
Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
3. Results
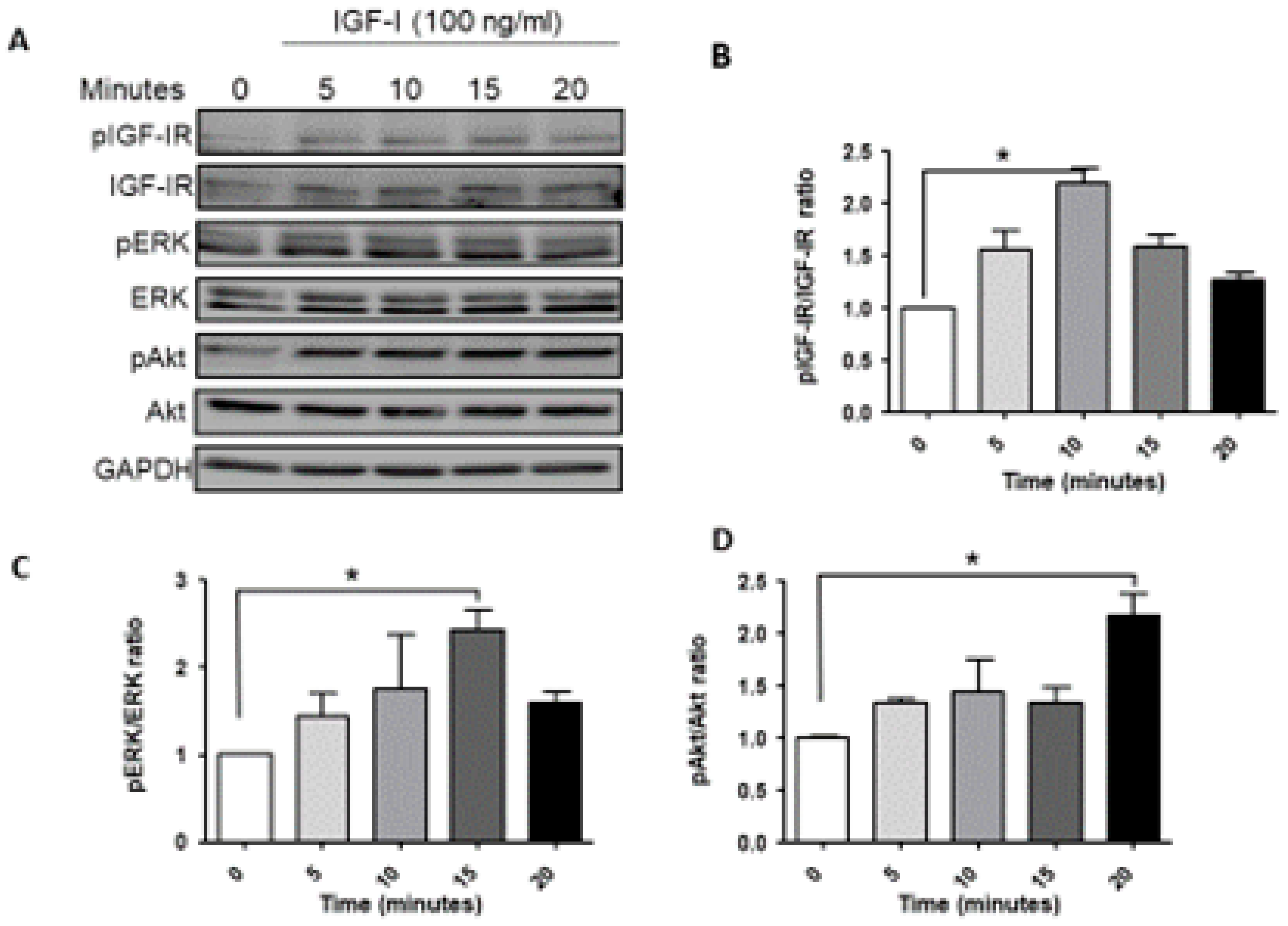
3.1. Ligand Induced IGF-1R Signaling in MDA-MB-231 Cells
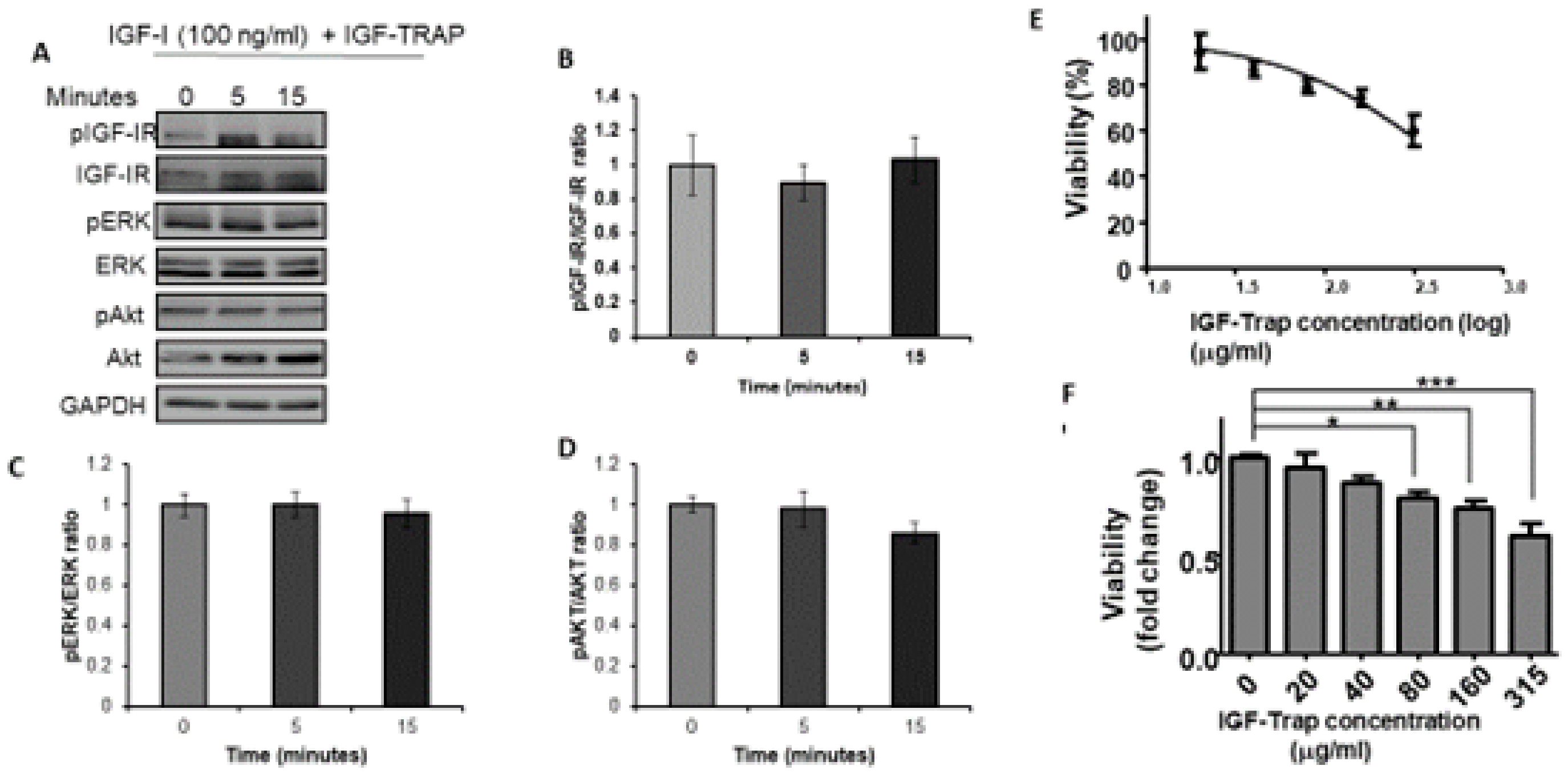
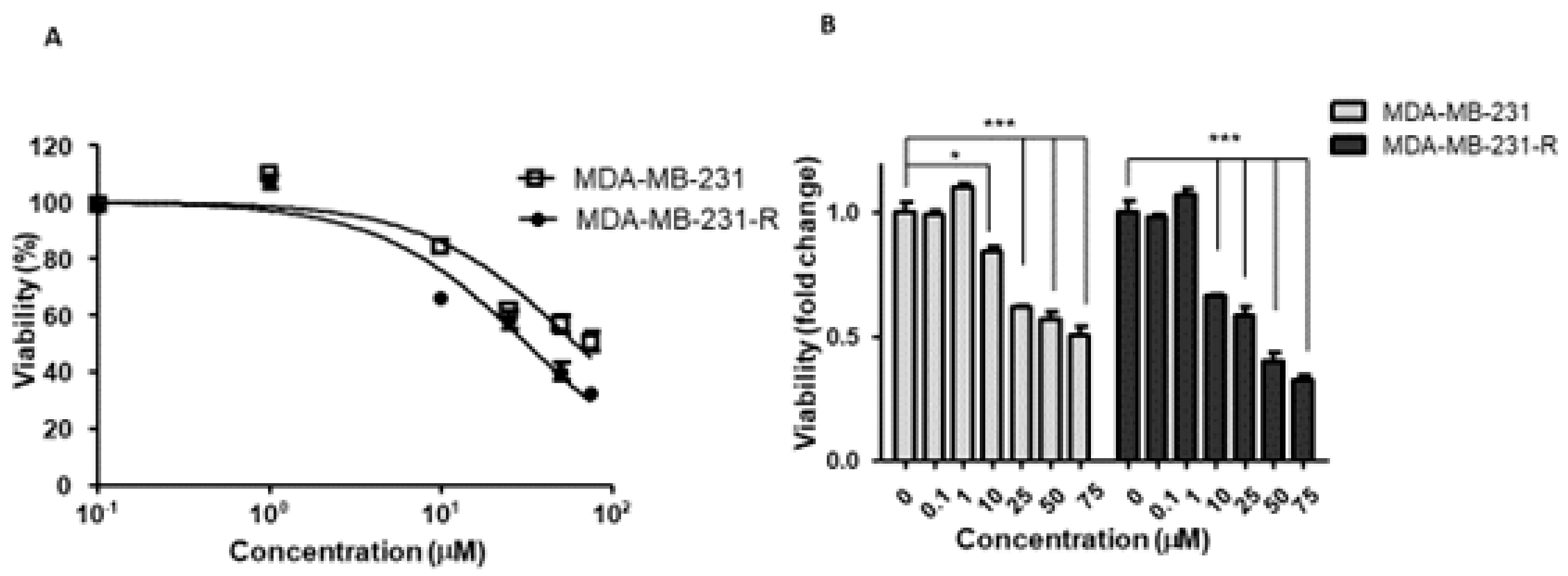
3.2. The IGF-Trap Blocks IGF-1 Signaling and Inhibits MDA-MB-231 Proliferation
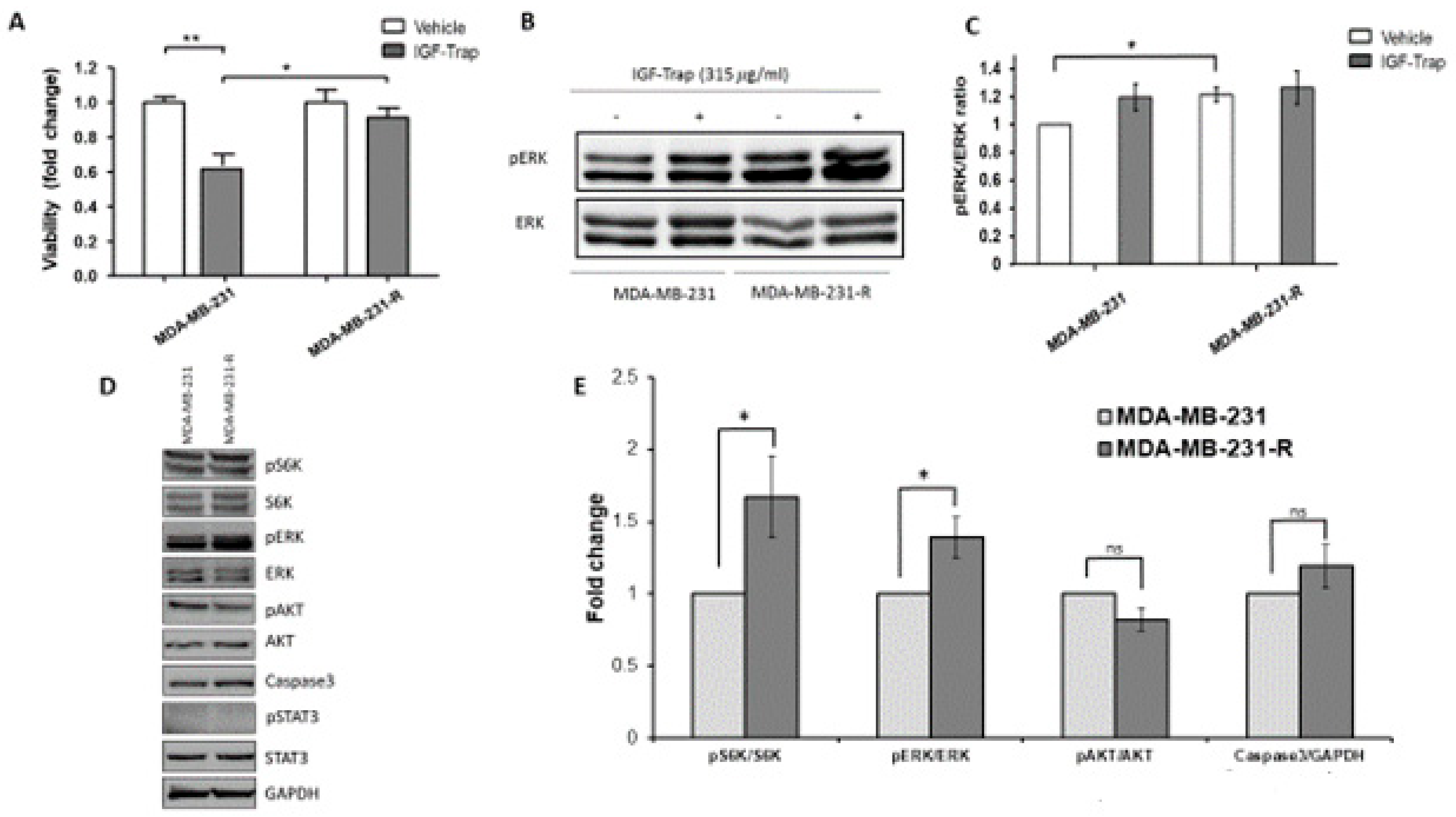
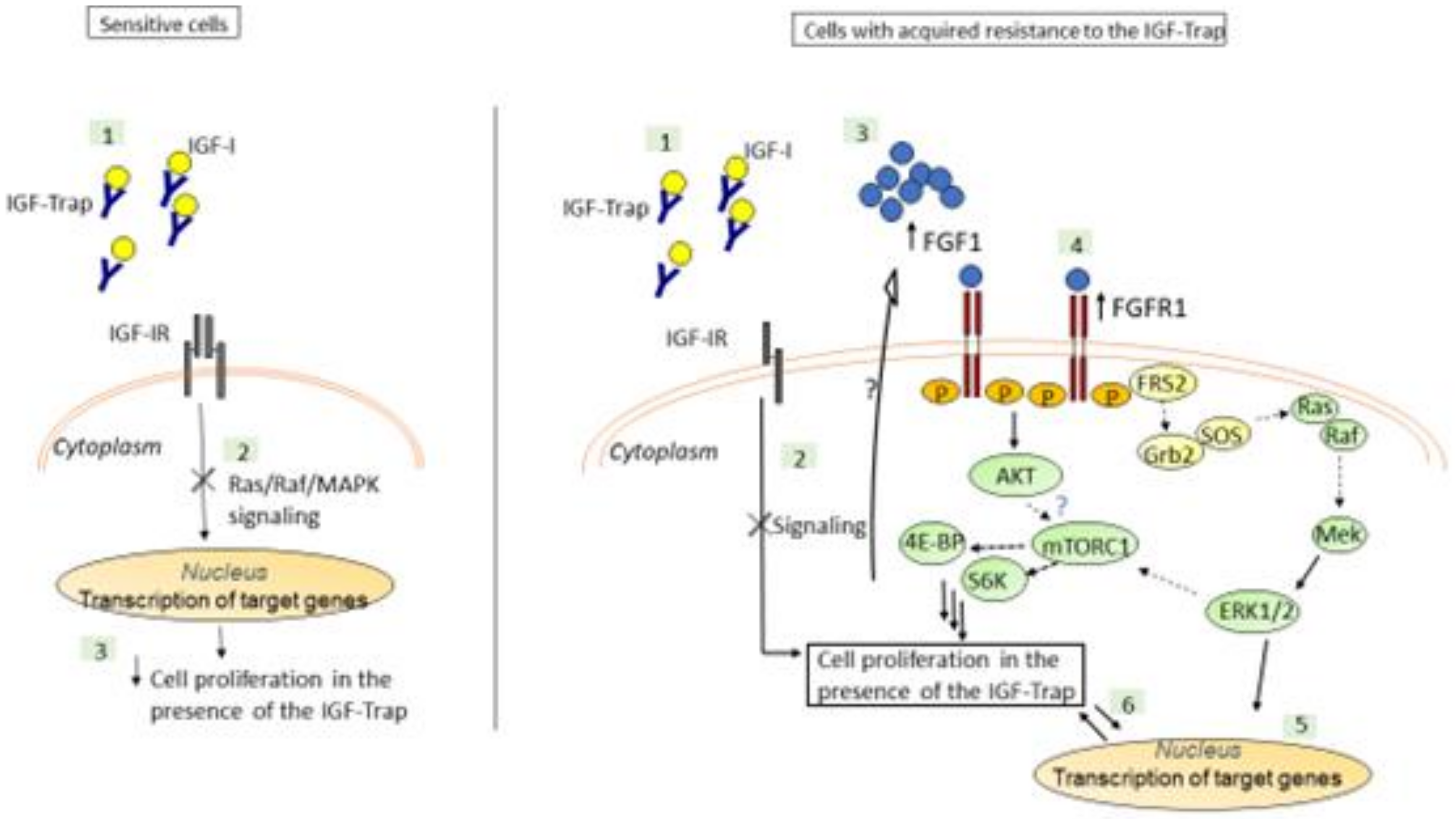
3.3. Increased Constitutive ERK Activation Contributes to an Acquired Resistance of MDA-MB-231 Cells to the IGF-Trap
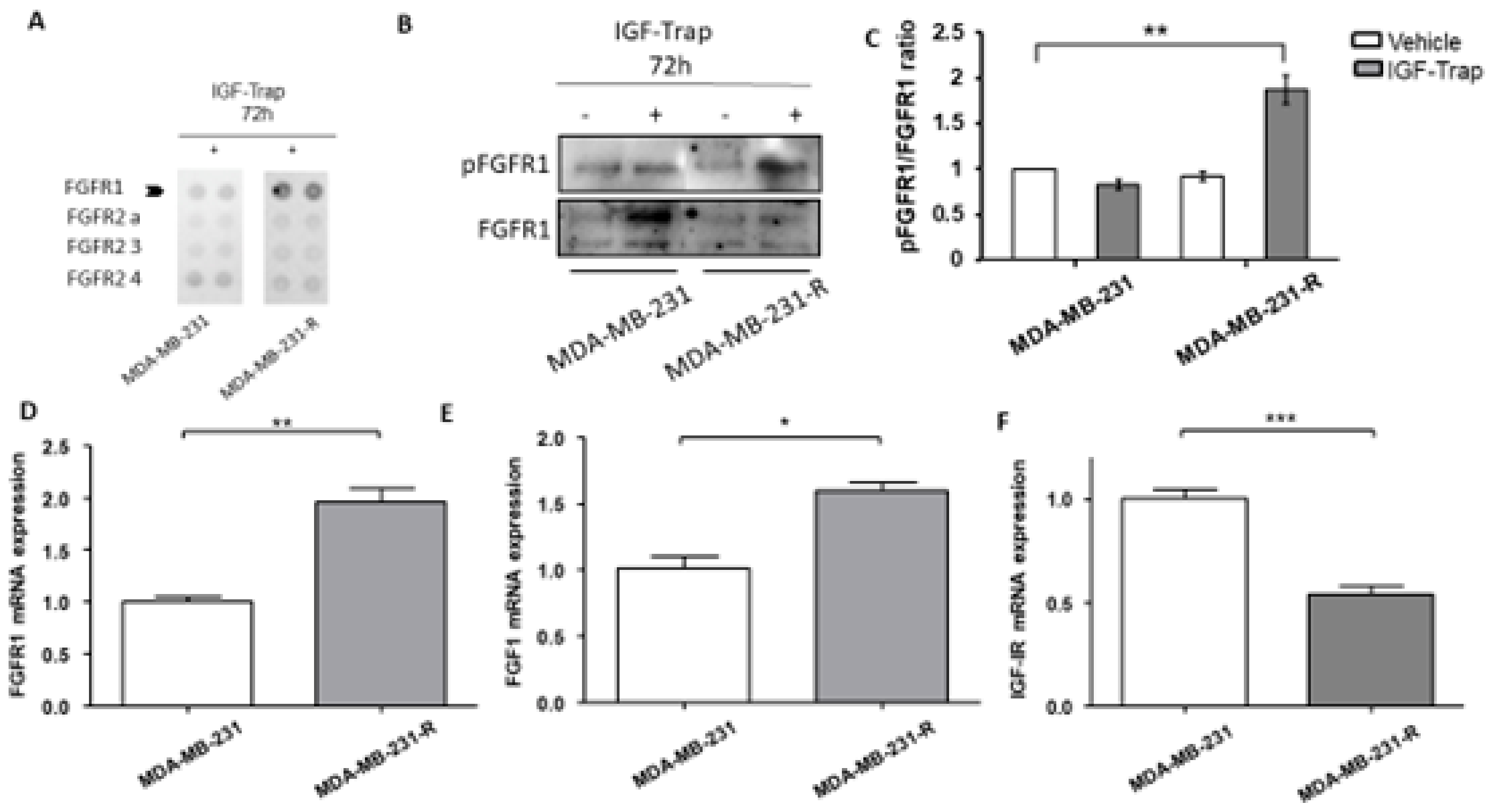
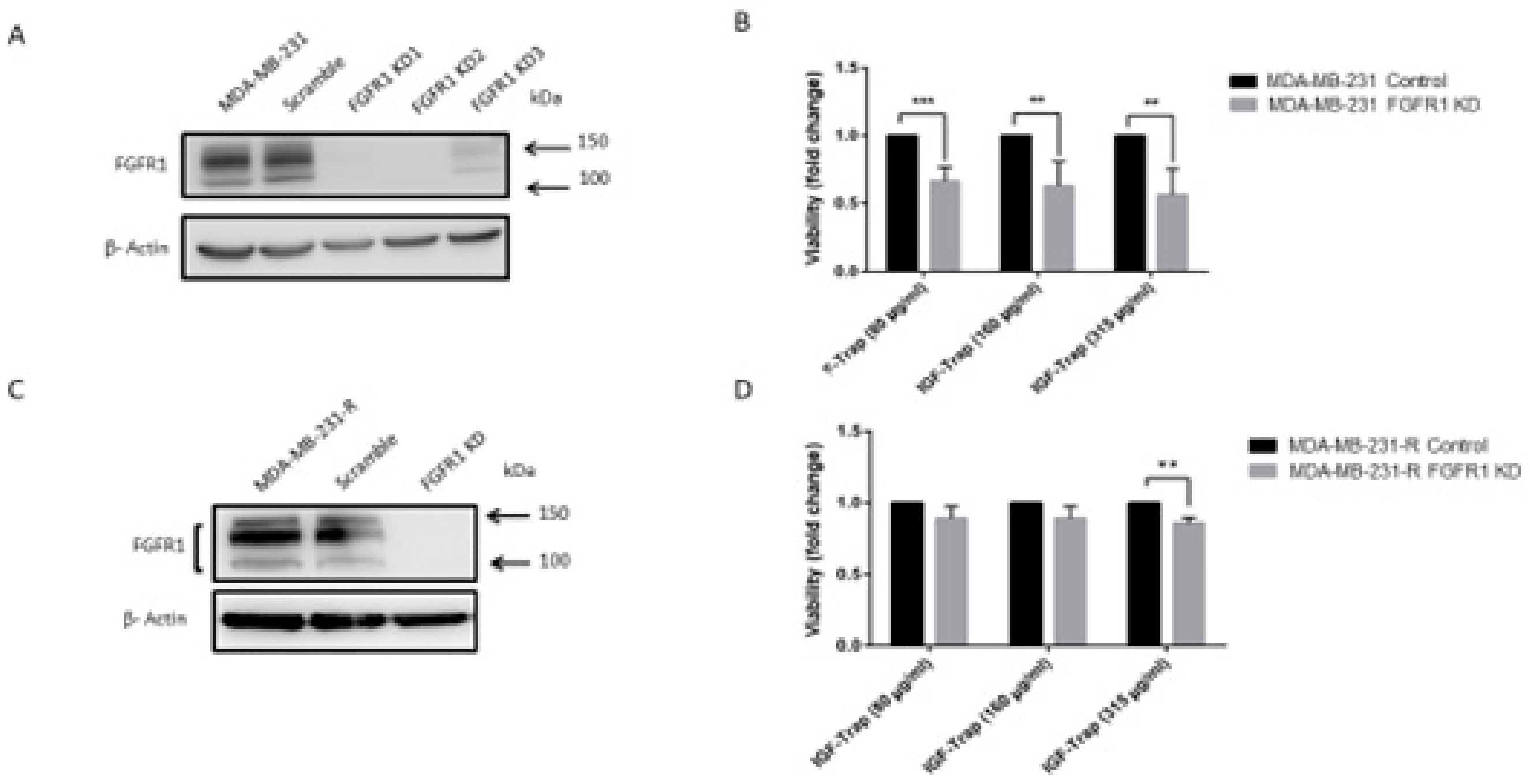
3.4. Expression and Activation Levels of FGFR1 Are Upregulated in MDA-MB-231-R Cells
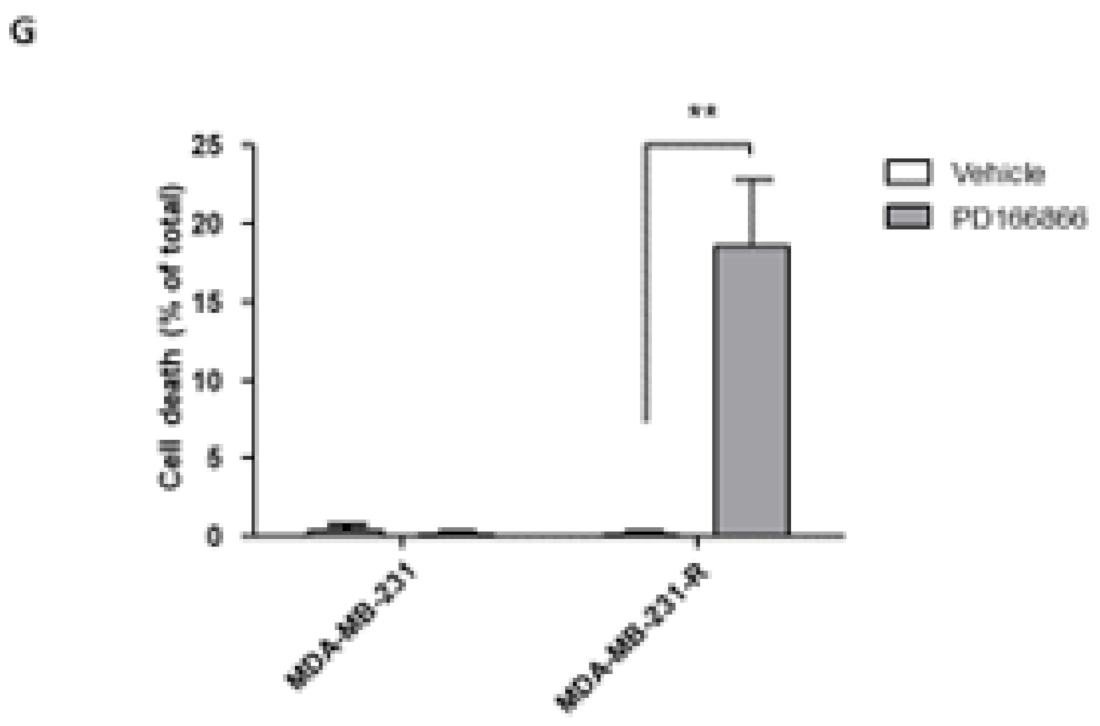
3.5. The Selective FGFR1 Inhibitor PD166866 Decreases Proliferation and Triggers Cell Cycle Arrest in MDA-MB-231 Cells
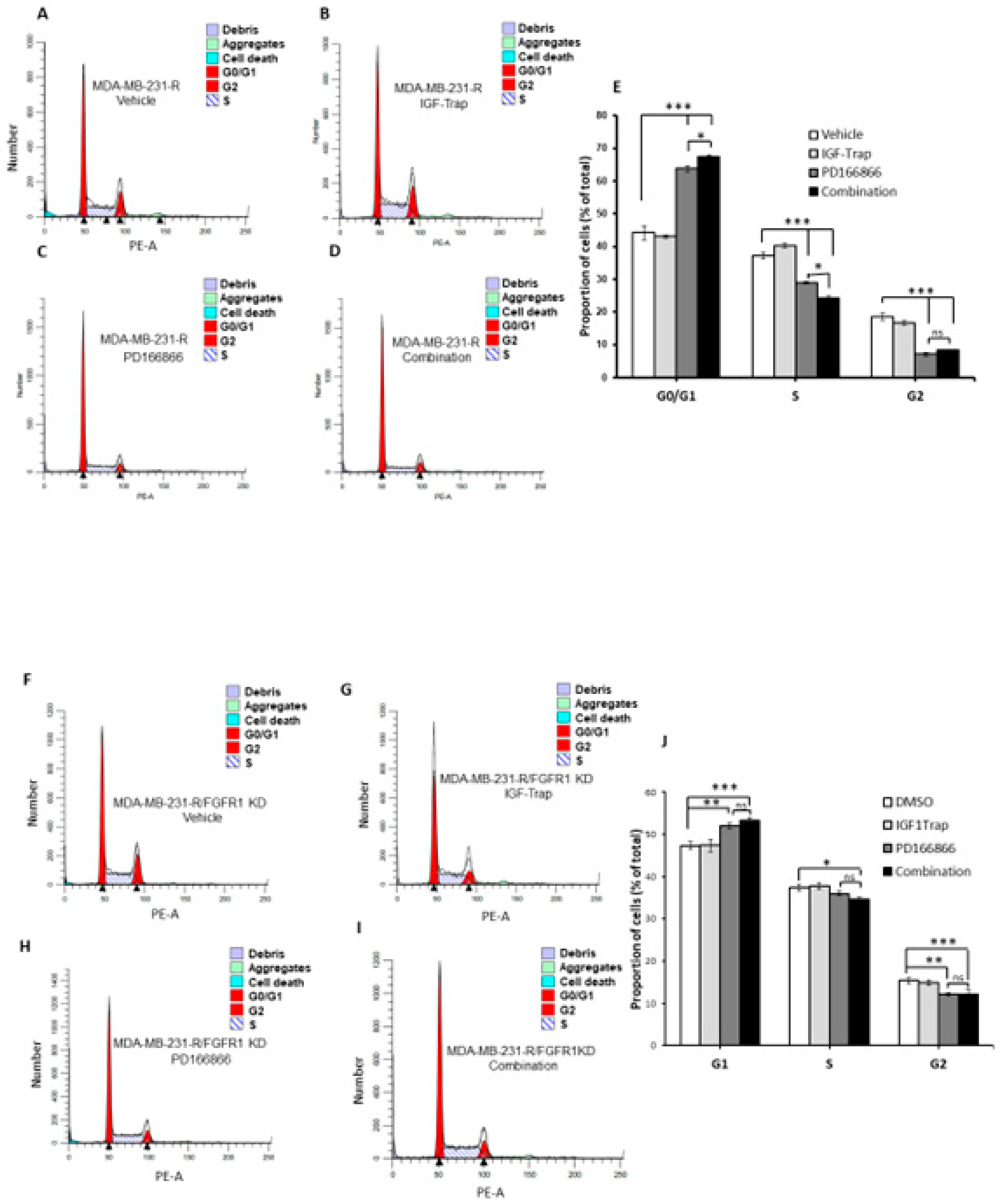
3.6. The Selective FGFR1 Inhibitor PD166866 Causes Cell Cycle Arrest in MDA-MB-231 Cells
3.7. FGFR1 Silencing Increases the Sensitivity of MDA-MB-231 Cells to the IGF-Trap
3.8. Combinatorial Therapy Increases Cell Cycle Arrest in MDA-MB-231-R Cells
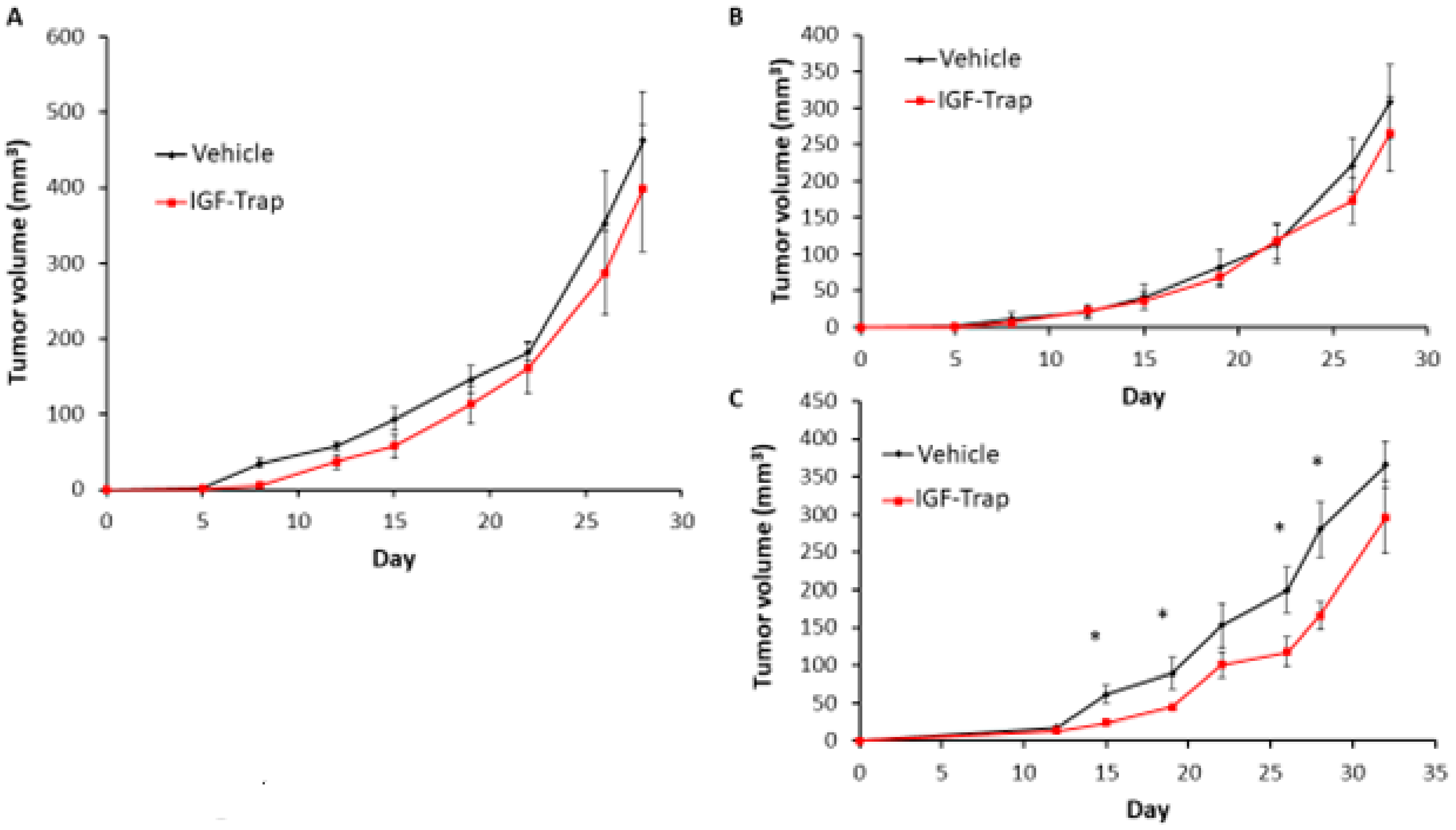
3.9. FGFR1 Silencing Increases the Sensitivity of MDA-MB-231 Cells to IGF-Trap Treatment In Vivo
4. Discussion
5. Conclusions
6. Patents
- (1)
- SOLUBLE IGF RECEPTOR Fc FUSION PROTEINS AND USES THEREOF Application No. 12857915.8 Case Ref. 05001770-467EP European Patent Office. Granted: February 2019.
- (2)
- SOLUBLE IGF RECEPTOR Fc FUSION PROTEINS AND USES THEREOF—US—International (PCT) Patent Application No. PCT/CA2012/050899. United States. 2014-06-16. Granted: September 2019.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and tri-ple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- A Carey, L.; Winer, E.P.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef]
- Kalimutho, M.; Parsons, K.; Mittal, D.; López, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, proges-terone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A popula-tion-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; A Bauer, J.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Stead, L.A.; Lash, T.L.; Sobieraj, J.E.; Chi, D.D.; Westrup, J.L.; Charlot, M.; Blanchard, R.A.; Lee, J.C.; King, T.C.; Rosenberg, C.L. Triple-negative breast cancers are increased in black women regardless of age or body mass index. Breast Cancer Res. 2009, 11, R18. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human tri-ple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients with Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Brodt, P.; Samani, A.; Navab, R. Inhibition of the type I insulin-like growth factor receptor expression and signaling: Novel strategies for antimetastatic therapy. Biochem. Pharmacol. 2000, 60, 1101–1107. [Google Scholar] [CrossRef]
- Haisa, M. The type 1 insulin-like growth factor receptor signalling system and targeted tyrosine kinase inhibition in cancer. J. Int. Med. Res. 2013, 41, 253–264. [Google Scholar] [CrossRef]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and re-cent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- Seccareccia, E.; Brodt, P. The role of the insulin-like growth factor-I receptor in malignancy: An update. Growth Horm. IGF Res. 2012, 22, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Yee, D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol. Cancer Ther. 2007, 6, 1–12. [Google Scholar] [CrossRef]
- Davison, Z.; de Blacquière, G.E.; Westley, B.R.; May, F.E. Insulin-like Growth Factor-Dependent Proliferation and Survival of Triple-Negative Breast Cancer Cells: Implications for Therapy. Neoplasia 2011, 13, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Tan, Q.; Brito, C.; De León, M. Insulin-like growth factors I and II receptors in the breast cancer survival disparity among African–American women. Growth Horm. IGF Res. 2010, 20, 245–254. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.C.; Stratford, A.L.; Park, E.; Gee, J.M.W.; Finlay, P.; Jones, H.E.; et al. Phosphorylated Insulin-Like Growth Factor-I/Insulin Receptor Is Present in All Breast Cancer Subtypes and Is Related to Poor Survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef]
- Bahhnassy, A.; Mohanad, M.; Shaarawy, S.; Ismail, M.F.; El-Bastawisy, A.; Ashmawy, A.M.; Zekri, A.R. Transforming growth factor-beta, insulin-like growth factor I/insulin-like growth factor I receptor and vascular endothelial growth factor-A: Prog-nostic and predictive markers in triple-negative and non-triple-negative breast cancer. Mol. Med. Rep. 2015, 12, 851–864. [Google Scholar] [CrossRef]
- Avnet, S.; Sciacca, L.; Salerno, M.; Gancitano, G.; Cassarino, M.F.; Longhi, A.; Zakikhani, M.; Carboni, J.M.; Gottardis, M.; Giunti, A.; et al. Insulin Receptor Isoform A and Insulin-like Growth Factor II as Additional Treatment Targets in Human Osteosarcoma. Cancer Res. 2009, 69, 2443–2452. [Google Scholar] [CrossRef]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth fac-tor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef]
- Buck, E.; Mulvihill, M. Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert Opin. Investig. Drugs 2011, 20, 605–621. [Google Scholar] [CrossRef]
- Vaniotis, G.; Moffett, S.; Sulea, T.; Wang, N.; Elahi, S.M.; Lessard, E.; Baardsnes, J.; Perrino, S.; Durocher, Y.; Frystyk, J.; et al. Enhanced anti-metastatic bioactivity of an IGF-TRAP re-engineered to improve physicochemical properties. Sci. Rep. 2018, 8, 17361. [Google Scholar] [CrossRef]
- Wang, N.; Rayes, R.F.; Elahi, S.M.; Lu, Y.; Hancock, M.A.; Massie, B.; Rowe, G.E.; Aomari, H.; Hossain, S.; Durocher, Y.; et al. The IGF-Trap: Novel Inhibitor of Carcinoma Growth and Metastasis. Mol. Cancer Ther. 2015, 14, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Qi, S.; Perrino, S.; Hashimoto, M.; Brodt, P. Targeting the IGF-Axis for Cancer Therapy: Development and Valida-tion of an IGF-Trap as a Potential Drug. Cells 2020, 9, 1098. [Google Scholar] [CrossRef] [PubMed]
- Honegger, A.; E Humbel, R. Insulin-like growth factors I and II in fetal and adult bovine serum. Purification, primary structures, and immunological cross-reactivities. J. Biol. Chem. 1986, 261, 569–575. [Google Scholar]
- Burnier, J.V.; Wang, N.; Michel, R.P.; Hassanain, M.; Li, S.; Lu, Y.; Metrakos, P.; Antecka, E.; Burnier, M.N.; Ponton, A.; et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene 2011, 30, 3766–3783. [Google Scholar] [CrossRef] [PubMed]
- Vaniotis, G.; Rayes, R.F.; Qi, S.; Milette, S.; Wang, N.; Perrino, S.; Bourdeau, F.; Nyström, H.; He, Y.; Lamarche-Vane, N.; et al. Collagen IV-conveyed signals can regulate chemokine production and promote liver metastasis. Oncogene 2018, 37, 3790–3805. [Google Scholar] [CrossRef] [PubMed]
- Seccareccia, E.; Pinard, M.; Wang, N.; Li, S.; Burnier, J.; Dankort, D.; Brodt, P. The inhibitor of kappa B kinase-epsilon regulates MMP-3 expression levels and can promote lung metastasis. Oncogenesis 2014, 3, e116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Risuleo, G.; Ciacciarelli, M.; Castelli, M.; Galati, G. The synthetic inhibitor of fibroblast growth factor receptor PD166866 con-trols negatively the growth of tumor cells in culture. J. Exp. Clin. Cancer Res. 2009, 28, 151. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.-L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The GAMMA trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef]
- Langer, C.J.; Novello, S.; Park, K.; Krzakowski, M.; Karp, D.D.; Mok, T.; Benner, R.J.; Scranton, J.R.; Olszanski, A.J.; Jassem, J. Randomized, Phase III Trial of First-Line Figitumumab in Combination with Paclitaxel and Carboplatin Versus Paclitaxel and Carboplatin Alone in Patients With Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2014, 32, 2059–2066. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Bondarenko, I.; Blackhall, F.; Barlesi, F.; Hsia, T.C.; Jassem, J.; Milanowski, J.; Popat, S.; Sanchez-Torres, J.M.; Novello, S.; et al. Randomized, phase III trial of figitumumab in combina-tion with erlotinib versus erlotinib alone in patients with nonadenocarcinoma nonsmall-cell lung cancer. Ann. Oncol. 2015, 26, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, X.; Li, X.; Wang, P.; Jing, Q.; Yue, J.; Liu, Y.; Cheng, Z.; Li, J.; Song, H.; et al. FGFR antagonist in-duces protective autophagy in FGFR1-amplified breast cancer cell. Biochem. Biophys. Res. Commun. 2016, 474, 1–7. [Google Scholar] [CrossRef]
- Shi, Y.; Ma, Z.; Cheng, Q.; Wu, Y.; Parris, A.B.; Kong, L.; Yang, X. FGFR1 overexpression renders breast cancer cells resistant to metformin through activation of IRS1/ERK signaling. Biochim. Biophys. Acta (BBA) Bioenergy 2021, 1868, 118877. [Google Scholar] [CrossRef]
- Park, S.-B.; Yu, K.-R.; Jung, J.-W.; Lee, S.-R.; Roh, K.-H.; Seo, M.-S.; Park, J.-R.; Kang, S.-K.; Lee, Y.-S.; Kang, K.-S. bFGF enhances the IGFs-mediated pluripotent and differentiation potentials in multipotent stem cells. Growth Factors 2009, 27, 425–437. [Google Scholar] [CrossRef]
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.T.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.S.; Ashworth, A.; et al. FGFR Signaling Promotes the Growth of Triple-Negative and Basal-Like Breast Cancer Cell Lines Both In Vitro and In Vivo. Clin. Cancer Res. 2011, 17, 5275–5286. [Google Scholar] [CrossRef]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular Profiling of the Residual Disease of Triple-Negative Breast Cancers after Neoadjuvant Chemotherapy Identifies Actionable Therapeutic Targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.L.; Thike, A.A.; Tan, S.Y.J.; Chua, P.J.; Bay, B.H.; Tan, P.H. Expression of FGFR1 is an independent prognostic factor in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 151, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Hurlburt, W.; Greer, A.; Reeves, K.A.; Hillerman, S.; Chang, H.; Fargnoli, J.; Finckenstein, F.G.; Gottardis, M.M.; Carboni, J.M. Differential Mechanisms of Acquired Resistance to Insulin-like Growth Factor-I Receptor Antibody Therapy or to a Small-Molecule Inhibitor, BMS-754807, in a Human Rhabdomyosarcoma Model. Cancer Res. 2010, 70, 7221–7231. [Google Scholar] [CrossRef]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.-C.; Wullenkord, R.; Lujambio, A.; De Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nat. Cell Biol. 2016, 534, 647–651. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsui, J.; Qi, S.; Perrino, S.; Leibovitch, M.; Brodt, P. Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells. Biomolecules 2021, 11, 527. https://doi.org/10.3390/biom11040527
Tsui J, Qi S, Perrino S, Leibovitch M, Brodt P. Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells. Biomolecules. 2021; 11(4):527. https://doi.org/10.3390/biom11040527
Chicago/Turabian StyleTsui, Jennifer, Shu Qi, Stephanie Perrino, Matthew Leibovitch, and Pnina Brodt. 2021. "Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells" Biomolecules 11, no. 4: 527. https://doi.org/10.3390/biom11040527
APA StyleTsui, J., Qi, S., Perrino, S., Leibovitch, M., & Brodt, P. (2021). Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells. Biomolecules, 11(4), 527. https://doi.org/10.3390/biom11040527