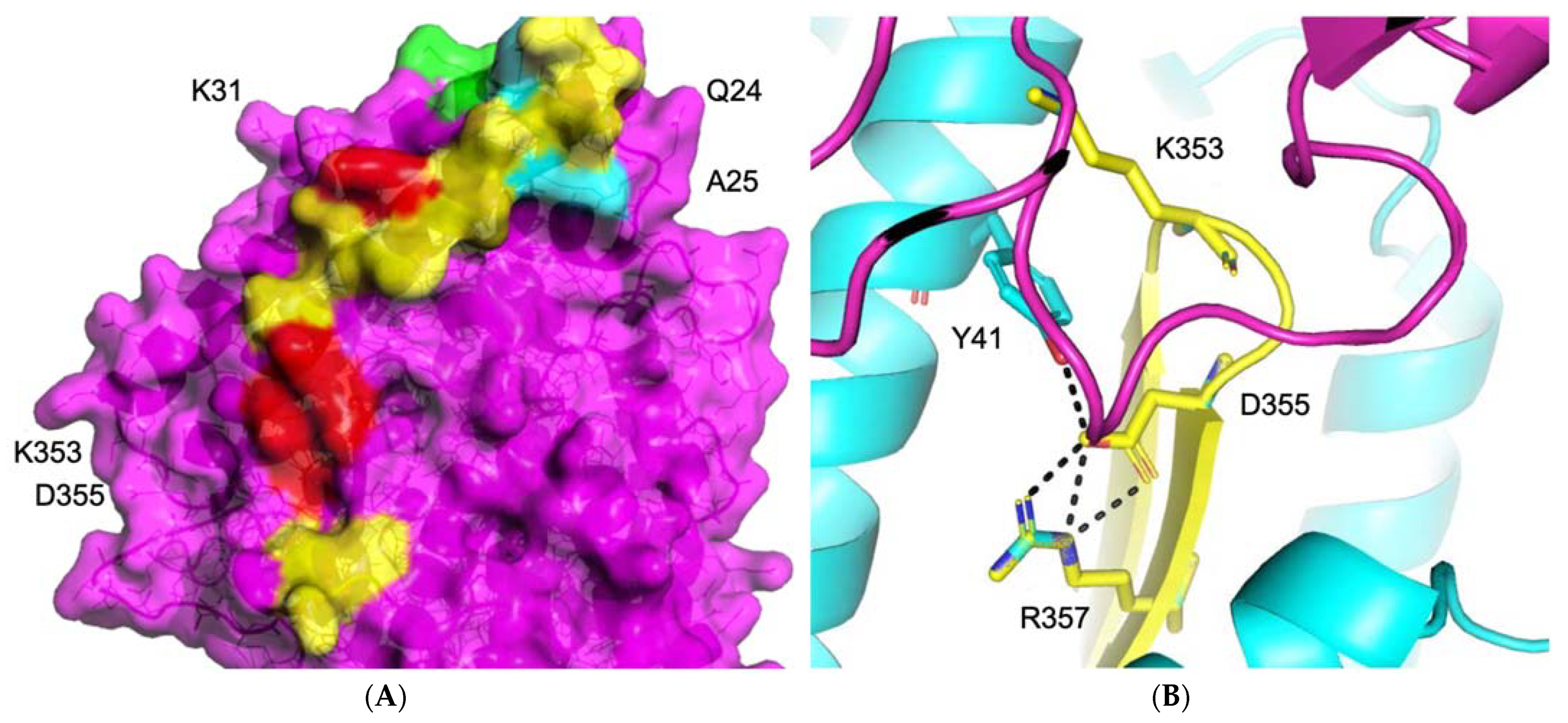
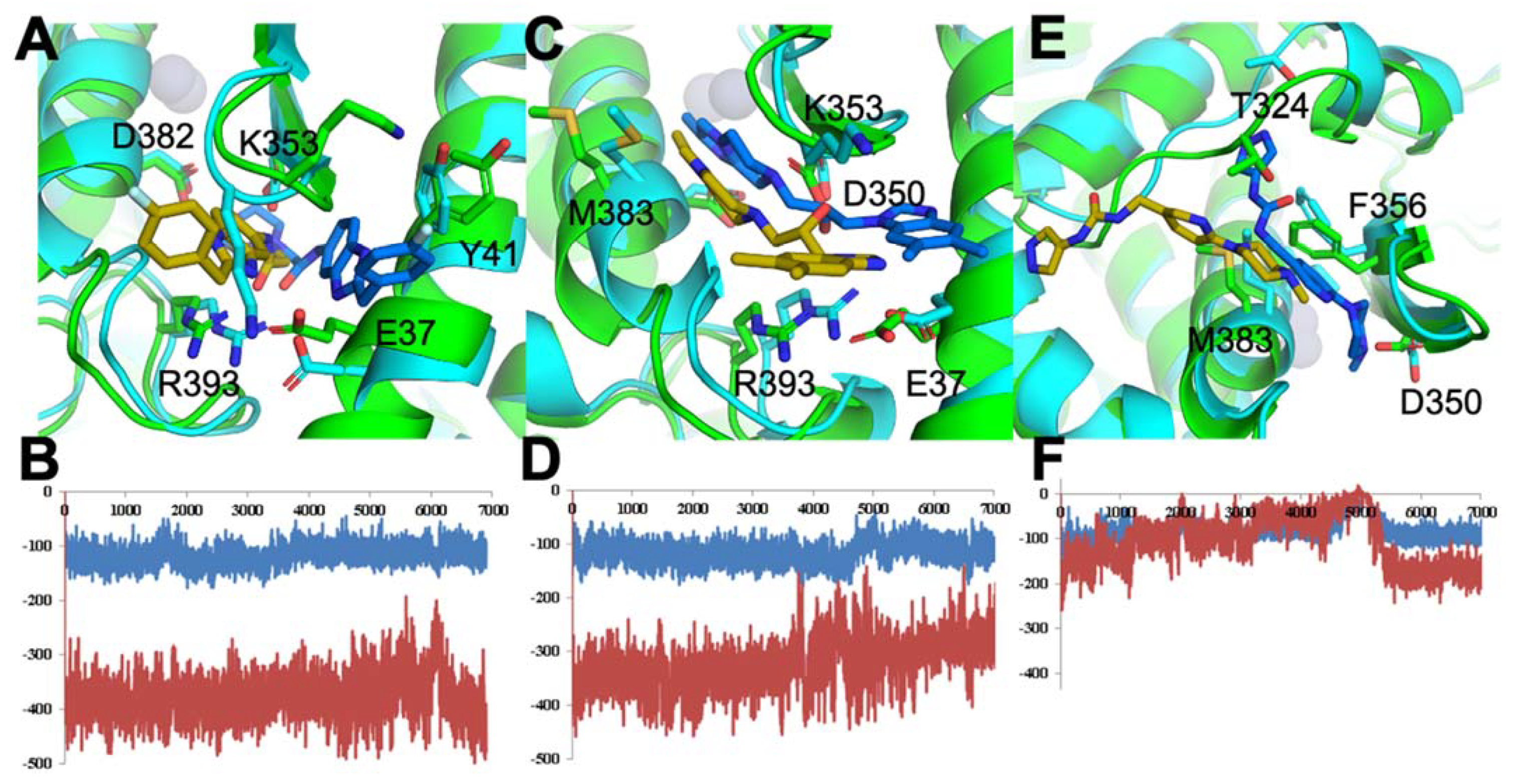
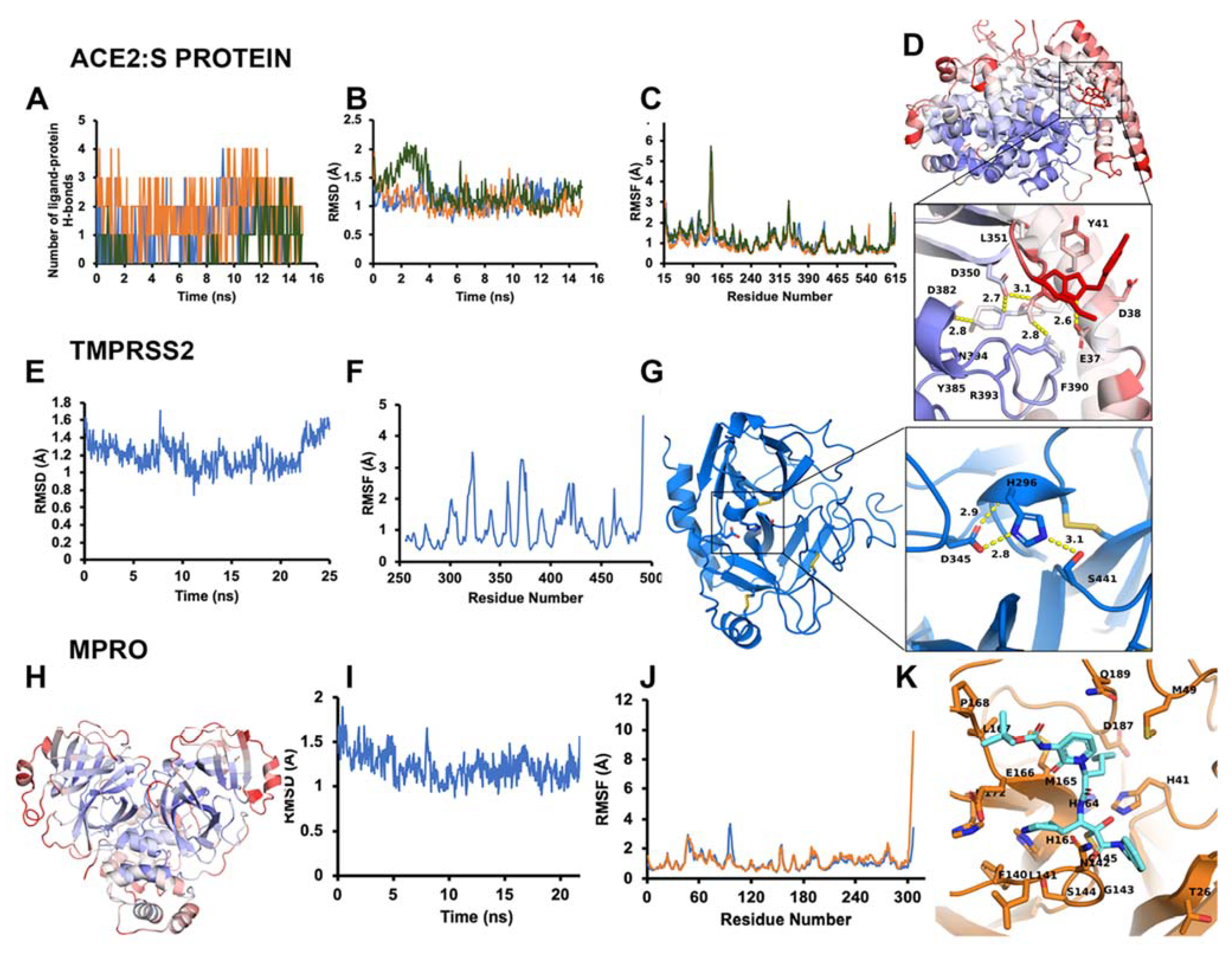
3.1.1. ACE2–S Protein Interaction Requires Dynamics to Reveal Binding Site
3.1.2. Identification of Predicted Inhibitors to Interrupt ACE2–S Protein PPI via Docking
3.1.3. Optimal Inhibitor Binding for TMPRSS2 and Mpro Revealed via Dynamics
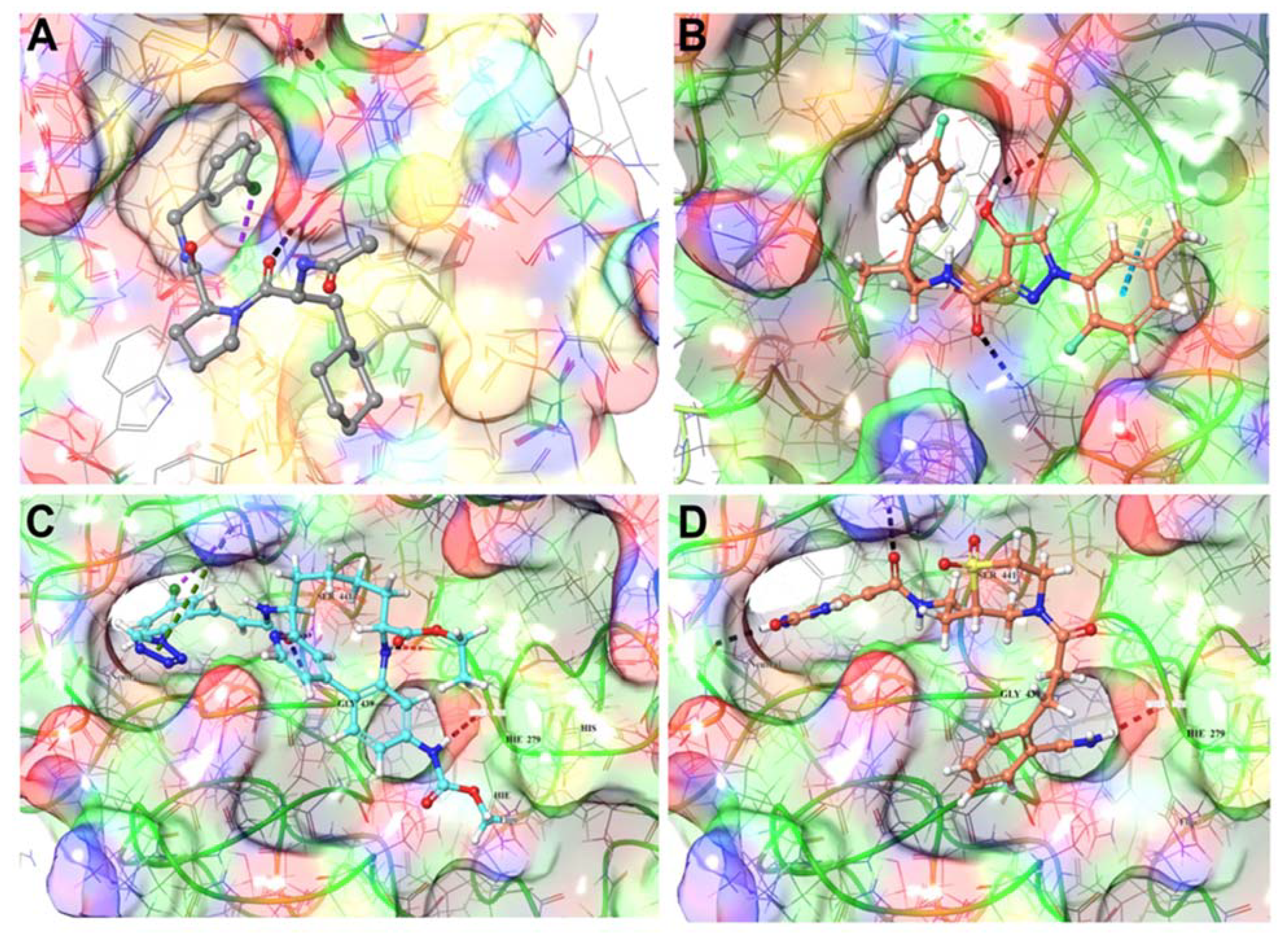
3.1.4. TMPRSS2 Inhibitors Identified
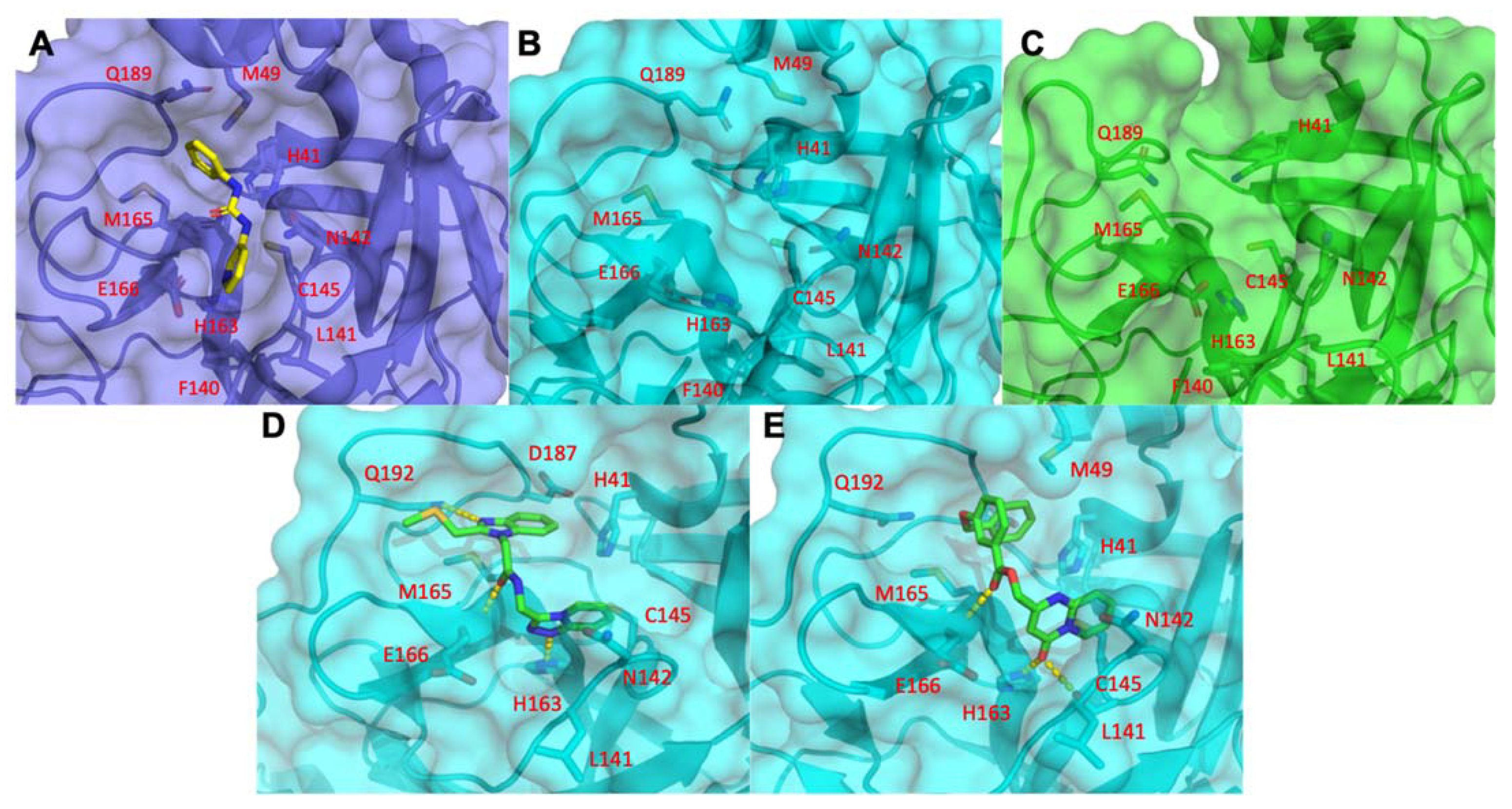
3.1.5. Mpro Inhibitors Identified
3.2. Analysis of Identified Compounds
3.3. Screening FDA-Approved Drugs for Repurposing
3.3.1. ACE2 Repurposing Drugs (FDA Set)
3.3.2. Mpro Repurposing Drugs (FDA Set)
3.3.3. TMPRSS2 Repurposing Drugs (FDA Set)
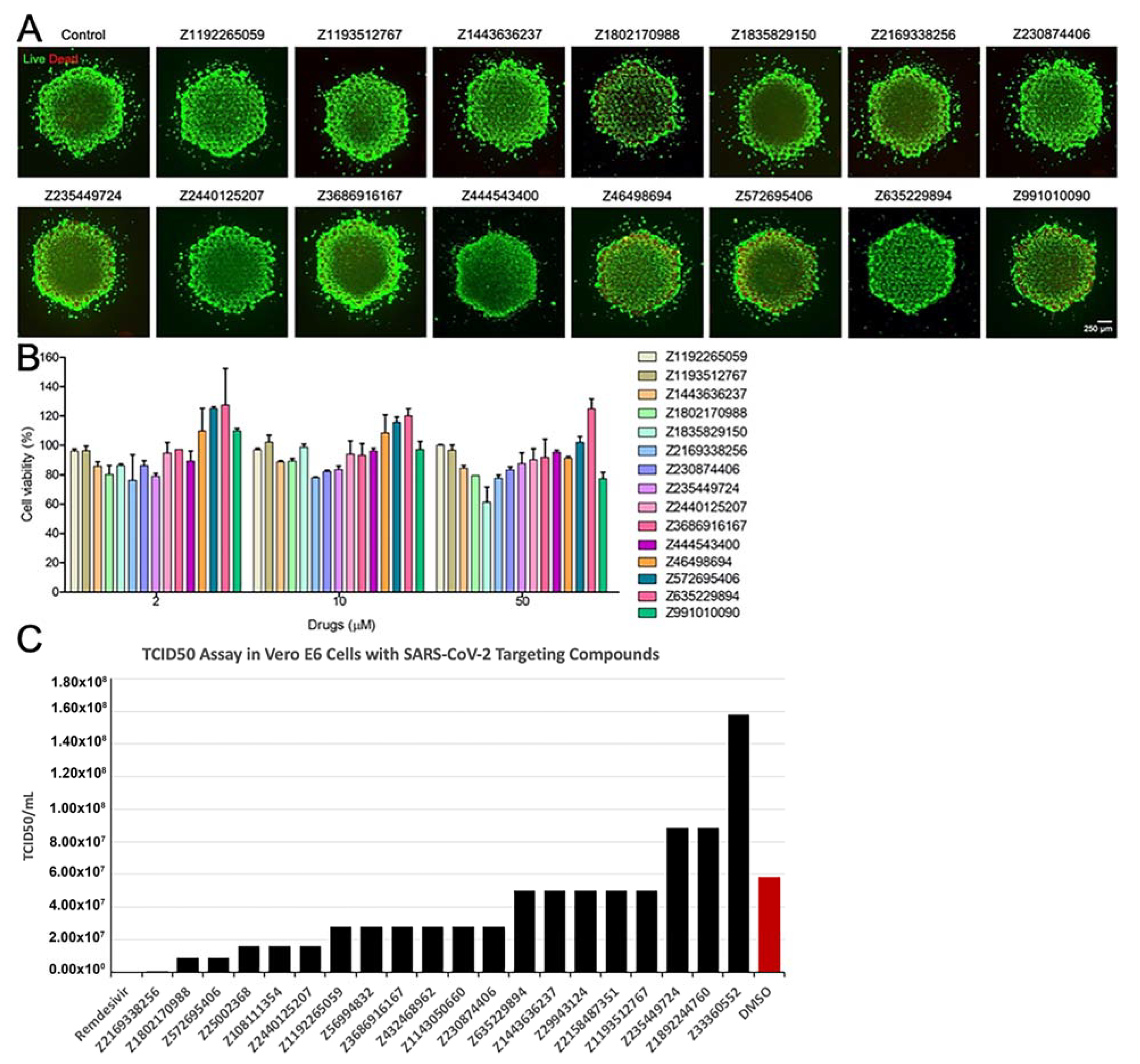
3.4. Results of In Vitro Assays for New Chemical Entities (Novel Compounds)
4. Discussion
4.1. Clinical Unmet Need for COVID-19 Acute Therapeutics
4.2. Comparison of FDA-Approved Compounds Identified from Another Recent Screen
4.3. Selective AI-SARS-Cov-2-Targeting and Drug Repurposing Data—ACE2, TMPRSS2, Mpro
4.4. NCE Set of Compounds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020. [Google Scholar] [CrossRef]
- Abdollahi, E.; Champredon, D.; Langley, J.M.; Galvani, A.P.; Moghadas, S.M. Temporal estimates of case-fatality rate for COVID-19 outbreaks in Canada and the United States. Can. Med. Assoc. J. 2020, 192, E666–E670. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, W.; Yao, J.; Nicholson, C.J.; Li, R.H.; Sigurslid, H.H.; Wooster, L.; Rotter, J.I.; Guo, X.; Malhotra, R. Predictors of mortality in hospitalized COVID-19 patients: A systematic review and meta-analysis. J. Med. Virol. 2020, 92, 1875–1883. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, Z.; Chen, W.; Cai, Z.; Ge, X.; Zhu, H.; Jiang, T.; Tan, W.; Peng, Y. Predicting the receptor-binding domain usage of the coronavirus based on kmer frequency on spike protein. Infect. Genet. Evol. 2018, 61, 183–184. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2013, 88, 1293–1307. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; Rubin, E.J. Covid-19—The Search for Effective Therapy. N. Engl. J. Med. 2020, 382, 1851–1852. [Google Scholar] [CrossRef] [PubMed]
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing Covid-19 Vaccines at Pandemic Speed. N. Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Lagier, J.-C.; Gautret, P.; Colson, P.; Fournier, P.-E.; Amrane, S.; Hocquart, M.; Mailhe, M.; Esteves-Vieira, V.; Doudier, B.; et al. Early treatment of COVID-19 patients with hydroxychloroquine and azithromycin: A retrospective analysis of 1061 cases in Marseille, France. Travel Med. Infect. Dis. 2020, 35, 101738. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef] [PubMed]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Sevestre, J.; Mailhe, M.; Doudier, B.; Aubry, C.; Amrane, S.; et al. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: A pilot observational study. Travel Med. Infect. Dis. 2020, 34, 101663. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Dufort, E.M.; Udo, T.; Wilberschied, L.A.; Kumar, J.; Tesoriero, J.; Weinberg, P.; Kirkwood, J.; Muse, A.; DeHovitz, J.; et al. Association of Treatment with Hydroxychloroquine or Azithromycin With In-Hospital Mortality in Patients With COVID-19 in New York State. JAMA 2020, 323, 2493–2502. [Google Scholar] [CrossRef]
- Mehra, M.R.; Desai, S.S.; Ruschitzka, F.; Patel, A.N. Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: A multinational registry analysis. Lancet 2020, 20, S0140. [Google Scholar]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19. JAMA 2021, 325, 632–644. [Google Scholar] [CrossRef]
- Freedman, M.S.; Ault, K.; Bernstein, H. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older—United States, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 193–196. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Espeseth, A.S.; Cejas, P.J.; Citron, M.P.; Wang, D.; DiStefano, D.J.; Callahan, C.; Donnell, G.O.; Galli, J.D.; Swoyer, R.; Touch, S.; et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccines 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Hammer, S.M.; Katzenstein, D.A.; Hughes, M.D.; Gundacker, H.; Schooley, R.T.; Haubrich, R.H.; Henry, W.K.; Lederman, M.M.; Phair, J.P.; Niu, M.T.; et al. A Trial Comparing Nucleoside Monotherapy with Combination Therapy in HIV-Infected Adults with CD4 Cell Counts from 200 to 500 per Cubic Millimeter. N. Engl. J. Med. 1996, 335, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Ahuja, S.D.; Akkerman, O.W.; Alffenaar, J.-W.C.; Anderson, L.F.; Baghaei, P.; Bang, D.; Barry, P.M.; Bastos, M.L.; Behera, D.; et al. Treatment correlates of successful outcomes in pulmonary multidrug-resistant tuberculosis: An individual patient data meta-analysis. Lancet 2018, 392, 821–834. [Google Scholar] [CrossRef]
- Wang, X.; Fan, X.; Deng, H.; Zhang, X.; Zhang, K.; Li, N.; Han, Q.; Lv, Y.; Liu, Z. Efficacy and safety of glecaprevir/pibrentasvir for chronic hepatitis C virus genotypes 1–6 infection: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2019, 54, 780–789. [Google Scholar] [CrossRef]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef]
- Dror, O.; Shulman-Peleg, A.; Nussinov, R.; Wolfson, H.J. Predicting molecular interactions in silico: I. A guide to pharmacophore identification and its applications to drug design. Curr. Med. Chem. 2004, 11, 71–90. [Google Scholar] [CrossRef]
- Jahn, A.; Hinselmann, G.; Fechner, N.; Zell, A. Optimal assignment methods for ligand-based virtual screening. J. Chemin. 2009, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.G.; Doucet, J.P.; Petitjean, M.; Fan, B.-T. Molecular similarity and diversity in chemoinformatics: From theory to applications. Mol. Divers. 2006, 10, 39–79. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Renault, N.; Lagorce, D.; Montes, M.; Miteva, M.A. Free Resources to Assist Structure-Based Virtual Ligand Screening Experiments. Curr. Protein Pept. Sci. 2007, 8, 381–411. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Recio, J.; Totrov, M.; Skorodumov, C.; Abagyan, R. Optimal docking area: A new method for predicting protein-protein interaction sites. Proteins: Struct. Funct. Bioinform. 2004, 58, 134–143. [Google Scholar] [CrossRef]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. Four-Dimensional Docking: A Fast and Accurate Account of Discrete Receptor Flexibility in Ligand Docking. J. Med. Chem. 2008, 52, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.M.; Evers, A.; Krüger, D.M. Comparison of Structure- and Ligand-Based Virtual Screening Protocols Considering Hit List Complementarity and Enrichment Factors. ChemMedChem 2010, 5, 148–158. [Google Scholar] [CrossRef]
- Bender, A. Compound bioactivities go public. Nat. Chem. Biol. 2010, 6, 309. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC–A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bernard, P.; Medina-Franco, J.L.; Maggiora, G.M. Large compound databases for structure-activity relationships studies in drug discovery. Mini-Rev. Med. Chem. 2007, 7, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Dueñas-González, A.; García-López, P.; Herrera, L.A.; Medina-Franco, J.L.; Gonzalez-Fierro, A.; Candelaria, M. The prince and the pauper. A tale of anticancer targeted agents. Mol. Cancer 2008, 7, 82. [Google Scholar] [CrossRef]
- O’Connor, K.A.; Roth, B.L. Finding New Tricks for Old Drugs: An Efficient Route For Public-Sector Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 1005–1014. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2007, 36, D901–D906. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Gozalbes, R.; Simon, L.; Froloff, N.; Sartori, E.; Monteils, C.; Baudelle, R. Development and Experimental Validation of a Docking Strategy for the Generation of Kinase-Targeted Libraries. J. Med. Chem. 2008, 51, 3124–3132. [Google Scholar] [CrossRef]
- Plewczyński, D.; Grotthuss, M.; Rychlewski, L.; Ginalski, K. Virtual High Throughput Screening Using Combined Random Forest and Flexible Docking. Comb. Chem. High Throughput Screen. 2009, 12, 484–489. [Google Scholar] [CrossRef]
- Lee, H.S.; Choi, J.; Kufareva, I.; Abagyan, R.; Filikov, A.; Yang, Y.; Yoon, S. Optimization of High Throughput Virtual Screening by Combining Shape-Matching and Docking Methods. J. Chem. Inf. Model. 2008, 48, 489–497. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, A.J.-F.; Cornell, W.D. Comparison of Topological, Shape, and Docking Methods in Virtual Screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef]
- Klon, A. Bayesian Modeling in Virtual High Throughput Screening. Comb. Chem. High Throughput Screen. 2009, 12, 469–483. [Google Scholar] [CrossRef]
- Jiang, W.; Phillips, J.C.; Huang, L.; Fajer, M.; Meng, Y.; Gumbart, J.C.; Luo, Y.; Schulten, K.; Roux, B. Generalized scalable multiple copy algorithms for molecular dynamics simulations in NAMD. Comput. Phys. Commun. 2014, 185, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Caulfield, T. An Induced-Fit Docking Method for Refining Drug-Receptor Interactions Derived from Maxwellian-Assessor Nanoprobes (Quantum Mechanics-Based Criterion Assessment) Placed Over Adaptive Intervals of Molecular Dynamics Sampling. Biophys. J. 2012, 102, 171a–172a. [Google Scholar] [CrossRef]
- Caulfield, T.R. Inter-ring rotation of apolipoprotein A-I protein monomers for the double-belt model using biased molecular dynamics. J. Mol. Graph. Model. 2011, 29, 1006–1014. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Kalid, O.; Warshaviak, D.T.; Shechter, S.; Sherman, W.; Shacham, S. Consensus Induced Fit Docking (cIFD): Methodology, validation, and application to the discovery of novel Crm1 inhibitors. J. Comput. Mol. Des. 2012, 26, 1217–1228. [Google Scholar] [CrossRef]
- Caulfield, T.; Coban, M.; Tek, A.; Flores, S.C. Tek Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of –1 Frameshifting by tRNASer3. Biomolecule 2019, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Kayode, O.; Wang, R.; Pendlebury, D.F.; Cohen, I.; Henin, R.D.; Hockla, A.; Soares, A.S.; Papo, N.; Caulfield, T.R.; Radisky, E.S. An Acrobatic Substrate Metamorphosis Reveals a Requirement for Substrate Conformational Dynamics in Trypsin Proteolysis. J. Biol. Chem. 2016, 291, 26304–26319. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.R.; Fiesel, F.C.; Moussaud-Lamodière, E.L.; Dourado, D.F.A.R.; Flores, S.; Springer, W. Phosphorylation by PINK1 Releases the UBL Domain and Initializes the Conformational Opening of the E3 Ubiquitin Ligase Parkin. PLoS Comput. Biol. 2014, 10, e1003935. [Google Scholar] [CrossRef]
- Caulfield, T.R.; Devkota, B.; Rollins, G.C. Examinations of tRNA Range of Motion Using Simulations of Cryo-EM Microscopy and X-Ray Data. J. Biophys. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef]
- Janes, J.; Young, M.E.; Chen, E.; Rogers, N.H.; Burgstaller-Muehlbacher, S.; Hughes, L.D.; Love, M.S.; Hull, M.V.; Kuhen, K.L.; Woods, A.K.; et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2018, 115, 10750–10755. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M.; Sankaranarayanan, M. A medicinal chemistry perspective of drug repositioning: Recent advances and challenges in drug discovery. Eur. J. Med. Chem. 2020, 195, 112275. [Google Scholar] [CrossRef]
- Ekins, S.; Freundlich, J.S.; Coffee, M. A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000Research 2014, 3, 277. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, J.C.B.; Arantes, M.E.; Andrade, C.H.; Silva, L.A.; Neves, B.J. In silico REPOSITIONING OF NEW DRUGS AGAINST Schistosoma mansoni. Rev. Patol. Trop. J. Trop. Pathol. 2018, 47, 159–166. [Google Scholar] [CrossRef]
- Lagarde, N.; Rey, J.; Gyulkhandanyan, A.; Tufféry, P.; Miteva, M.A.; Villoutreix, B.O. Online structure-based screening of purchasable approved drugs and natural compounds: Retrospective examples of drug repositioning on cancer targets. Oncotarget 2018, 9, 32346–32361. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Burgstaller-Muehlbacher, S.; Pache, L.; de Jesus, P.P.; Hull, M.V.; Chang, M.; et al. A Large-scale Drug Repositioning Survey for SARS-CoV-2 Antivirals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nat. Cell Biol. 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Baum, B.; Muley, L.; Heine, A.; Smolinski, M.; Hangauer, D.; Klebe, G. Think Twice: Understanding the High Potency of Bis(phenyl)methane Inhibitors of Thrombin. J. Mol. Biol. 2009, 391, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins: Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Hooft, R.; Sander, C.; Scharf, M.; Vriend, G. The PDBFINDER database: A summary of PDB, DSSP and HSSP information with added value. Bioinformatics 1996, 12, 525–529. [Google Scholar] [CrossRef][Green Version]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nat. Cell Biol. 1996, 381, 272. [Google Scholar] [CrossRef]
- King, R.D.; Sternberg, M.J.E. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 1996, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Elber, R. SSALN: An alignment algorithm using structure-dependent substitution matrices and gap penalties learned from structurally aligned protein pairs. Proteins: Struct. Funct. Bioinform. 2005, 62, 881–891. [Google Scholar] [CrossRef]
- Laskowski, R.; MacArthur, M.W.; Moss, D.S.; Thornton, J. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Caulfield, T.; Devkota, B. Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins Struct. Funct. Bioinform. 2012, 80, 2489–2500. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Abraham, M.J.; Gready, J.E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J. Comput. Chem. 2011, 32, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Chem. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Mooers, B.H.M. Simplifying and enhancing the use of PyMOL with horizontal scripts. Protein Sci. 2016, 25, 1873–1882. [Google Scholar] [CrossRef]
- Bhachoo, J.; Beuming, T. Investigating Protein–Peptide Interactions Using the Schrödinger Computational Suite. Methods Mol. Biology 2017, 1561, 235–254. [Google Scholar] [CrossRef]
- Maestro; Schrödinger, LLC: New York, NY, USA, 2014.
- Unger, M.; Eichhoff, A.M.; Schumacher, L.; Strysio, M.; Menzel, S.; Schwan, C.; Alzogaray, V.; Zylberman, V.; Seman, M.; Brandner, J.; et al. Selection of Nanobodies that Block the Enzymatic and Cytotoxic Activities of the Binary Clostridium Difficile Toxin CDT. Sci. Rep. 2015, 5, srep07850. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Vivoli, M.; Caulfield, T.R.; Martínez-Mayorga, K.; Johnson, A.T.; Jiao, G.-S.; Lindberg, I. Inhibition of Prohormone Convertases PC1/3 and PC2 by 2,5-Dideoxystreptamine Derivatives. Mol. Pharmacol. 2011, 81, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Loving, K.; Salam, N.K.; Sherman, W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel?an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Reumers, J.; Schymkowitz, J.; Ferkinghoff-Borg, J.; Stricher, F.; Serrano, L.; Rousseau, F. SNPeffect: A database mapping molecular phenotypic effects of human non-synonymous coding SNPs. Nucleic Acids Res. 2004, 33, D527–D532. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Rousseau, F.; Martins, I.C.; Ferkinghoff-Borg, J.; Stricher, F.; Serrano, L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc. Natl. Acad. Sci. USA 2005, 102, 10147–10152. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Caulfield, T.; Xu, Y.-F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef]
- Abdul-Hay, S.O.; Lane, A.L.; Caulfield, T.R.; Claussin, C.; Bertrand, J.; Masson, A.; Choudhry, S.; Fauq, A.H.; Maharvi, G.M.; Leissring, M.A. Optimization of Peptide Hydroxamate Inhibitors of Insulin-Degrading Enzyme Reveals Marked Substrate-Selectivity. J. Med. Chem. 2013, 56, 2246–2255. [Google Scholar] [CrossRef]
- Ando, M.; Fiesel, F.C.; Hudec, R.; Caulfield, T.R.; Ogaki, K.; Górka-Skoczylas, P.; Koziorowski, D.; Friedman, A.; Chen, L.; Dawson, V.L.; et al. The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 2017, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.R.; Fiesel, F.C.; Springer, W. Activation of the E3 ubiquitin ligase Parkin. Biochem. Soc. Trans. 2015, 43, 269–274. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Ando, M.; Hudec, R.; Hill, A.R.; Castanedes-Casey, M.; Caulfield, T.R.; Moussaud-Lamodière, E.L.; Stankowski, J.N.; Bauer, P.O.; Lorenzo-Betancor, O.; et al. (Patho-)physiological relevance of PINK 1-dependent ubiquitin phosphorylation. EMBO Rep. 2015, 16, 1114–1130. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Caulfield, T.R.; Moussaud-Lamodière, E.L.; Ogaki, K.; Dourado, D.F.; Flores, S.; Ross, O.A.; Springer, W. Structural and Functional Impact of Parkinson Disease-Associated Mutations in the E3 Ubiquitin Ligase Parkin. Hum. Mutat. 2015, 36, 774–786. [Google Scholar] [CrossRef]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Baugh, E.H.; Lyskov, S.; Weitzner, B.D.; Gray, J.J. Real-Time PyMOL Visualization for Rosetta and PyRosetta. PLoS ONE 2011, 6, e21931. [Google Scholar] [CrossRef]
- Dilip, A.; Lešnik, S.; Štular, T.; Janežič, D.; Konc, J. Ligand-based virtual screening interface between PyMOL and LiSiCA. J. Chem. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Janson, G.; Zhang, C.; Prado, M.G.; Paiardini, A. PyMod 2.0: Improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 2016, 33, 444–446. [Google Scholar] [CrossRef] [PubMed]
- Makarewicz, T.; Kaźmierkiewicz, R. Molecular Dynamics Simulation by GROMACS Using GUI Plugin for PyMOL. J. Chem. Inf. Model. 2013, 53, 1229–1234. [Google Scholar] [CrossRef]
- Makarewicz, T.; Kaźmierkiewicz, R. Improvements in GROMACS plugin for PyMOL including implicit solvent simulations and displaying results of PCA analysis. J. Mol. Model. 2016, 22, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pi, Q.; Maharjan, S.; Yan, X.; Liu, X.; Singh, B.; Van Genderen, A.M.; Robledo-Padilla, F.; Parra-Saldivar, R.; Hu, N.; Jia, W.; et al. Digitally Tunable Microfluidic Bioprinting of Multilayered Cannular Tissues. Adv. Mater. 2018, 30, e1706913. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Schuurmans, C.C.L.; Van Genderen, A.M.; Cao, X.; Li, W.; Cheng, F.; He, J.J.; López, A.; Huerta, V.; Manríquez, J.; et al. Complexation-induced resolution enhancement of 3D-printed hydrogel constructs. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Miri, A.K.; Nieto, D.; Iglesias, L.; Hosseinabadi, H.G.; Maharjan, S.; Ruiz-Esparza, G.U.; Khoshakhlagh, P.; Manbachi, A.; Dokmeci, M.R.; Chen, S.; et al. Microfluidics-Enabled Multimaterial Maskless Stereolithographic Bioprinting. Adv. Mater. 2018, 30, e1800242. [Google Scholar] [CrossRef]
- Von Roemeling, C.A.; Caulfield, T.R.; Marlow, L.; Bok, I.; Wen, J.; Miller, J.L.; Hughes, R.; Hazlehurst, L.; Pinkerton, A.B.; Radisky, D.C.; et al. Accelerated bottom-up drug design platform enables the discovery of novel stearoyl-CoA desaturase 1 inhibitors for cancer therapy. Oncotarget 2017, 9, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.R.; Harvey, S.C. Conformational fitting of atomic models to cryogenic-electron microscopy maps using Maxwell’s demon molecular dynamics. Biophys. J. 2007, 1735-Plat, 368A. [Google Scholar]
- Ko, C.-J.; Huang, C.-C.; Lin, H.-Y.; Juan, C.-P.; Lan, S.-W.; Shyu, H.-Y.; Wu, S.-R.; Hsiao, P.-W.; Huang, H.-P.; Shun, C.-T.; et al. Androgen-Induced TMPRSS2 Activates Matriptase and Promotes Extracellular Matrix Degradation, Prostate Cancer Cell Invasion, Tumor Growth, and Metastasis. Cancer Res. 2015, 75, 2949–2960. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The Androgen-Regulated Protease TMPRSS2 Activates a Proteolytic Cascade Involving Components of the Tumor Microenvironment and Promotes Prostate Cancer Metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.K.; Greer, B.; Hooper, J.D.; Zijlstra, A.; Walker, B.; Quigley, J.P.; Hawthorne, S.J. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem. J. 2005, 388, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, E.; Kyriakos, G.; Quiles-Sanchez, L.V.; Farmaki, P.; Troupis, T. The Anti-Inflammatory Effects of Statins on Coronary Artery Disease: An Updated Review of the Literature. Curr. Cardiol. Rev. 2017, 13, 209–216. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Isemura, M. Catechin in Human Health and Disease. Molecules 2019, 24, 528. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Salmean, G.; Ciaraldi, T.P.; Nogueira, L.; Barboza, J.; Taub, P.R.; Hogan, M.C.; Henry, R.R.; Meaney, E.; Villarreal, F.; Ceballos, G.; et al. Effects of (−)-epicatechin on molecular modulators of skeletal muscle growth and differentiation. J. Nutr. Biochem. 2014, 25, 91–94. [Google Scholar] [CrossRef]
- Gokulan, K.; Kolluru, P.; Cerniglia, C.E.; Khare, S. Dose-Dependent Effects of Aloin on the Intestinal Bacterial Community Structure, Short Chain Fatty Acids Metabolism and Intestinal Epithelial Cell Permeability. Front. Microbiol. 2019, 10, 474. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-Y.; Wu, S.-L.; Chen, J.-C.; Li, C.-C.; Hsiang, C.-Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef]
- Ingólfsdóttir, K. Usnic acid. Phytochemistry 2002, 61, 729–736. [Google Scholar] [CrossRef]
- Hajimehdipoor, H.; Saeidnia, S.; Gohari, A.R.; Hamedani, M.P.; Shekarchi, M. Comparative study of rosmarinic acid content in some plants of Labiatae family. Pharmacogn. Mag. 2012, 8, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.; Kuntz, H.-D. Biochemistry and Functions of the Liver. Hepatology 2002, 25–62. [Google Scholar] [CrossRef]
- Guguenguillouzo, C.; Campion, J.; Brissot, P.; Glaise, D.; Launois, B.; Bourel, M.; Guillouzo, A. High yield preparation of isolated human adult hepatocytes by enzymatic perfusion of the liver. Cell Biol. Int. Rep. 1982, 6, 625–628. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Shaegh, S.A.M.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef]
- Sun, L.; Yang, H.; Wang, Y.; Zhang, X.; Jin, B.; Xie, F.; Jin, Y.; Pang, Y.; Zhao, H.; Lu, X.; et al. Application of a 3D Bioprinted Hepatocellular Carcinoma Cell Model in Antitumor Drug Research. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Kizawa, H.; Nagao, E.; Shimamura, M.; Zhang, G.; Torii, H. Scaffold-free 3D bio-printed human liver tissue stably maintains metabolic functions useful for drug discovery. Biochem. Biophys. Rep. 2017, 10, 186–191. [Google Scholar] [CrossRef]
- Lam, T.; Ruppelt, A.; Thomas, A.; Amler, A.-K.; Noichl, B.P.; Lauster, R.; Kloke, L. Bioprinting Perfusion-Enabled Liver Equivalents for Advanced Organ-on-a-Chip Applications. Genes 2018, 9, 176. [Google Scholar] [CrossRef]
- Knowlton, S.; Tasoglu, S. A Bioprinted Liver-on-a-Chip for Drug Screening Applications. Trends Biotechnol. 2016, 34, 681–682. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, J.; Zhu, W.; Tang, M.; Lawrence, N.; Yu, C.; Gou, M.; Chen, S. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Adv. Drug Deliv. Rev. 2018, 132, 235–251. [Google Scholar] [CrossRef]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Zhang, Y.S.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8, 014101. [Google Scholar] [CrossRef]
- Massa, S.; Sakr, M.A.; Seo, J.; Bandaru, P.; Arneri, A.; Bersini, S.; Zare-Eelanjegh, E.; Jalilian, E.; Cha, B.-H.; Antona, S.; et al. Bioprinted 3D vascularized tissue model for drug toxicity analysis. Biomicrofluidics 2017, 11, 044109. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Jiang, N.; Yu, C.; Zhang, Y.S. Three-dimensional bioprinting of gelatin methacryloyl (GelMA). Bio-Design Manuf. 2018, 1, 215–224. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Sivaraman, A.; Leach, J.; Townsend, S.; Iida, T.; Hogan, B.; Stolz, D.; Fry, R.; Samson, L.; Tannenbaum, S.; Griffith, L. A Microscale In Vitro Physiological Model of the Liver: Predictive Screens for Drug Metabolism and Enzyme Induction. Curr. Drug Metab. 2005, 6, 569–591. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Lim, H.-K.; Duczak, N.; Brougham, L.; Elliot, M.; Patel, K.; Chan, K. Automated Screening with Confirmation of Mechanism-Based Inactivation of CYP3A4, CYP2C9, CYP2C19, CYP2D6, and CYP1A2 in Pooled Human Liver Microsomes. Drug Metab. Dispos. 2005, 33, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.F.; Nafziger, A.N.; Bertino, J.S. Pharmacogenetics affects dosing, efficacy, and toxicity of cytochrome P450–metabolized drugs. Am. J. Med. 2002, 113, 746–750. [Google Scholar] [CrossRef]
- Zhou, S.-F. Polymorphism of Human Cytochrome P450 2D6 and Its Clinical Significance. Clin. Pharmacokinet. 2009, 48, 689–723. [Google Scholar] [CrossRef]
- Ma, X.; Qu, X.; Zhu, W.; Li, Y.-S.; Yuan, S.; Zhang, H.; Liu, J.; Wang, P.; Lai, C.S.E.; Zanella, F.; et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc. Natl. Acad. Sci. USA 2016, 113, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.; Pfeffer, M.A.; Solomon, S.D. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Jarcho, J.A.; Ingelfinger, J.R.; Hamel, M.B.; D’Agostino, R.B.; Harrington, D.P. Inhibitors of the Renin–Angiotensin–Aldosterone System and Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin–Angiotensin–Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Mehta, N.; Kalra, A.; Nowacki, A.S.; Anjewierden, S.; Han, Z.; Bhat, P.; Carmona-Rubio, A.E.; Jacob, M.; Procop, G.W.; Harrington, S.; et al. Association of Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1020. [Google Scholar] [CrossRef]
- Patel, A.B.; Verma, A. COVID-19 and Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers. JAMA 2020, 323, 1769–1770. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Synonyms | Predicted Protein | In Silico Score | Target | CAS |
|---|---|---|---|---|---|
| Metaproterenol Sulfate | Orciprenaline Sulfate | ACE2 | −8.05 | Others | 5874-97-5 |
| Isoprenaline HCl | Isuprel, Isadrine, Euspiran, Proternol, NSC 37745, NSC 89747 | ACE2 | −7.44 | Adrenergic Receptor | 51-30-9 |
| Epinephrine HCl | N/A | ACE2 | −7.12 | Adrenergic Receptor | 55-31-2 |
| Levosulpiride | N/A | ACE2 | −6.87 | Dopamine Receptor | 23672-07-3 |
| Metaraminol Bitartrate | Metaradrine Bitartrate | ACE2 | −6.84 | Others | 33402-03-8 |
| Valganciclovir HCl | N/A | ACE2 | −6.58 | Antifection (Anti-Infection) | 175865-59-5 |
| Isoprenaline HCl | Isuprel, Isadrine, Euspiran, Proternol, NSC 37745, NSC 89747 | ACE2 | −6.45 | Adrenergic Receptor | 51-30-9 |
| S4817 Atenolol | Tenormin, Normiten, Blokium | ACE2 | −6.35 | β1 Receptor, β2 Receptor | 29122-68-7 |
| S3783 Echinacoside | N/A | ACE2 | −6.09 | Others | 82854-37-3 |
| Propafenone | Rythmol SR, Rytmonorm | ACE2 | −6.04 | Sodium Channel | 34183-22-7 |
| Amikacin sulfate | BB-K8 | ACE2 | −5.98 | Antifection | 39831-55-5 |
| Pro-Chlorperazine Dimaleate Salt | Prochlorperazin, Compazine, Capazine, Stemetil | ACE2 | −5.79 | Dopamine Receptor | 30718 |
| Isoetharine Mesylate | N/A | ACE2 | −5.47 | Others | 7279-75-6 |
| Levosulpiride | N/A | ACE2 | −6.87 | Dopamine Receptor | 23672-07-3 |
| S5023 Nadolol | Corgard, Solgol, Anabet | ACE2 | −5.16 | Androgen Receptor | 42200-33-9 |
| Benserazide HCl | Ro-4-4602 | ACE2 | −5.93 | Dopamine Receptor | 14919-77-8 |
| S3694 Glucosamine (HCl) | 2-Amino-2-Deoxy-Glucose HCl | ACE2 | −5.57 | Others | 66-84-2 |
| S4701 2-Deoxy-d-Glucose | 2-Deoxyglucose, NSC 15193 | ACE2 | −5.18 | Others | 154-17-6 |
| Inulin | N/A | ACE2 | −5.18 | Others | 9005-80-5 |
| Cephalexin | Alcephin, Cefablan, Keflex, Cefadin, Tepaxin | ACE2 | −5.11 | Antifection | 15686-71-2 |
| S4722 (+)-Catechin | Cianidanol, Catechinic Acid, Catechuic Acid | MPro | −6.73 | Others | 154-23-4 |
| S4723 (−) Epicatechin | l-Epicatechin, (−)-Epicatechol | MPro | −6.32 | Others | 490-46-0 |
| S5105 Proanthocyanidins | Condensed Tannins | MPro | −6.19 | Others | 20347-71-1 |
| Carbenicillin Disodium | N/A | MPro | −5.78 | Antifection | 4800-94-6 |
| AG-120 (Ivosidenib) | N/A | MPro | −5.52 | Dehydrogenase | 1448347-49-6 |
| Atorvastatin Calcium | N/A | MPro | −5.39 | HMG-CoA Reductase | 134523-03-8 |
| Bezafibrate | N/A | MPro | −4.93 | PPAR | 41859-67-0 |
| PF299804 | N/A | MPro | −4.34 | EGFR | 1110813-31-4 |
| Bumetanide | Bumex | TMPRSS2 | −6.5 | Others | 28395-03-1 |
| Aloin | Barbaloin | TMPRSS2 | −6.45 | Tyrosinase | 1415-73-2 |
| Salbutamol Sulfate | Ventolin, Asthalin, Asthavent | TMPRSS2 | −6.1 | Adrenergic Receptor | 51022-70-9 |
| S4953 Usnic Acid | Usniacin | TMPRSS2 | −5.8 | Others | 125-46-2 |
| Avanafil | N/A | TMPRSS2 | −5.62 | PDE | 330784-47-9 |
| S3612 Rosmarinic Acid | Rosemary Acid | TMPRSS2 | −5.6 | IKK-β | 20283-92-5 |
| S5105 Proanthocyanidins | Condensed Tannins | TMPRSS2 | −5.51 | Others | 20347-71-1 |
| Ractopamine HCl | N/A | TMPRSS2 | −5.22 | Others | 90274-24-1 |
| Neohesperidin Dihydrochalcone | Neohesperidin DHC | TMPRSS2 | −5.2 | Others | 20702-77-6 |
| Cidofovir | Vistide | TMPRSS2 | −5.18 | Others | 113852-37-2 |
| Zidovudine | Azidothymidine | TMPRSS2 | −5.02 | Reverse Transcriptase | 30516-87-1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coban, M.A.; Morrison, J.; Maharjan, S.; Hernandez Medina, D.H.; Li, W.; Zhang, Y.S.; Freeman, W.D.; Radisky, E.S.; Le Roch, K.G.; Weisend, C.M.; et al. Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement. Biomolecules 2021, 11, 787. https://doi.org/10.3390/biom11060787
Coban MA, Morrison J, Maharjan S, Hernandez Medina DH, Li W, Zhang YS, Freeman WD, Radisky ES, Le Roch KG, Weisend CM, et al. Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement. Biomolecules. 2021; 11(6):787. https://doi.org/10.3390/biom11060787
Chicago/Turabian StyleCoban, Mathew A., Juliet Morrison, Sushila Maharjan, David Hyram Hernandez Medina, Wanlu Li, Yu Shrike Zhang, William D. Freeman, Evette S. Radisky, Karine G. Le Roch, Carla M. Weisend, and et al. 2021. "Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement" Biomolecules 11, no. 6: 787. https://doi.org/10.3390/biom11060787
APA StyleCoban, M. A., Morrison, J., Maharjan, S., Hernandez Medina, D. H., Li, W., Zhang, Y. S., Freeman, W. D., Radisky, E. S., Le Roch, K. G., Weisend, C. M., Ebihara, H., & Caulfield, T. R. (2021). Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement. Biomolecules, 11(6), 787. https://doi.org/10.3390/biom11060787