
Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine



Abstract
1. Introduction
2. Materials and Methods
2.1. Mathematical Modelling
2.2. Describing the Theoretical Analysis
3. Results
3.1. KRAS Mutant Cancer Cells Exhibit High Levels of Both GLUT1 and Autophagy
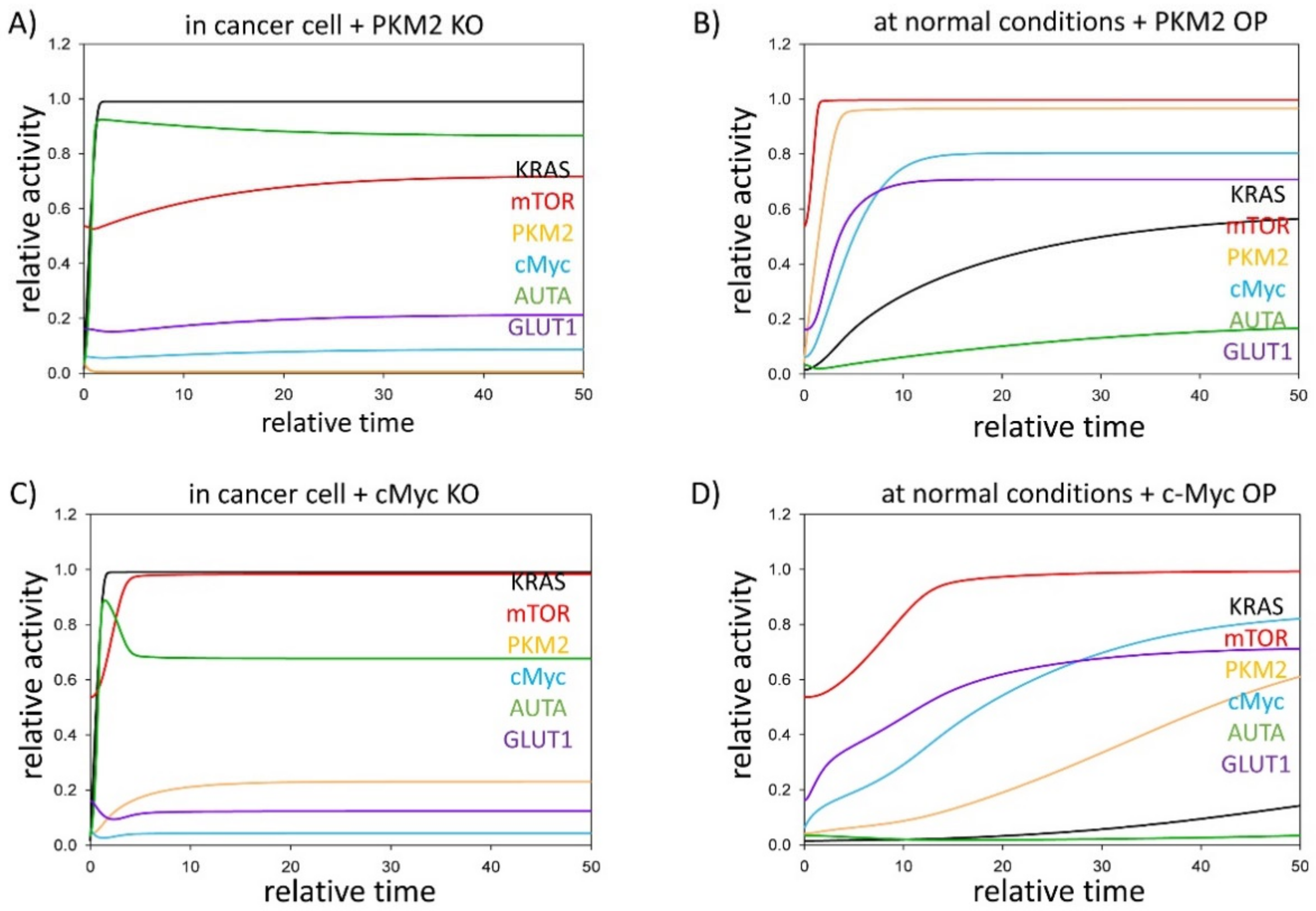
3.2. PKM2 Level Directly and Indirectly Affects Stress–Response Mechanisms
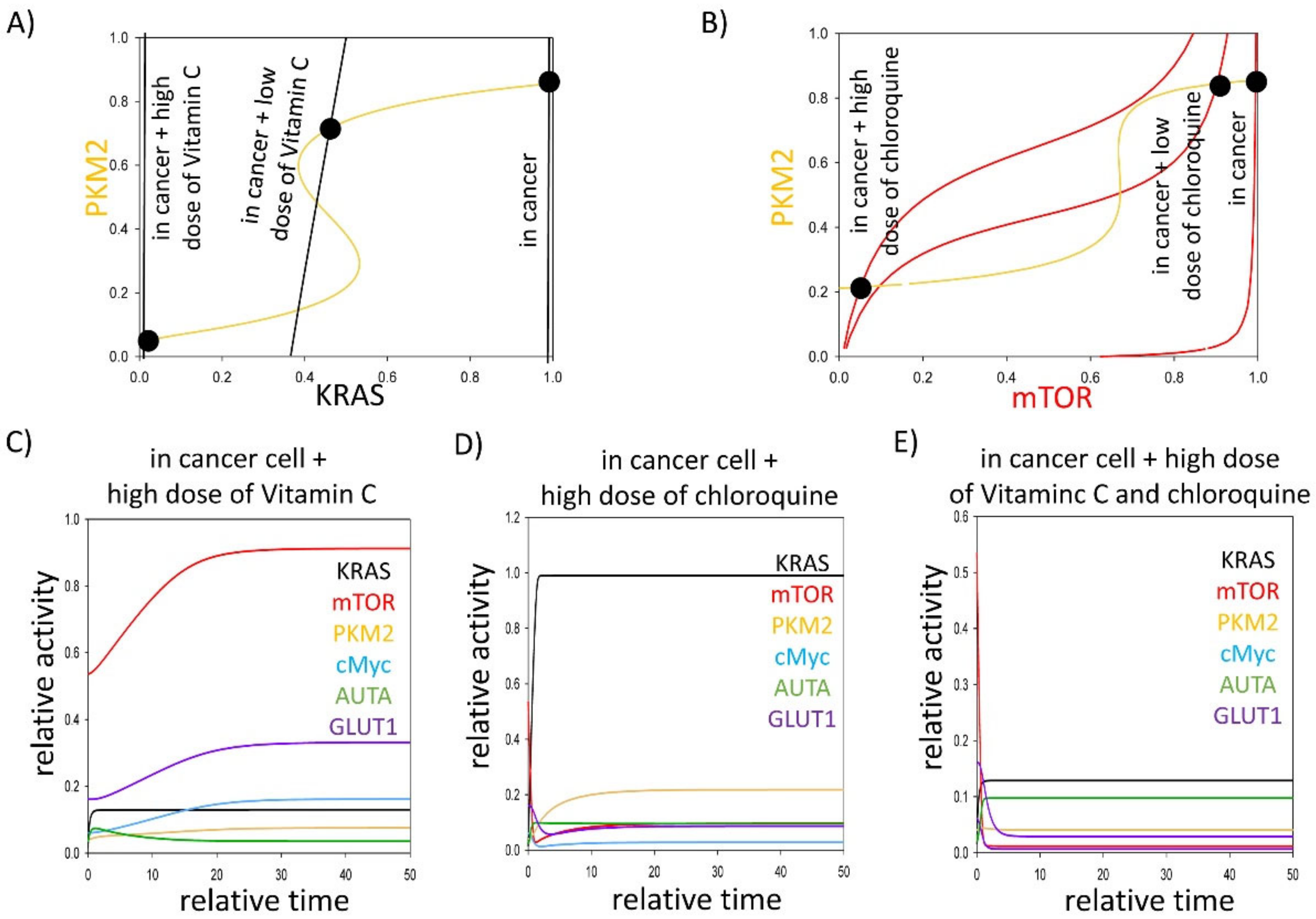
3.3. Either KRAS- or mTOR Pathway Inhibition Downregulates GLUT1 Level in Cancer Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| AMP | adenosine monophosphate |
| ATP | adenosine triphosphate |
| ERK1/2 | extracellular signal-regulated kinases |
| GLUT1–4 | glucose transporter isozymes 1–4 |
| GSH | glutathione |
| HK 1–4 | hexokinase isozymes 1–4 |
| LDHA | lactate dehydrogenase A |
| mTOR | mammalian target of rapamycin |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| ODE | ordinary differential equations |
| PET | positron emission tomography |
| PFK1/2 | phosphofructokinase isozymes 1/2 |
| PI3K | phosphoinositide 3-kinase |
| PK | pyruvate kinase |
| PKM1/2 | pyruvate kinase isozymes M1/M2 |
| SGLT1 | sodium-glucose linked transporter |
| TCA cycle | tricarboxylic acid cycle |
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Elements of the Theoretical Models | Description | Parameter Value |
|---|---|---|
| KRAS | The active form of KRAS | variable |
| KRAST | Total level of KRAS | 1 |
| PKM2 | The active form of pyruvate kinase 2 | variable |
| PKM2T | Total level of pyruvate kinase 2 | 1 |
| mTOR | The active form of mTOR | variable |
| mTORT | Total level of mTOR | 1 |
| cMyc | The active form of cMyc | variable |
| cMycT | Total level of cMyc | 1 |
| AUTA | The active autophagy activator complex | variable |
| AUTAT | Total amount of autophagy | 1 |
| GLUT1 | The active form of GLUT1 | variable |
| VitC | Vitamin C | 1 |
| CHL | Chloroquine | 1 |
| Constants of the Theoretical Models | Description | Parameter Value |
|---|---|---|
| karas | Background activation of KRAS | 0.01; 1 |
| karas’ | PKM2-dependent activation of KRAS | 0.1 |
| kiras | Background inactivation of KRAS | 0.1 |
| kiras’ | Vitamin C-dependent inactivation of KRAS | 0; 1.5 |
| Jkras | Michaelis constant of KRAS | 0.1 |
| kamr | Background activation of mTOR | 0.01 |
| kamr’ | KRAS-dependent activation of mTOR | 0.1 |
| kamr” | PKM2-dependent activation of mTOR | 2.25 |
| kimr | Background inactivation of mTOR | 0.1; 0.2 |
| kimr’ | Chloroquine-dependent inactivation of mTOR | 0; 1; 1.5 |
| Jmr | Michaelis constant of mTOR | 0.1 |
| kapkm | Background activation of PKM2 | 0.05 |
| kapkm’ | KRAS-dependent activation of PKM2 | 0.1 |
| kapkm” | cMyc-dependent activation of PKM2 | 0.2 |
| kipkm | Background activation of autophagy | 0.2 |
| Jpkm | Michaelis constant of PKM2 | 0.1 |
| kamyc | Background activation of cMyc | 0.01 |
| kamyc’ | PKM2-dependent activation of cMyc | 0.2 |
| kamyc” | mTOR-dependent inactivation of cMyc | 0.2 |
| kimyc | Background inactivation of cMyc | 0.3 |
| Jmyc | Michaelis constant of cMyc | 0.1 |
| kaau | Background activation of autophagy | 1 |
| kaau’ | KRAS-dependent activation of autophagy | 7.5 |
| kiau | Background inactivation of autophagy | 0.01 |
| kiau’ | mTOR-dependent inactivation of autophagy | 7.5 |
| Jau | Michaelis constant of autophagy | 0.1 |
| kaglu | Background activation of GLUT1 | 0.01 |
| kaglu’ | cMyc-dependent activation of GLUT1 | 3 |
| kiglu | Background inactivation of GLUT1 | 1 |
| Jglu | Michaelis constant of GLUT1 | 0.01 |
Appendix B
| #1 CODE |
| # a model to generate PKM2–KRAS signal–response curves |
| # initial conditions |
| init KRAS=0, PKM2=0 |
| # differential equations |
| # KRAS represents the active form of KRAS |
| KRAS’= (karas + karas’*PKM2)*(KRAST-KRAS)/(Jkras + KRAST-KRAS)−(kiras + kiras’*VitC)*KRAS/(Jkras + KRAS) |
| # PKM2 represents the active form of PKM2 |
| PKM2’ = (kapkm + kapkm’*KRAS + kapkm”*cMyc)*(PKM2T-PKM2)/(Jpkm + PKM2T-PKM2)−kipkm*PKM2/(Jpkm + PKM2) |
| # steady state functions |
| # mTOR represents the active form of mTOR |
| mTOR = mTORT*GK(kamr + kamr’*KRAS + kamr”*PKM2,kimr + kimr’*CHL,Jmr,Jmr) |
| # cMyc represents the active form of cMyc |
| cMyc = cMycT*GK(kamyc + kamyc’*PKM2 + kamyc”*mTOR,kimyc,Jmyc,Jmyc) |
| # ‘Goldbeter-Koshand’ function (GK) |
| GB(arg1,arg2,arg3,arg4) = arg2-arg1+arg2*arg3+arg1*arg4 |
| GK(arg1,arg2,arg3,arg4) = 2*arg1*arg4/(GB(arg1,arg2,arg3,arg4)+sqrt(GB(arg1,arg2,arg3,arg4)^2-4*(arg2-arg1)*arg1*arg4)) |
| # parameters |
| # to simulate cancer: karas=1 |
| # to simulate chloroquine treatment in cancer: karas=1, kimr’=1.5 |
| # to simulate Vitamin C treatment in cancer: karas=1, kiras’=1.5 |
| # to simulate PKM2 over-expression: karas=0.01, kapkm=0.5 |
| # to simulate PKM2 depletion: karas=0.01, PKM2T=0.01 |
| # to simulate cMyc over-expression: karas=1, cMycT=10 |
| # to simulate cMyc depletion: karas=1, cMycT=0.1 |
| p karas=0.01, karas’=0.1, kiras=0.1, kiras’=0, KRAST=1, Jkras=0.1 |
| p kamr=0.01, kamr’=0.1, kamr”=2.25, kimr=0.1, kimr’=0, mTORT=1, Jmr=0.1 |
| p kapkm=0.05, kapkm’=0.1, kapkm”=0.2, kipkm=0.2, PKM2T=1, Jpkm=0.1 |
| p kamyc=0.01, kamyc’=0.2, kamyc”=0.2, kimyc=0.3, cMycT=1, Jmyc=0.1 |
| p CHL=1, VitC=1 |
| done |
| #2 CODE |
| # a model to generate mTOR–KRAS signal–response curves |
| # initial conditions |
| init KRAS=0, PKM2=0 |
| # differential equations |
| # mTOR represents the active form of mTOR |
| mTOR’ = (kamr + kamr’*KRAS + kamr”*PKM2)*(mTORT-mTOR)/(Jmr + mTORT-mTOR)−(kimr + kimr’*CHL)*mTOR/(Jmr + mTOR) |
| # PKM2 represents the active form of PKM2 |
| PKM2’ = (kapkm + kapkm’*KRAS + kapkm”*cMyc)*(PKM2T-PKM2)/(Jpkm + PKM2T-PKM2)−kipkm*PKM2/(Jpkm + PKM2) |
| # steady state functions |
| # KRAS represents the active form of KRAS |
| KRAS = KRAST*GK(karas + karas’*PKM2,kiras + kiras’*VitC,Jkras,Jkras) |
| # cMyc represents the active form of cMyc |
| cMyc = cMycT*GK(kamyc + kamyc’*PKM2 + kamyc”*mTOR,kimyc,Jmyc,Jmyc) |
| # ‘Goldbeter-Koshand’ function (GK) |
| GB(arg1,arg2,arg3,arg4) = arg2-arg1+arg2*arg3+arg1*arg4 |
| GK(arg1,arg2,arg3,arg4) = 2*arg1*arg4/(GB(arg1,arg2,arg3,arg4)+sqrt(GB(arg1,arg2,arg3,arg4)^2-4*(arg2-arg1)*arg1*arg4)) |
| # parameters |
| # to simulate cancer: karas=1 |
| # to simulate chloroquine treatment in cancer: karas=1, kimr’=1.5 |
| # to simulate Vitamin C treatment in cancer: karas=1, kiras’=1.5 |
| # to simulate PKM2 over-expression: karas=0.01, kapkm=0.5 |
| # to simulate PKM2 depletion: karas=0.01, PKM2T=0.01 |
| # to simulate cMyc over-expression: karas=1, cMycT=10 |
| # to simulate cMyc depletion: karas=1, cMycT=0.1 |
| p karas=0.01, karas’=0.1, kiras=0.1, kiras’=0, KRAST=1, Jkras=0.1 |
| p kamr=0.01, kamr’=0.1, kamr”=2.25, kimr=0.2, kimr’=0, mTORT=1, Jmr=0.1 |
| p kapkm=0.05, kapkm’=0.1, kapkm”=0.2, kipkm=0.2, PKM2T=1, Jpkm=0.1 |
| p kamyc=0.01, kamyc’=0.2, kamyc”=0.2, kimyc=0.3, cMycT=1, Jmyc=0.1 |
| p CHL=1, VitC=1 |
| done |
| #3 CODE |
| # a model to simulate time courses |
| # initial conditions, |
| init KRAS= 0.0146, mTOR= 0.5365, PKM2= 0.0405, cMyc= 0.0607, AUTA= 0.0332, GLUT1= 0.1612 |
| # differential equations |
| # KRAS represents the active form of KRAS |
| KRAS’= (karas + karas’*PKM2)*(KRAST-KRAS)/(Jkras + KRAST-KRAS)−(kiras + kiras’*VitC)*KRAS/(Jkras + KRAS) |
| # mTOR represents the active form of mTOR |
| mTOR’ = (kamr + kamr’*KRAS + kamr”*PKM2)*(mTORT-mTOR)/(Jmr + mTORT-mTOR)−(kimr + kimr’*CHL)*mTOR/(Jmr + mTOR) |
| # PKM2 represents the active form of PKM2 |
| PKM2’ = (kapkm + kapkm’*KRAS + kapkm”*cMyc)*(PKM2T-PKM2)/(Jpkm + PKM2T-PKM2)−kipkm*PKM2/(Jpkm + PKM2) |
| # cMyc represents the active form of cMyc |
| cMyc’ = (kamyc + kamyc’*PKM2 + kamyc”*mTOR)*(cMycT-cMyc)/(Jmyc + cMycT-cMyc)−kimyc*cMyc/(Jmyc + cMyc) |
| # AUTA represents the active autophagy |
| AUTA’ = (kaau + kaau’*KRAS)*(AUTAT-AUTA)/(Jau + AUTAT -AUTA)−(kiau + kiau’*mTOR)*AUTA/(Jau + AUTA) |
| # represents the active form of GLUT1 |
| GLUT1’ =(kaglu + kaglu’*cMyc)*(1-GLUT1)/(Jglu + 1-GLUT1)−kiglu*GLUT1/(Jglu + 1-GLUT1) |
| # parameters |
| # to simulate cancer: karas=1 |
| # to simulate chloroquine treatment in cancer: karas=1, kimr’=1, AUTAT=0.1 |
| # to simulate Vitamin C treatment in cancer: karas=1, kiras’=1 |
| # to simulate PKM2 over-expression: karas=0.01, kapkm=0.5 |
| # to simulate PKM2 depletion: karas=0.01, PKM2T=0.01 |
| # to simulate cMyc over-expression: karas=1, cMycT=10 |
| # to simulate cMyc depletion: karas=1, cMycT=0.1 |
| p karas=0.01, karas’=0.1, kiras=0.1, kiras’=0, KRAST=1, Jkras=0.1 |
| p kamr=0.01, kamr’=0.1, kamr”=2.25, kimr=0.1, kimr’=0, mTORT=1, Jmr=0.1 |
| p kapkm=0.05, kapkm’=0.1, kapkm”=0.2, kipkm=0.2, PKM2T=1, Jpkm=0.1 |
| p kamyc=0.01, kamyc’=0.2, kamyc”=0.2, kimyc=0.3, cMycT=1, Jmyc=0.1 |
| p kaau=1, kaau’=7.5 kiau=0.01, kiau’=7.5, Jau=0. 1, |
| p kaglu=0.01, kaglu’=3, kiglu=1, Jglu=0.01 |
| p CHL=1, VitC=1 |
| done |
References
- Warburg, O.; Wind, F.; Negelein, E. Über den Stoffwechsel von Tumoren im Körper. J. Mol. Med. 1926, 5, 829–832. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Über den heutigen Stand des Carcinomproblems. In Über die Katalytischen Wirkungen der Lebendigen Substanz; Warburg, O., Ed.; Springer: Berlin/Heidelberg, Germany, 1928; pp. 501–509. [Google Scholar]
- Chance, B.; Castor, L.N. Some Patterns of the Respiratory Pigments of Ascites Tumors of Mice. Science 1952, 116, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Hess, B. Spectroscopic Evidence of Metabolic Control: Rapid measurements of intracellular events afford new evidence on mechanisms for metabolic control. Science 1959, 129, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, S.; Warburg, O.; Burk, D.; Schade, A.L. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 267–272. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation | Science. Available online: https://science.sciencemag.org/content/324/5930/1029/tab-e-letters (accessed on 27 August 2020).
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Omar, M.F.M.; Soong, R. The Warburg effect and drug resistance. Br. J. Pharmacol. 2016, 173, 970–979. [Google Scholar] [CrossRef]
- Szarka, A.; Kapuy, O.; Lőrincz, T.; Bánhegyi, G. Vitamin C and Cell Death. Antioxid. Redox Signal. 2021, 34, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Hoffman, J.M.; Johnson, B.; Scher, H.I.; Siegel, B.A.; Cheng, E.Y.; Cheson, B.D.; O’Shaughnessy, J.; Guyton, K.Z.; Mankoff, D.A.; et al. Progress and Promise of FDG-PET Imaging for Cancer Patient Management and Oncologic Drug Development. Clin. Cancer Res. 2005, 11, 2785–2808. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Onodera, Y.; Nam, J.-M.; Bissell, M.J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 2014, 124, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.L.; Palacios-Callender, M.; Frakich, N.; De Leon, J.; Schmitt, C.A.; Boorn, L.; Davis, N.; Moncada, S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 18868–18873. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.A.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3343–3347. [Google Scholar] [CrossRef]
- Unterlass, J.E.; Curtin, N.J. Warburg and Krebs and related effects in cancer. Expert Rev. Mol. Med. 2019, 21, e4. [Google Scholar] [CrossRef]
- Serna-Blasco, R.; Sanz-Álvarez, M.; Aguilera, Ó.; García-Foncillas, J. Targeting the RAS-dependent chemoresistance: The Warburg connection. Semin. Cancer Biol. 2019, 54, 80–90. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish, A.; Pandey, U.; Mishra, S.P. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef]
- Komatsu, N.; Fujita, Y.; Matsuda, M.; Aoki, K. mTORC1 upregulation via ERK-dependent gene expression change confers intrinsic resistance to MEK inhibitors in oncogenic KRas-mutant cancer cells. Oncogene 2015, 34, 5607–5616. [Google Scholar] [CrossRef]
- Dey, P.; Kundu, A.; Sachan, R.; Park, J.H.; Ahn, M.Y.; Yoon, K.; Lee, J.; Kim, N.D.; Kim, I.S.; Lee, B.M.; et al. PKM2 Knockdown Induces Autophagic Cell Death via AKT/mTOR Pathway in Human Prostate Cancer Cells. Cell. Physiol. Biochem. 2019, 52, 1535–1552. [Google Scholar] [CrossRef] [PubMed]
- He, C.-L.; Bian, Y.-Y.; Xue, Y.; Liu, Z.-X.; Zhou, K.-Q.; Yao, C.-F.; Lin, Y.; Zou, H.-F.; Luo, F.-X.; Qu, Y.-Y.; et al. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci. Rep. 2016, 6, 21524. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5, 2078. [Google Scholar] [CrossRef] [PubMed]
- Iovanna, J.L. Autophagy Induced during Pancreatitis Promotes KRAS-Dependent Transformation in the Pancreas. Front. Oncol. 2016, 6, 226. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Shamsi, M.; Saghafian, M.; Dejam, M.; Sanati-Nezhad, A. Mathematical Modeling of the Function of Warburg Effect in Tumor Microenvironment. Sci. Rep. 2018, 8, 8903. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Bucci, V.; Akkari, L.; Deforet, M.; Joyce, J.A.; Xavier, J.B. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc. Natl. Acad. Sci. USA 2013, 110, 19402–19407. [Google Scholar] [CrossRef]
- Astanin, S.; Preziosi, L. Mathematical modelling of the Warburg effect in tumour cords. J. Theor. Biol. 2009, 258, 578–590. [Google Scholar] [CrossRef][Green Version]
- Schuster, S.; Boley, D.; Möller, P.; Stark, H.; Kaleta, C. Mathematical models for explaining the Warburg effect: a review focussed on ATP and biomass production. Biochem. Soc. Trans. 2015, 43, 1187–1194. [Google Scholar] [CrossRef]
- O’Brien, C.; Allman, A.; Daoutidis, P.; Hu, W.-S. Kinetic model optimization and its application to mitigating the Warburg effect through multiple enzyme alterations. Metab. Eng. 2019, 56, 154–164. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.C.; Novak, B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Strogatz, S.H. Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry, and Engineering; Perseus Books Publishing: New York, NY, USA, 1994. [Google Scholar]
- Kaplan, D.; Glass, L. Understanding Nonlinear Dynamics. Available online: https://www.springer.com/gp/book/9780387944234 (accessed on 27 August 2020).
- Holczer, M.; Márton, M.; Kurucz, A.; Bánhegyi, G.; Kapuy, O. A Comprehensive Systems Biological Study of Autophagy-Apoptosis Crosstalk during Endoplasmic Reticulum Stress. BioMed Res. Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Novak, B.; Tyson, J.J. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J. Cell Sci. 1993, 106, 1153–1168. [Google Scholar] [CrossRef]
- Tyson, J.J.; Baumann, W.T.; Chen, C.; Verdugo, A.; Tavassoly, I.; Wang, Y.; Weiner, L.M.; Clarke, R. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 2011, 11, 523–532. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.; Novak, B. Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2001, 2, 908–916. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Available online: https://www.wiley.com/en-us/Enzyme+Kinetics%3A+Behavior+and+Analysis+of+Rapid+Equilibrium+and+Steady+State+Enzyme+Systems-p-9780471303091 (accessed on 31 August 2020).
- Goldbeter, A.; Koshland, D.E. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. USA 1981, 78, 6840–6844. [Google Scholar] [CrossRef]
- Tyson, J.J.; Csikasz-Nagy, A.; Novak, B. The dynamics of cell cycle regulation. BioEssays 2002, 24, 1095–1109. [Google Scholar] [CrossRef]
- Ferrell, J.E. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 1996, 21, 460–466. [Google Scholar] [CrossRef]
- Kapuy, O.; Barik, D.; Sananes, M.R.D.; Tyson, J.J.; Novák, B. Bistability by multiple phosphorylation of regulatory proteins. Prog. Biophys. Mol. Biol. 2009, 100, 47–56. [Google Scholar] [CrossRef]
- Cai, Y.; Cai, J.; Ma, Q.; Xu, Y.; Zou, J.; Xu, L.; Wang, D.; Guo, X. Chloroquine affects autophagy to achieve an anticancer effect in EC109 esophageal carcinoma cells in�vitro. Oncol. Lett. 2017, 15, 1143–1148. [Google Scholar] [CrossRef]
- Xu, Y.; Parmar, A.; Roux, E.; Balbis, A.; Dumas, V.; Chevalier, S.; Posner, B.I. Epidermal Growth Factor-induced Vacuolar (H+)-ATPase Assembly. J. Biol. Chem. 2012, 287, 26409–26422. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.D.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy, Metabolism, and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; RajeshKumar, N.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Gao, S.; Chen, M.; Wei, W.; Zhang, X.; Zhang, M.; Yao, Y.; Lv, Y.; Ling, T.; Wang, L.; Zou, X. Crosstalk of mTOR/PKM2 and STAT3/c-Myc signaling pathways regulate the energy metabolism and acidic microenvironment of gastric cancer. J. Cell. Biochem. 2019, 120, 1193–1202. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Kawada, K.; Nakamoto, Y.; Itatani, Y.; Inamoto, S.; Toda, K.; Kimura, H.; Sasazuki, T.; Shirasawa, S.; Okuyama, H.; et al. Regulation of 18F-FDG Accumulation in Colorectal Cancer Cells with Mutated KRAS. J. Nucl. Med. 2014, 55, 2038–2044. [Google Scholar] [CrossRef]
- Kawada, K.; Nakamoto, Y.; Kawada, M.; Hida, K.; Matsumoto, T.; Murakami, T.; Hasegawa, S.; Togashi, K.; Sakai, Y. Relationship between 18F-Fluorodeoxyglucose Accumulation and KRAS/BRAF Mutations in Colorectal Cancer. Clin. Cancer Res. 2012, 18, 1696–1703. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.D.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.P.; Fako, V.; Dang, H.; Dominguez, D.A.; Khatib, S.; Ma, L.; Wang, H.; Zheng, W.; Wang, X.W. PKM2 inhibition may reverse therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 99. [Google Scholar] [CrossRef] [PubMed]



| Model Organism (Mutant) | Treatment | Observed Phenomenon (after Treatment) | Reference |
|---|---|---|---|
| Xenografts (KRAS mutant G12D) | The mutated KRAS maintains tumour growth by the stimulation of glucose uptake and channels glucose intermediates into the hexosamine biosynthesis pathway and PPP to provide further biosynthetic precursors | [59] | |
| HCT116 and DLD-1 (KRAS mutants); clinical CRC samples | CRC cells with mutated KRAS increased 18F-FDG accumulation by upregulating GLUT1 | [60] | |
| HCT116 and DLD-1 (KRAS mutants); xenografts | Glucose deprivation can drive the acquisition of KRAS pathway mutations in human cancer cells | [62] | |
| U251 human glioblastoma multiforme cells (ERK2, MEK1 mutants), U87/EGFR cells (PKM2 mutants) | EGF stimulation | Nuclear translocation of PKM2 is a cause of the Warburg effect; nuclear PKM2 induces c-Myc expression, resulting in the upregulation of GLUT1, LDHA, and, in a positive feedback loop, PTB-dependent PKM2 expression | [51] |
| U87/EGFR GBM cells, U251 GBM cells, and human embryonic kidney 293T | EGF treatment | Monomeric PKM2 translocates into the nucleus, where it functions as a histone kinase and upregulates the expression of c-Myc and cyclin D1 | [20] |
| U87/EGFR cells | EGF treatment | PKM2 upregulates the expression of c-Myc and cyclin D1 | [19] |
| AGS and HGC-27 gastric cancer cells | PKM2 and c-Myc lentivirus | PKM2 and c-Myc were upregulated in human gastric cancer; knockdown of c-Myc in gastric cancer cells suppressed cell proliferation capacity and glycolysis level, the inhibitory effects on gastric cancer cells upon co-knockdown of PKM2 and c-Myc were more obvious compared with knockout of PKM2 or c-Myc alone | [55] |
| HCT116 and DLD1 (KRAS mutants) xenografts derived from parental HCT116 and VACO432 cell lines | High dose Vitamin C | Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells | [63] |
| SW480 (KRAS mutation (G12V); LoVo cancer cells (KRAS mutant G13D); xenograft (SW480) | Pharmacologic ascorbate | c-Myc is able to upregulate PKM2; in KRAS mutants, Vitamin C induces strong downregulation of the glucose transporter (GLUT1) and PKM2-PTB-dependent protein expression | [24] |
| NCI-H460, A549, HCT116 (KRAS mutants) | mTOR (mammalian target of rapamycin) is also activated in KRAS mutant | [25] | |
| DU145 prostate cancer cells | Prostate cancer cells show increased proliferation because of PKM2 overexpression; PKM2 knockdown downregulated β-catenin and c-MYC expression, which in turn decreased the expression of glycolytic enzymes LDHA, GLUT1, and of HIF1α; PKM2 knockdown inhibits the Akt/mTOR pathway | [26] | |
| HEK293T and HeLa cells | Overexpression of PKM2 activates mTORC1 signalling | [27] | |
| MEFs, PANC-1, PC3, and HepG2 | mTOR pathway also enhances aerobic glycolysis by upregulating PKM2 via c-Myc induction | [28] | |
| T24, H1299, H460, PC-3, PANC-1, HCT116 | Chloroquine | Expression of an H-rasV12 or K-rasV12 oncogene up-regulates basal autophagy, which is required for tumour cell survival in starvation and in tumorigenesis; high level of autophagy in Ras expressing cells occurred despite active mTOR, which suppresses autophagy and is activated by Ras, which suggests that the high basal autophagy caused by Ras must result from an mTOR-independent mechanism | [52] |
| PDAC cell lines (8988T, Panc1, pl45, HupT3, 8902, BXPC3, 10.05, 2.03); xenografts; primary tumours | Chloroquine | The basal level of autophagy was found to be elevated in several PDAC cell lines and primary tumours; PDAC cell lines exhibit a marked sensitivity to chloroquine; chloroquine treatment leads to robust tumour regression and prolonged survival in pancreatic cancer xenografts | [58] |
| Pa01C, Pa02C, Pa04C, Pa14C and Pa16C; PANC-1; HPAC; HPAF-II; MIA PaCa-2; SW1990; CFPAC-1; BxPC3; HEK-293T; RIE-1 cell lines; xenografts | KRAS; MEK; ERK1/2 inhibitors; hydroxychloroquine | Genetic or pharmacologic inhibition of specific autophagy regulators synergistically enhanced the ability of ERK inhibitors to mediate anti-tumour activity in KRAS-driven PDAC | [56] |
| Mia-PaCa2; BxPC3; PDX220; xenografts | chloroquine/ hydroxychloroquine; ARS-853 (inhibitor of KRASG12C), trametinib or cobimetinib (MEK1/2 inhibitors), or SCH772984 (ERK1/2 inhibitor) | Trametinib and chloroquine are synergistically cytotoxic to PDA cell lines in vitro and promote regression of RAS→RAF→MEK→ERK driven cancers | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapuy, O.; Makk-Merczel, K.; Szarka, A. Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine. Biomolecules 2021, 11, 652. https://doi.org/10.3390/biom11050652
Kapuy O, Makk-Merczel K, Szarka A. Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine. Biomolecules. 2021; 11(5):652. https://doi.org/10.3390/biom11050652
Chicago/Turabian StyleKapuy, Orsolya, Kinga Makk-Merczel, and András Szarka. 2021. "Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine" Biomolecules 11, no. 5: 652. https://doi.org/10.3390/biom11050652
APA StyleKapuy, O., Makk-Merczel, K., & Szarka, A. (2021). Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine. Biomolecules, 11(5), 652. https://doi.org/10.3390/biom11050652