
Interaction of Ligands for PET with the Dopamine D3 Receptor: In Silico and In Vitro Methods

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
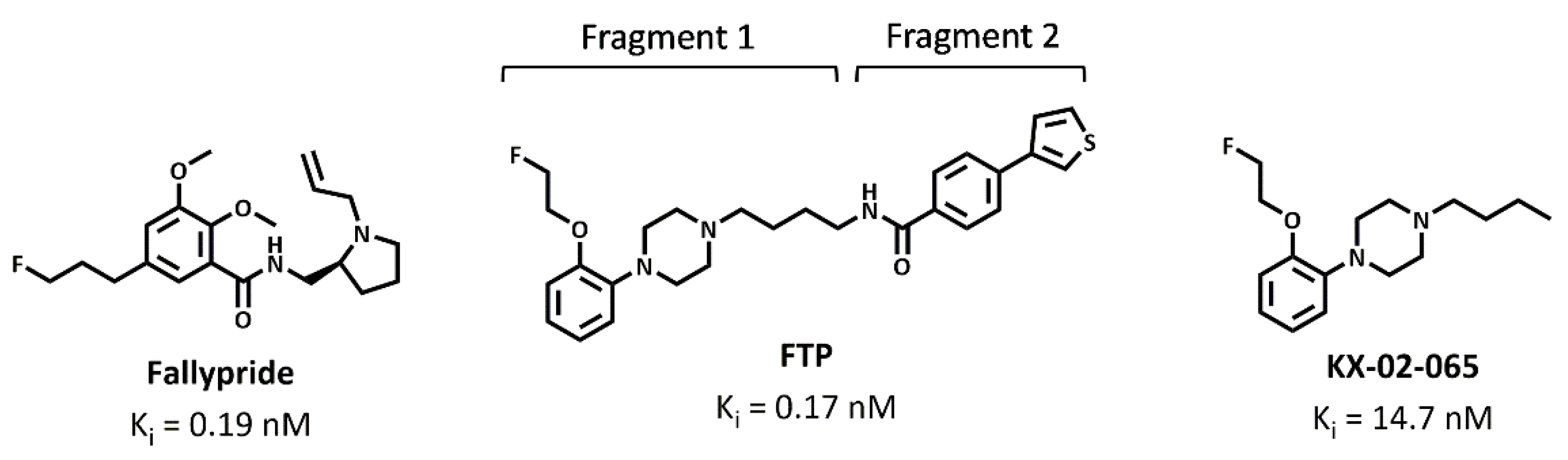
2.1. D3 Ligands
2.2. β-Arrestin Recruitment Assay
2.3. Molecular Docking
2.4. Molecular Dynamics Simulation
2.4.1. Building Protein-Ligand Complex
2.4.2. Equilibration and Production Simulations
2.4.3. Molecular Dynamics Simulation Analysis
3. Results
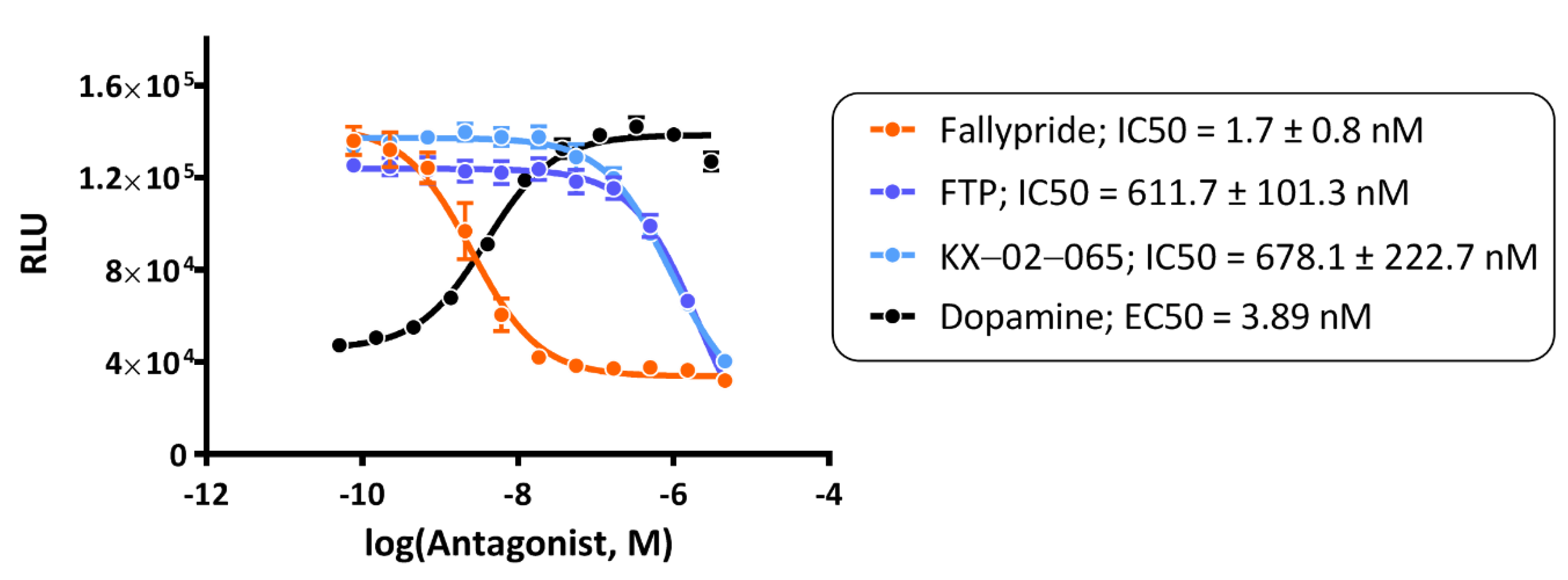
3.1. β-Arrestin Recruitment Assays
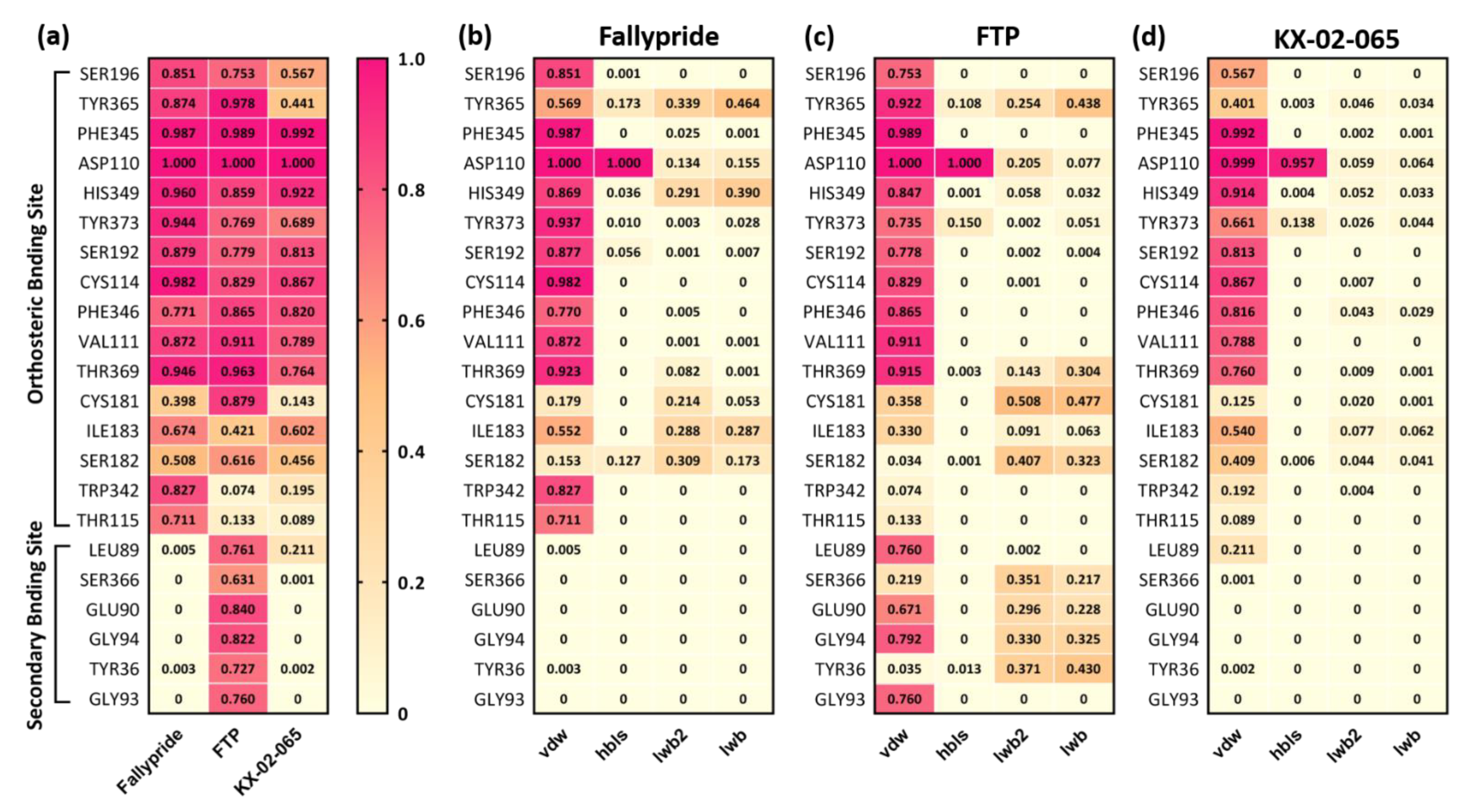
3.2. Molecular Docking
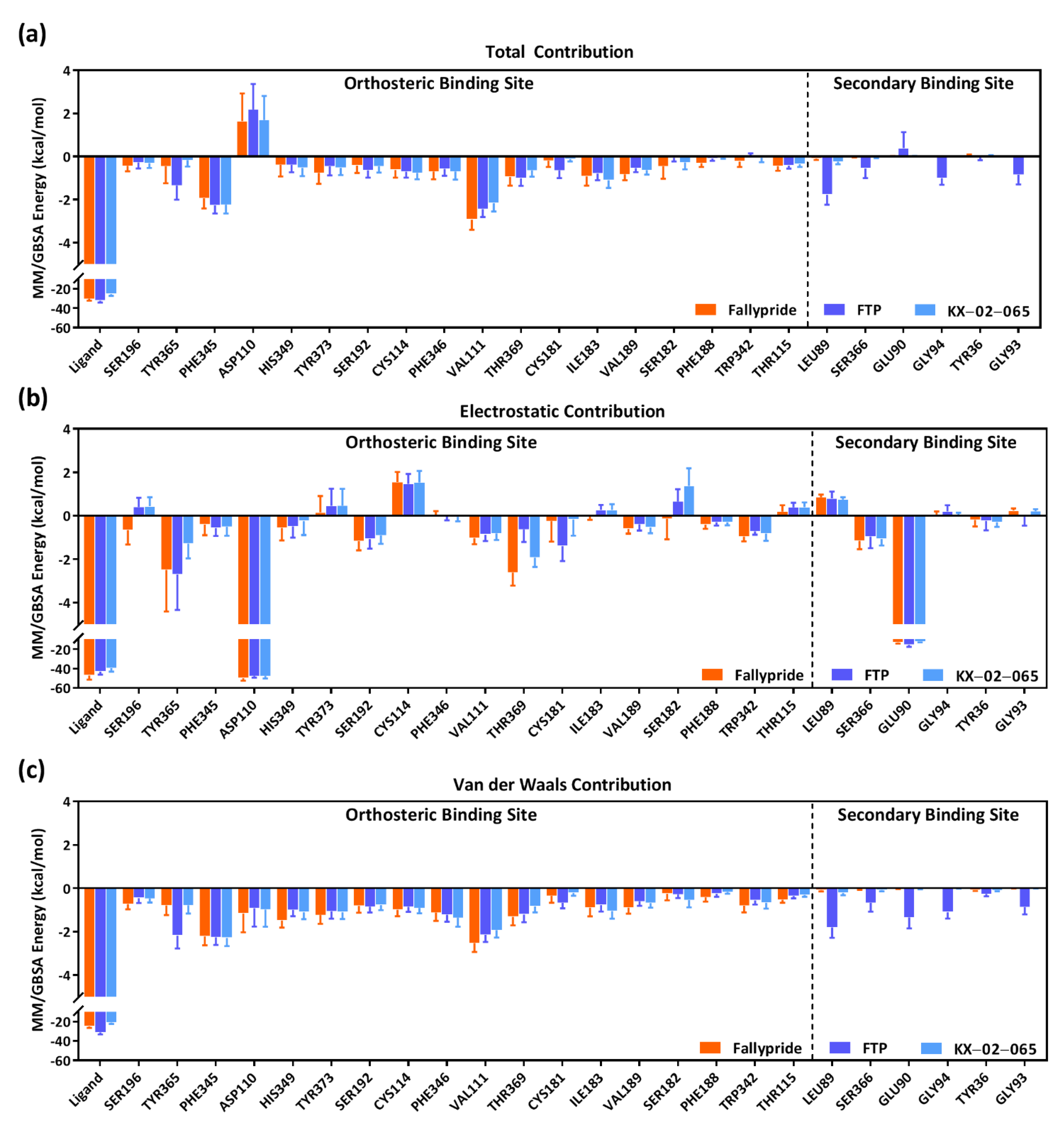
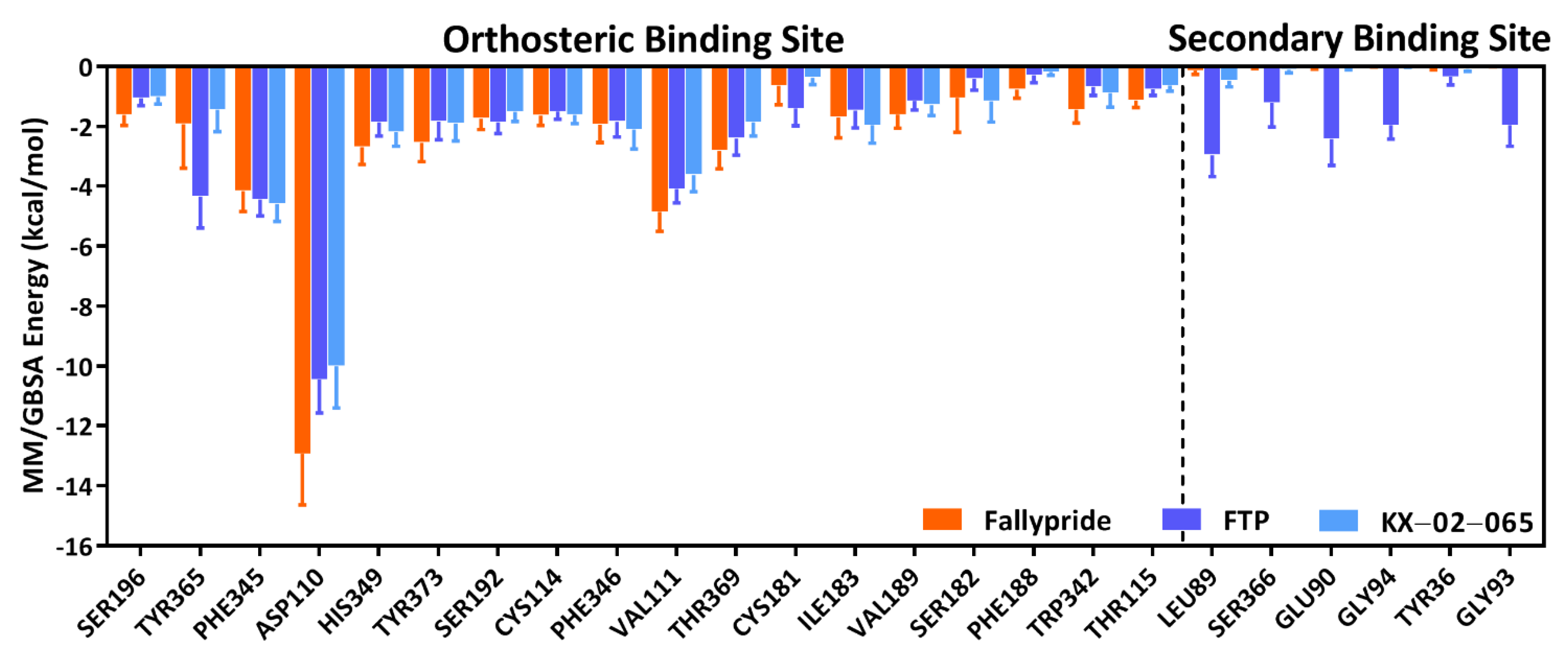
3.3. Molecular Dynamics Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volkow, N.; Fowler, J.; Wang, G.; Baler, R.; Telang, F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 2009, 56, 3–8. [Google Scholar] [CrossRef]
- Arakawa, R.; Takano, A.; Halldin, C. PET technology for drug development in psychiatry. Neuropsychopharmacol. Rep. 2020, 40, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A. From “bedside” to “bench” and back: A translational approach to studying dopamine dysfunction in schizophrenia. Neurosci. Biobehav. Rev. 2020, 110, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Mach, R.H.; Luedtke, R.R. Challenges in the development of dopamine D2-and D3-selective radiotracers for PET imaging studies. J. Label. Compd. Radiopharm. 2018, 61, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, R.; Rangel-Barajas, C.; Malik, M.; Reichert, D.; Mach, R. Bitropic D3 Dopamine Receptor Selective Compounds s Potential Antipsychotics. Curr. Pharm. Des. 2015, 21, 3700–3724. [Google Scholar] [CrossRef] [PubMed]
- Kügler, F.; Sihver, W.; Ermert, J.; Hübner, H.; Gmeiner, P.; Prante, O.; Coenen, H.H. Evaluation of18F-Labeled Benzodioxine Piperazine-Based Dopamine D4Receptor Ligands: Lipophilicity as a Determinate of Nonspecific Binding. J. Med. Chem. 2011, 54, 8343–8352. [Google Scholar] [CrossRef]
- Kügler, F.; Ermert, J.; Kaufholz, P.; Coenen, H.H. 4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-Arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to Build-Up of the Dopamine D4 Ligand [18F]FAUC 316. Molecules 2014, 20, 470–486. [Google Scholar] [CrossRef]
- Doot, R.K.; Dubroff, J.G.; Labban, K.J.; Mach, R.H. Selectivity of probes for PET imaging of dopamine D3 receptors. Neurosci. Lett. 2019, 691, 18–25. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Cairns, N.J.; Perlmutter, J.S.; Mach, R.H. Dopamine D1, D2, D3 Receptors, Vesicular Monoamine Transporter Type-2 (VMAT2) and Dopamine Transporter (DAT) Densities in Aged Human Brain. PLoS ONE 2012, 7, e49483. [Google Scholar] [CrossRef]
- Sun, J.; Cairns, N.J.; Perlmutter, J.S.; Mach, R.H.; Xu, J. Regulation of dopamine D3 receptor in the striatal regions and substantia nigra in diffuse Lewy body disease. Neuroscience 2013, 248, 112–126. [Google Scholar] [CrossRef]
- Orio, L.; Wee, S.; Newman, A.H.; Pulvirenti, L.; Koob, G.F. The dopamine D3 receptor partial agonist CJB090 and antagonist PG01037 decrease progressive ratio responding for methamphetamine in rats with extended-access. Addict. Biol. 2010, 15, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.H.; Blaylock, B.L.; Nader, M.A.; Bergman, J.; Sibley, D.R.; Skolnick, P. Medication discovery for addiction: Translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol. 2012, 84, 882–890. [Google Scholar] [CrossRef]
- Galaj, E.; Newman, A.H.; Xi, Z.-X. Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: Rationale, progress, and challenges. Neurosci. Biobehav. Rev. 2020, 114, 38–52. [Google Scholar] [CrossRef]
- Tu, Z.; Li, S.; Cui, J.; Xu, J.; Taylor, M.; Ho, D.; Luedtke, R.R.; Mach, R.H. Synthesis and Pharmacological Evaluation of Fluorine-Containing D3Dopamine Receptor Ligands. J. Med. Chem. 2011, 54, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Laforest, R.; Karimi, M.; Moerlein, S.M.; Xu, J.; Flores, H.P.; Bognar, C.; Li, A.; Mach, R.H.; Perlmutter, J.S.; Tu, Z. Absorbed radiation dosimetry of the D(3)-specific PET radioligand [18F]FluorTriopride estimated using rodent and nonhuman primate. Am. J. Nucl. Med. Mol. Imaging 2016, 6, 301–309. [Google Scholar]
- Mach, R.H.; Tu, Z.; Xu, J.; Li, S.; Jones, L.A.; Taylor, M.; Luedtke, R.R.; Derdeyn, C.P.; Perlmutter, J.S.; Mintun, M.A. Endogenous dopamine (DA) competes with the binding of a radiolabeled D3 receptor partial agonist in vivo: A positron emission tomography study. Synap 2011, 65, 724–732. [Google Scholar] [CrossRef]
- Christian, B.T.; Narayanan, T.K.; Shi, B.; Mukherjee, J. Quantitation of striatal and extrastriatal D-2 dopamine receptors using PET imaging of [18F]fallypride in nonhuman primates. Synap 2000, 38, 71–79. [Google Scholar] [CrossRef]
- Mukherjee, J.; Christian, B.T.; Narayanan, T.K.; Shi, B.; Collins, D. Measurement of d-amphetamine-induced effects on the binding of dopamine D-2/D-3 receptor radioligand, 18F-fallypride in extrastriatal brain regions in non-human primates using PET. Brain Res. 2005, 1032, 77–84. [Google Scholar] [CrossRef]
- Cropley, V.L.; Innis, R.B.; Nathan, P.J.; Brown, A.K.; Sangare, J.L.; Lerner, A.; Ryu, Y.H.; Sprague, K.E.; Pike, V.W.; Fujita, M. Small effect of dopamine release and no effect of dopamine depletion on [18F]fallypride binding in healthy humans. Synap 2008, 62, 399–408. [Google Scholar] [CrossRef]
- Hayatshahi, H.S.; Xu, K.; Griffin, S.A.; Taylor, M.; Mach, R.H.; Liu, J.; Luedtke, R.R. Analogues of Arylamide Phenylpiperazine Ligands To Investigate the Factors Influencing D3 Dopamine Receptor Bitropic Binding and Receptor Subtype Selectivity. ACS Chem. Neurosci. 2018, 9, 2972–2983. [Google Scholar] [CrossRef]
- Reilly, S.W.; Riad, A.A.; Hsieh, C.-J.; Sahlholm, K.; Jacome, D.A.; Griffin, S.; Taylor, M.; Weng, C.-C.; Xu, K.; Kirschner, N.; et al. Leveraging a Low-Affinity Diazaspiro Orthosteric Fragment to Reduce Dopamine D3 Receptor (D3R) Ligand Promiscuity across Highly Conserved Aminergic G-Protein-Coupled Receptors (GPCRs). J. Med. Chem. 2019, 62, 5132–5147. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jo, S.; Brooks III, C.L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell Jr, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Venable, R.M.; Sodt, A.J.; Rogaski, B.; Rui, H.; Hatcher, E.; MacKerell Jr, A.D.; Pastor, R.W.; Klauda, J.B. CHARMM all-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive curvature. Biophys. J. 2014, 107, 134–145. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for posi-tioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Case, D.; Walker, R.C.; Cheatham III, T.E.; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Darden, T.; Wang, J.; Duke, R.E.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference (SciPy), Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Malik, M.; Taylor, M.; Neve, K.A.; Mach, R.H.; Luedtke, R.R. Characterization of [3 H]LS-3-134, a novel arylamide phenylpiperazine D3 dopamine receptor selective radioligand. J. Neurochem. 2014, 131, 418–431. [Google Scholar] [CrossRef]
- Prante, O.; Maschauer, S.; Banerjee, A. Radioligands for the dopamine receptor subtypes. J. Label. Compd. Radiopharm. 2013, 56, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mach, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype Selectivity of Dopamine Receptor Ligands: Insights from Structure and Ligand-Based Methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delta Total | Van der Waals | Electrostatic | |
|---|---|---|---|
| Fallypride | −46.70 ± 4.38 | −49.89 ± 3.63 | −94.13 ± 9.41 |
| FTP | −51.76 ± 4.43 | −62.30 ± 4.91 | −85.64 ± 9.18 |
| KX-02-065 | −38.75 ± 4.19 | −41.95 ± 3.28 | −79.38 ± 9.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-J.; Riad, A.; Lee, J.Y.; Sahlholm, K.; Xu, K.; Luedtke, R.R.; Mach, R.H. Interaction of Ligands for PET with the Dopamine D3 Receptor: In Silico and In Vitro Methods. Biomolecules 2021, 11, 529. https://doi.org/10.3390/biom11040529
Hsieh C-J, Riad A, Lee JY, Sahlholm K, Xu K, Luedtke RR, Mach RH. Interaction of Ligands for PET with the Dopamine D3 Receptor: In Silico and In Vitro Methods. Biomolecules. 2021; 11(4):529. https://doi.org/10.3390/biom11040529
Chicago/Turabian StyleHsieh, Chia-Ju, Aladdin Riad, Ji Youn Lee, Kristoffer Sahlholm, Kuiying Xu, Robert R. Luedtke, and Robert H. Mach. 2021. "Interaction of Ligands for PET with the Dopamine D3 Receptor: In Silico and In Vitro Methods" Biomolecules 11, no. 4: 529. https://doi.org/10.3390/biom11040529
APA StyleHsieh, C.-J., Riad, A., Lee, J. Y., Sahlholm, K., Xu, K., Luedtke, R. R., & Mach, R. H. (2021). Interaction of Ligands for PET with the Dopamine D3 Receptor: In Silico and In Vitro Methods. Biomolecules, 11(4), 529. https://doi.org/10.3390/biom11040529