
The Phosphorylation Status of Drp1-Ser637 by PKA in Mitochondrial Fission Modulates Mitophagy via PINK1/Parkin to Exert Multipolar Spindles Assembly during Mitosis

 ,
,  ,
, 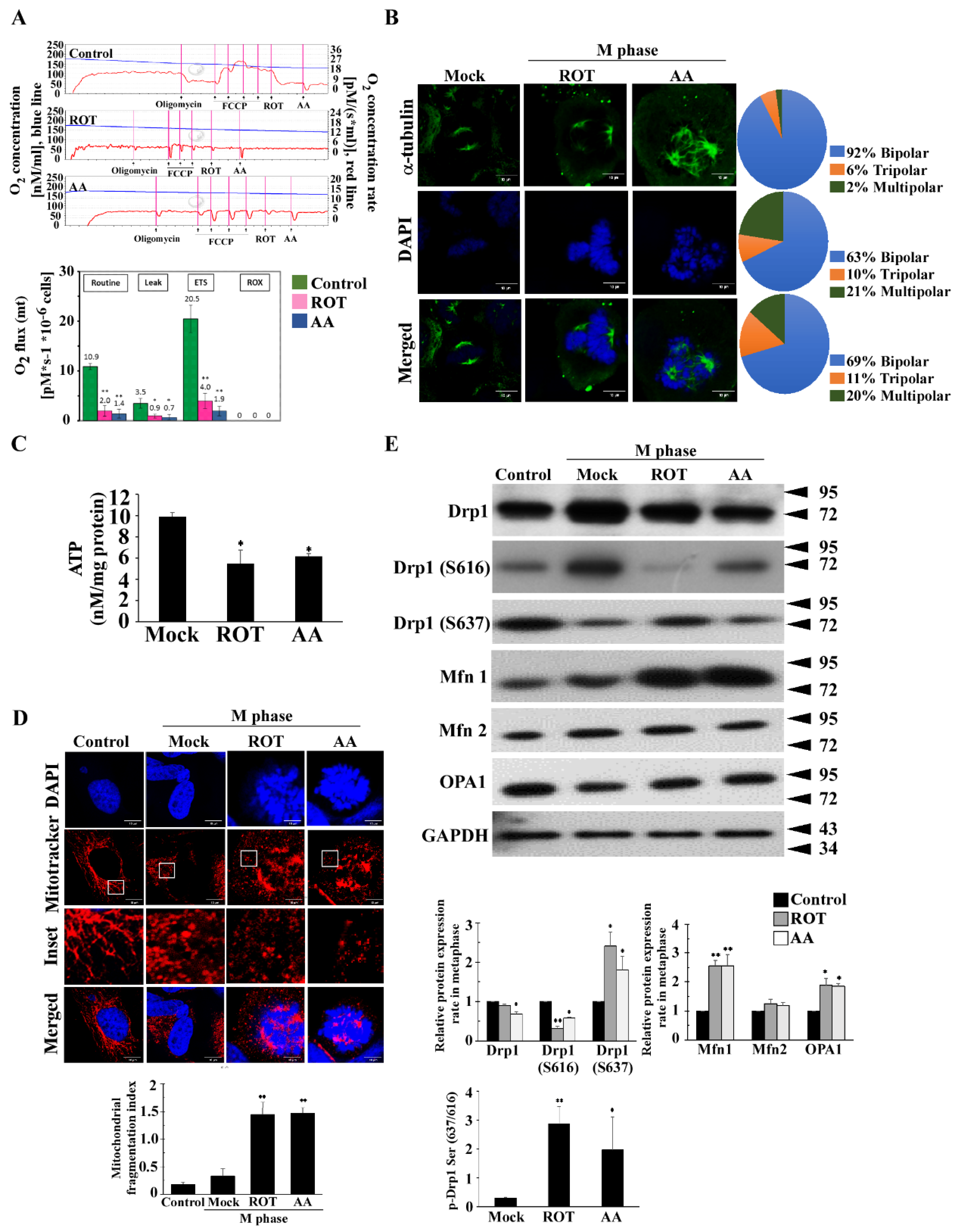{kind=link}
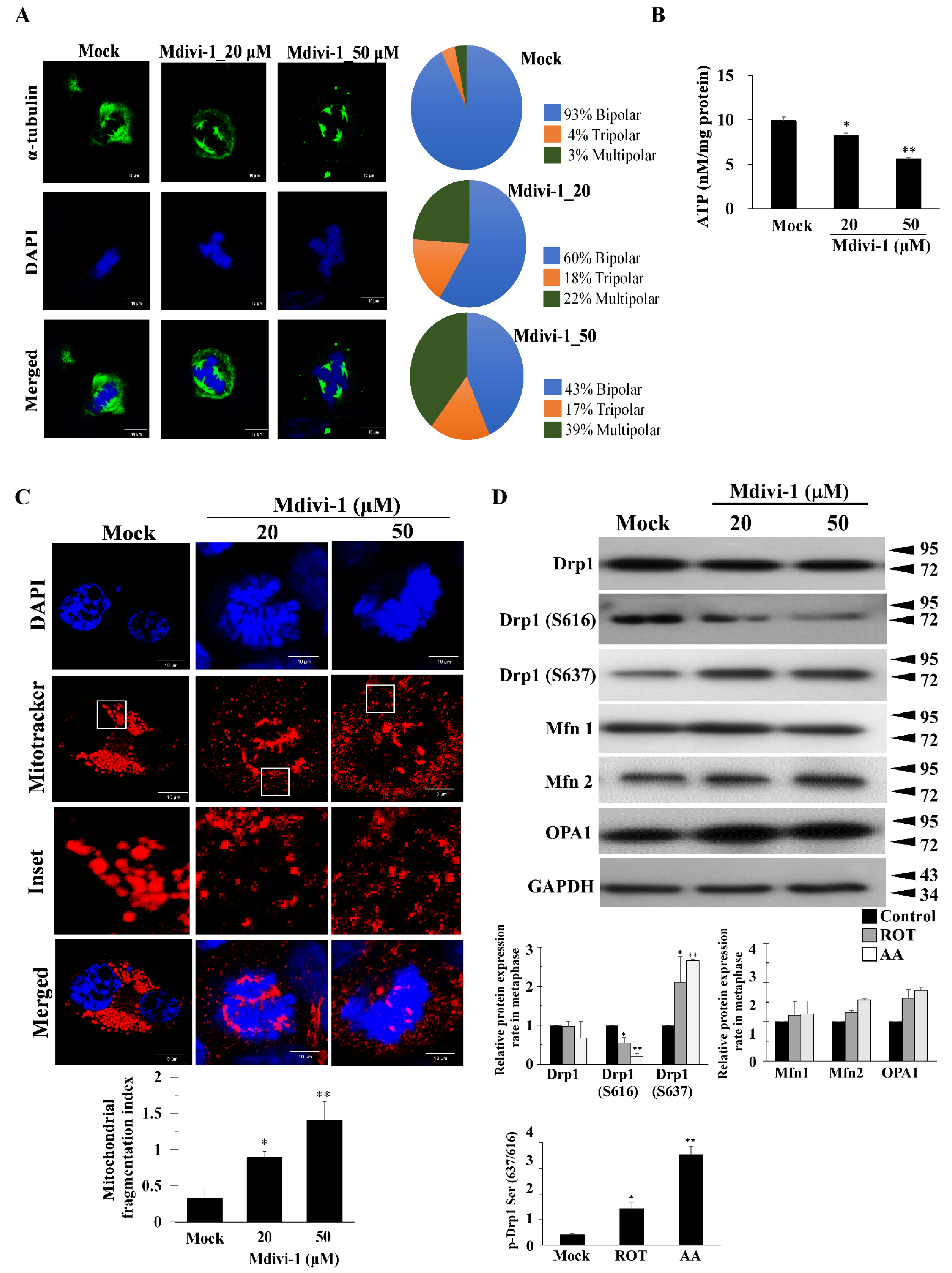{kind=link}
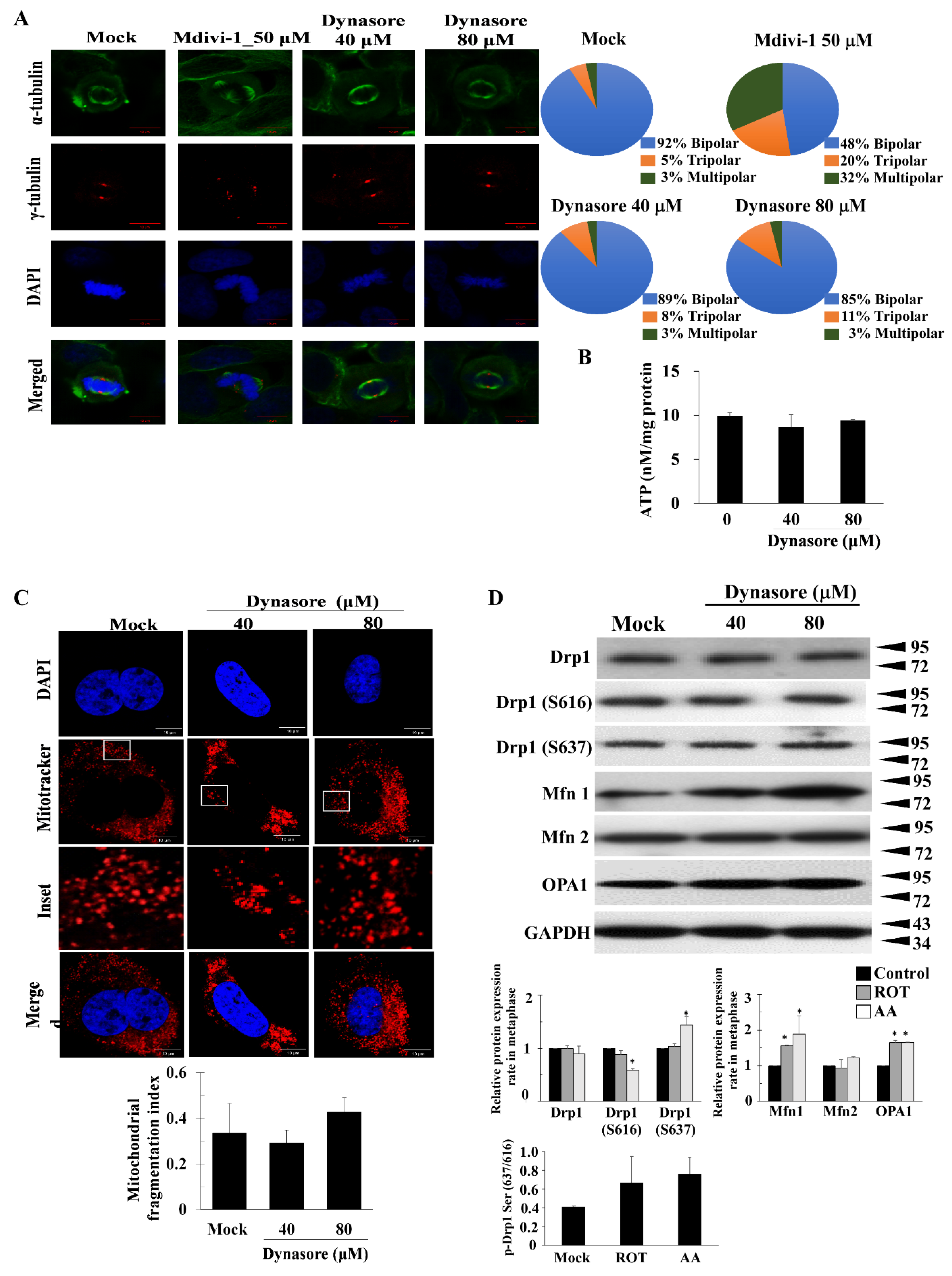{kind=link}
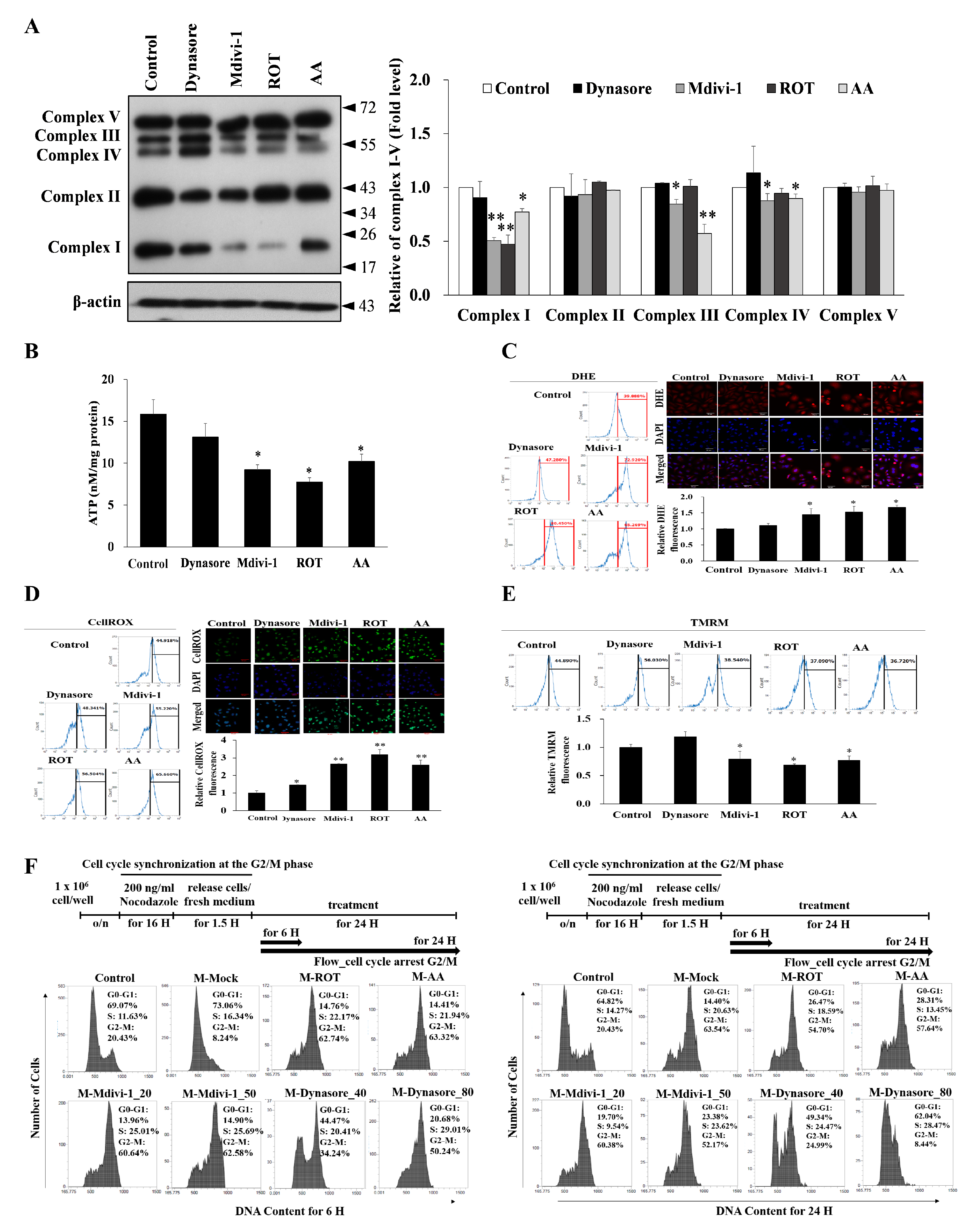{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cell Cycle Synchronization
2.2. In Vitro Cytotoxicity Assay
2.3. Oxygen Consumption Rate (OCR)
2.4. Measurement of Mitochondrial Membrane Potential (ΔΨm)
2.5. ATP Measurement
2.6. Flow Cytometry
2.7. Cell Cycle Analysis
2.8. Immunofluorescence Staining and Confocal Microscopy
2.9. Mitochondria Subcellular Fractionation and Preparation
2.10. Western Blot Analysis
2.11. Statistical Analysis
| Reagent Type (Species) or Resource | Designation | Source or Reference | Identifiers | Additional Information |
| Cell line (H. sapiens) | HeLa | ATCC (Manassas, VA, USA) | CCL-2 | Maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin and 1% L-Glutamine |
| Antibody | Rabbit anti-Aurora A | Abnova (Taipei, Taiwan) | PAB0359 | 1:1000 |
| Antibody | Rabbit anti-Aurora-A(T288) | Abnova (Taipei, Taiwan) | MAB7299 | 1:1000 |
| Antibody | Rabbit anti-AIBp | Abcam (Cambridge, MA, USA) | ab122014 | 1:500 |
| Antibody | Rabbit anti-Plk1 | Cell signaling (Beverly, MA, USA) | s4513 | 1:1000 |
| Antibody | Rabbit anti-Plk1(Thr210) | Cell signaling (Beverly, MA, USA) | S5472 | 1:1000 |
| Antibody | Mouse anti-Drp1 | BD (BD Biosciences, San Jose, CA, USA) | 611112 | 1:1000 |
| Antibody | Mouse anti-Drp1 (Ser 616) | Cell signaling (Beverly, MA, USA) | S3455 | 1:1000 |
| Antibody | Mouse anti-Drp1 (Ser 637) | Cell signaling (Beverly, MA, USA) | S4867 | 1:1000 |
| Antibody | Rabbit anti-Opa1 | Cell signaling (Beverly, MA, USA) | S67589 | 1:1000 |
| Antibody | Rabbit anti-Mfn1 | Cell signaling (Beverly, MA, USA) | S14739 | 1:1000 |
| Antibody | Rabbit anti-Mfn2 | Cell signaling (Beverly, MA, USA) | S9482 | 1:1000 |
| Antibody | Rabbit anti-cdc (Cdk1) | Cell signaling (Beverly, MA, USA) | S28439 | 1:1000 |
| Antibody | Rabbit anti-cdc2 (Thr14) | Cell signaling (Beverly, MA, USA) | S2543 | 1:1000 |
| Antibody | Rabbit anti-Cyclin B | Cell signaling (Beverly, MA, USA) | S12231 | 1:1000 |
| Antibody | Rabbit anti-Cyclin B (S133) | Cell signaling (Beverly, MA, USA) | S4133 | 1:1000 |
| Antibody | Rabbit anti-VDAC1 | Invitrogen (Life Technologies, CA, USA) | PA1-954A | 1:1000 |
| Antibody | Rabbit anti-ch-TOG (CKAP5) | GeneTex (Irvine, CA, USA) | GTX30693 | 1:1000 |
| Antibody | Rabbit anti-TACC3 | Santa Cruz (Santa Cruz, CA, USA) | SC-22773 | 1:500 |
| Antibody | Rabbit anti-TACC3 (Ser 558) | Cell signaling (Beverly, MA, USA) | S8842 | 1:1000 |
| Antibody | Mouse anti-P62 | Abcam (Cambridge, MA, USA) | Ab56416 | 1:2000 |
| Antibody | Rabbit anti-LC3B | Cell signaling (Beverly, MA, USA) | S3868 | 1:1000 |
| Antibody | Mouse anti-GAPDH | Arigo (Hsinchu, Taiwan) | ARG10112 | 1:5000 |
| Antibody | Rabbit anti-PINK1 | Elabscience (Houston, TX, USA) | E-AB-17371 | 1:1000 |
| Antibody | Rabbit anti-PRKN | Elabscience (Houston, TX, USA) | E-AB-63490 | 1:1000 |
| Antibody | Mouse anti-α-tubulin | Sigma-Aldrich (St. Louis, MO, USA) | T5168 | 1:5000 |
| Antibody | Rabbit anti-γ-tubulin | Sigma-Aldrich (St. Louis, MO, USA) | T5192 | 1:5000 |
| Antibody | Total OXPHOS rodent WB antibody cocktail (Mouse) | Abcam (Cambridge, MA, USA) | ab122014 | 1:1000 |
| Commercial assay or kit | Mitochondria Isolation Kit | Thermo Fisher (Waltham, MA, USA) | 89874 | |
| Commercial assay or kit | TMRM Reagent | Invitrogen (Life Technologies, CA, USA) | I34361 | Mitochondrial membrane potential indicator |
| Commercial assay or kit | CellROX™ Green Reagent | Invitrogen (Life Technologies, CA, USA) | C10444 | Oxidative stress detection |
| Commercial assay or kit | ATP Assay Kit | Sigma-Aldrich (St. Louis, MO, USA) | MAK190 | ATP Colorimetric/Fluorometric |
| Commercial assay or kit | Cell counting Kit-8 | Sigma-Aldrich (St. Louis, MO, USA) | 96992 | Proliferation and cytotoxicity assays |
| Commercial assay or kit | MitoTracker | Invitrogen (Life Technologies, CA, USA) | M7512 |
3. Results
3.1. Mitotic Spindles Multipolarity Induced by ROT and AA Shows Loss of Spindles Pole Integrity
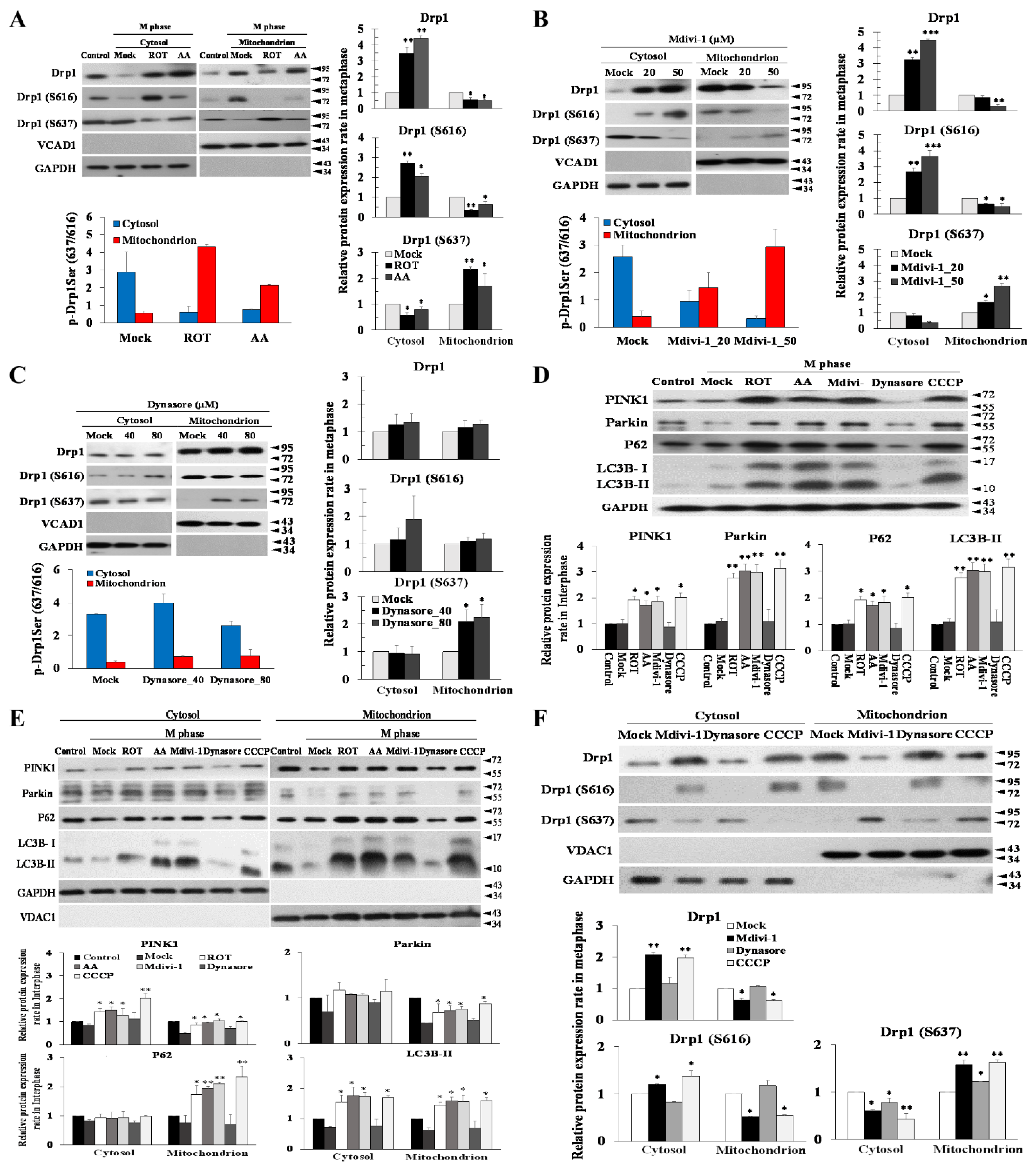
3.2. Mdivi-1 Induces Multipolar Spindles Formation by Increased Oxidative Stress due to Mitochondrial Fragmentation and Dysfunction
3.3. Dynasore Has a Slight Effect on Mitotic Defects, but Does Not Affect Mitochondrial Morphology
3.4. Multipolar Spindles Induction in Mitotic Cells Is Associated with Mitochondrial ETC Blockade
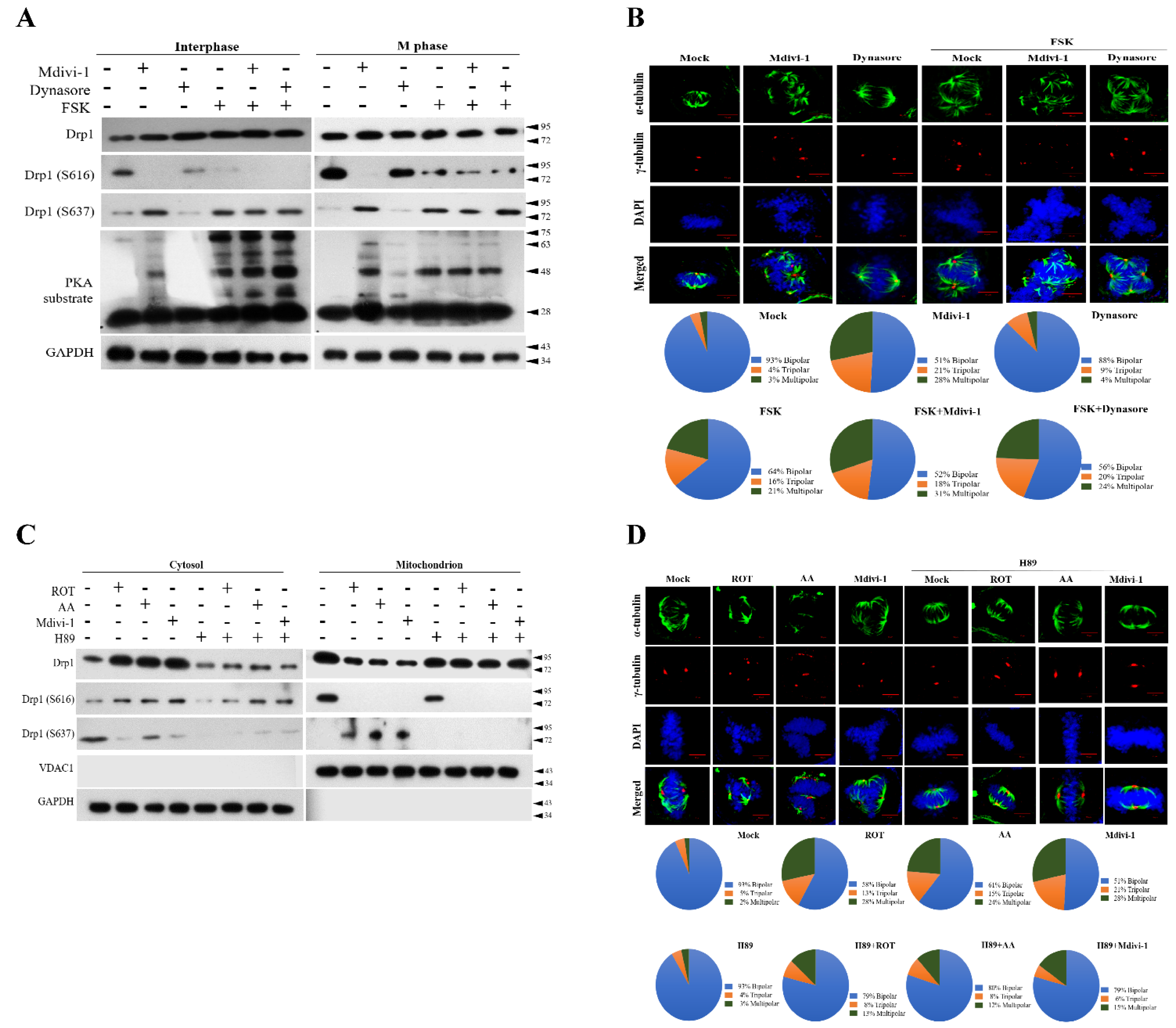
3.5. PKA Plays a Key Role to Switch the Phosphorylation Status of Mito-Drp1-Ser637 Rather than Mito-Drp1Ser616 and Triggers Multipolar Spindles Formation in Mitochondria during Mitotic Arrest
3.6. The Phosphorylation Status of Ser637, but Not Ser616, Modulates Mitophagy via the PINK/Parkin Pathway to Induce Multipolar Spindles Formation
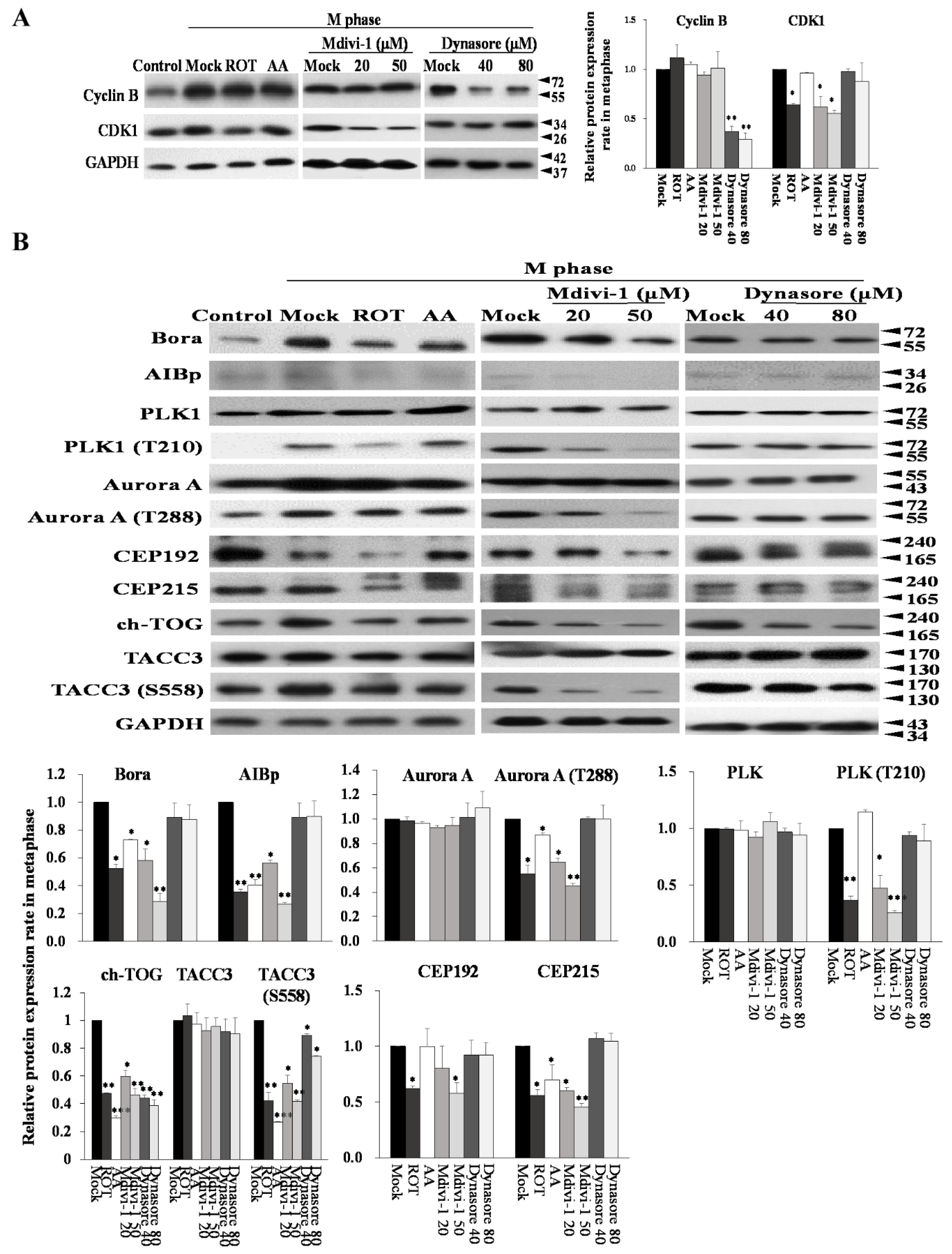
3.7. Mitotic Kinases Regulating Centrosome Maturation and Mitotic Spindles Assembly Are Associated with Mitochondrial ETC Blockade
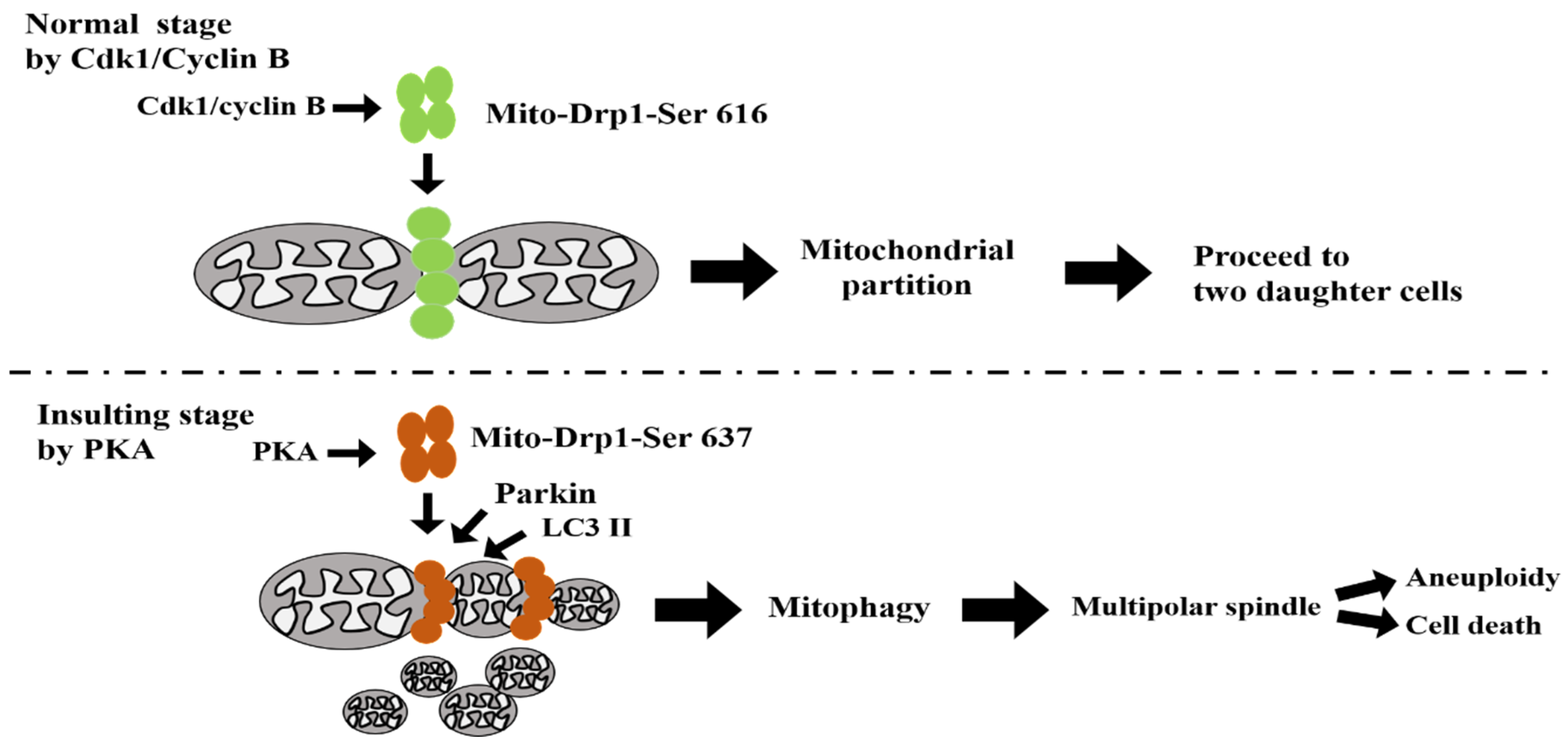
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, C.J.; Heng, H.H. Chromosomal instability and transcriptome dynamics in cancer. Cancer Metastasis Rev. 2013, 32, 391–402. [Google Scholar] [CrossRef]
- Tan, Z.; Chan, Y.J.A.; Chua, Y.J.K.; Rutledge, S.D.; Pavelka, N.; Cimini, D.; Rancati, G. Environmental stresses induce karyotypic instability in colorectal cancer cells. Mol. Biol. Cell 2019, 30, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef]
- Godinho, S.A.; Picone, R.; Burute, M.; Dagher, R.; Su, Y.; Leung, C.T.; Polyak, K.; Brugge, J.S.; Théry, M.; Pellman, D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature 2014, 510, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Contadini, C.; Monteonofrio, L.; Virdia, I.; Prodosmo, A.; Valente, D.; Chessa, L.; Musio, A.; Fava, L.L.; Rinaldo, C.; Di Rocco, G.; et al. p53 mitotic centrosome localization preserves centrosome integrity and works as sensor for the mitotic surveillance pathway. Cell Death Dis. 2019, 10, 850. [Google Scholar] [CrossRef] [PubMed]
- Donthamsetty, S.; Brahmbhatt, M.; Pannu, V.; Rida, P.C.; Ramarathinam, S.; Ogden, A.; Cheng, A.; Singh, K.K.; Aneja, R. Mitochondrial genome regulates mitotic fidelity by maintaining centrosomal homeostasis. Cell Cycle 2014, 13, 2056–2063. [Google Scholar] [CrossRef]
- Maiato, H.; Logarinho, E. Mitotic spindle multipolarity without centrosome amplification. Nat. Cell Biol. 2014, 16, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Dodson, H.; Bourke, E.; Jeffers, L.J.; Vagnarelli, P.; Sonoda, E.; Takeda, S.; Earnshaw, W.C.; Merdes, A.; Morrison, C. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 2004, 23, 3864–3873. [Google Scholar] [CrossRef]
- Doxsey, S. Duplicating Dangerously: Linking Centrosome Duplication and Aneuploidy. Mol. Cell 2002, 10, 439–440. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef]
- Pfau, S.J.; Amon, A. Chromosomal instability and aneuploidy in cancer: From yeast to man. EMBO Rep. 2012, 13, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, H.; Lengauer, C. Aneuploidy and cancer. Nature 2004, 432, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Wiggs, M.P.; Duarte, J.A.; Zergeroglu, A.M.; Demirel, H.A. Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E31–E39. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Srivastava, P.; Panda, D. Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding. FEBS J. 2007, 274, 4788–4801. [Google Scholar] [CrossRef]
- Moore, A.; Golden, A. Hypothesis: Bifunctional mitochondrial proteins have centrosomal functions. Environ. Mol. Mutagenes. 2009, 50, 637–648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.S.; Huh, S.; Liu, S.; Lee, D.Y.; Hong, S.H.; Yu, K.; Lu, B. Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca2+ Homeostasis in Neural Stem Cell Development. Dev. Cell 2016, 37, 174–189. [Google Scholar] [CrossRef]
- Ruan, W.; Lim, H.H.; Surana, U. Mapping Mitotic Death: Functional Integration of Mitochondria, Spindle Assembly Checkpoint and Apoptosis. Front. Cell Dev. Biol. 2019, 6, 177. [Google Scholar] [CrossRef]
- Zhu, J.; Tsai, H.-J.; Gordon, M.R.; Li, R. Cellular Stress Associated with Aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-F.; Dong, Q.; Bai, Y.; Yuan, J.; Xu, Q.; Cao, C.; Liu, X. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic. Biol. Med. 2016, 103, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 2019, 28, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, L.; Jiang, X.; Zhai, S.; Xing, D. The Essential Role of Drp1 and Its Regulation by S-Nitrosylation of Parkin in Dopaminergic Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 609–622. [Google Scholar] [CrossRef]
- Huang, M.; Chiang, S.; Kalinowski, D.; Bae, D.-H.; Sahni, S.; Richardson, D. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis. Oxidative Med. Cell. Longev. 2019, 2019, 6392763. [Google Scholar] [CrossRef]
- Chow, J.P.H.; Poon, R.Y.C. The CDK1 inhibitory kinase MYT1 in DNA damage checkpoint recovery. Oncogene 2013, 32, 4778–4788. [Google Scholar] [CrossRef]
- Chou, C.-H.; Loh, J.-K.; Yang, M.-C.; Lin, C.-C.; Hong, M.-C.; Cho, C.-L.; Chou, A.-K.; Wang, C.-H.; Lieu, A.-S.; Howng, S.-L.; et al. AIBp regulates mitotic entry and mitotic spindle assembly by controlling activation of both Aurora-A and Plk1. Cell Cycle 2015, 14, 2764–2776. [Google Scholar] [CrossRef][Green Version]
- Forkink, M.; Basit, F.; Teixeira, J.; Swarts, H.G.; Koopman, W.J.H.; Willems, P. Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol. 2015, 6, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Santana-Santos, L.; Shuda, M.; Sobol, R.W.; Van Houten, B.; Qian, W. A novel strategy for targeted killing of tumor cells: Induction of multipolar acentrosomal mitotic spindles with a quinazolinone derivative mdivi-1. Mol. Oncol. 2015, 9, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Preta, G.; Cronin, J.G.; Sheldon, I.M. Dynasore—Not just a dynamin inhibitor. Cell Commun. Signal. 2015, 13, 24. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef]
- You, Y.; He, Q.; Lu, H.; Zhou, X.; Chen, L.; Liu, H.; Lu, Z.; Liu, D.; Liu, Y.; Zuo, D.; et al. Silibinin Induces G2/M Cell Cycle Arrest by Activating Drp1-Dependent Mitochondrial Fission in Cervical Cancer. Front. Pharm. 2020, 11, 271. [Google Scholar] [CrossRef]
- Tanwar, D.K.; Parker, D.J.; Gupta, P.; Spurlock, B.; Alvarez, R.D.; Basu, M.K.; Mitra, K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget 2016, 7, 60021–60037. [Google Scholar] [CrossRef] [PubMed]
- Hirusaki, K.; Yokoyama, K.; Cho, K.; Ohta, Y. Temporal depolarization of mitochondria during M phase. Sci. Rep. 2017, 7, 16044. [Google Scholar] [CrossRef]
- Dai, W.; Wang, G.; Chwa, J.; Oh, M.E.; Abeywardana, T.; Yang, Y.; Wang, Q.A.; Jiang, L. Mitochondrial division inhibitor (mdivi-1) decreases oxidative metabolism in cancer. Br. J. Cancer 2020, 122, 1288–1297. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, G.; Bulteau, A.-L.; Alves-Guerra, M.-C.; Burel, A.; Lavault, M.-T.; Gavard, O.; Le Bras, S.; Gagné, J.-P.; Poirier, G.G.; Le Borgne, R.; et al. Aurora kinase A localises to mitochondria to control organelle dynamics and energy production. eLife 2018, 7, e38111. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef]
- Doménech, E.; Maestre, C.; Esteban-Martínez, L.; Partida, D.; Pascual, R.; Fernández-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.-B.; Lendahl, U.; Zhao, J.; Nistér, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, L.; Wu, S.; Xing, D. Drp1, Mff, Fis1, and MiD51 are coordinated to mediate mitochondrial fission during UV irradiation-induced apoptosis. FASEB J. 2016, 30, 466–476. [Google Scholar] [CrossRef]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, e49112. [Google Scholar] [CrossRef]
- Skroblin, P.; Grossmann, S.; Schäfer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of protein kinase A anchoring. Int. Rev. Cell Mol. Biol. 2010, 283, 235–330. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 235–330. [Google Scholar] [CrossRef] [PubMed]
- Bialy, H. Aneuploidy and cancer: Vintage wine in a new bottle? Nat. Biotechnol. 1998, 16, 137–138. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, H.-J.; Tsai, C.-Y.; Chiou, S.-J.; Lai, Y.-L.; Wang, C.-H.; Cheng, J.-T.; Chuang, T.-H.; Huang, C.-Y.F.; Kwan, A.-L.; Loh, J.-K.; et al. The Phosphorylation Status of Drp1-Ser637 by PKA in Mitochondrial Fission Modulates Mitophagy via PINK1/Parkin to Exert Multipolar Spindles Assembly during Mitosis. Biomolecules 2021, 11, 424. https://doi.org/10.3390/biom11030424
Ko H-J, Tsai C-Y, Chiou S-J, Lai Y-L, Wang C-H, Cheng J-T, Chuang T-H, Huang C-YF, Kwan A-L, Loh J-K, et al. The Phosphorylation Status of Drp1-Ser637 by PKA in Mitochondrial Fission Modulates Mitophagy via PINK1/Parkin to Exert Multipolar Spindles Assembly during Mitosis. Biomolecules. 2021; 11(3):424. https://doi.org/10.3390/biom11030424
Chicago/Turabian StyleKo, Huey-Jiun, Cheng-Yu Tsai, Shean-Jaw Chiou, Yun-Ling Lai, Chi-Huei Wang, Jiin-Tsuey Cheng, Tsung-Hsien Chuang, Chi-Ying F. Huang, Aij-Lie Kwan, Joon-Khim Loh, and et al. 2021. "The Phosphorylation Status of Drp1-Ser637 by PKA in Mitochondrial Fission Modulates Mitophagy via PINK1/Parkin to Exert Multipolar Spindles Assembly during Mitosis" Biomolecules 11, no. 3: 424. https://doi.org/10.3390/biom11030424
APA StyleKo, H.-J., Tsai, C.-Y., Chiou, S.-J., Lai, Y.-L., Wang, C.-H., Cheng, J.-T., Chuang, T.-H., Huang, C.-Y. F., Kwan, A.-L., Loh, J.-K., & Hong, Y.-R. (2021). The Phosphorylation Status of Drp1-Ser637 by PKA in Mitochondrial Fission Modulates Mitophagy via PINK1/Parkin to Exert Multipolar Spindles Assembly during Mitosis. Biomolecules, 11(3), 424. https://doi.org/10.3390/biom11030424