
Interfaces with Structure Dynamics of the Workhorses from Cells Revealed through Cross-Linking Mass Spectrometry (CLMS)

 ,
,  , ,
, , 
 and
and
Abstract
1. Introduction

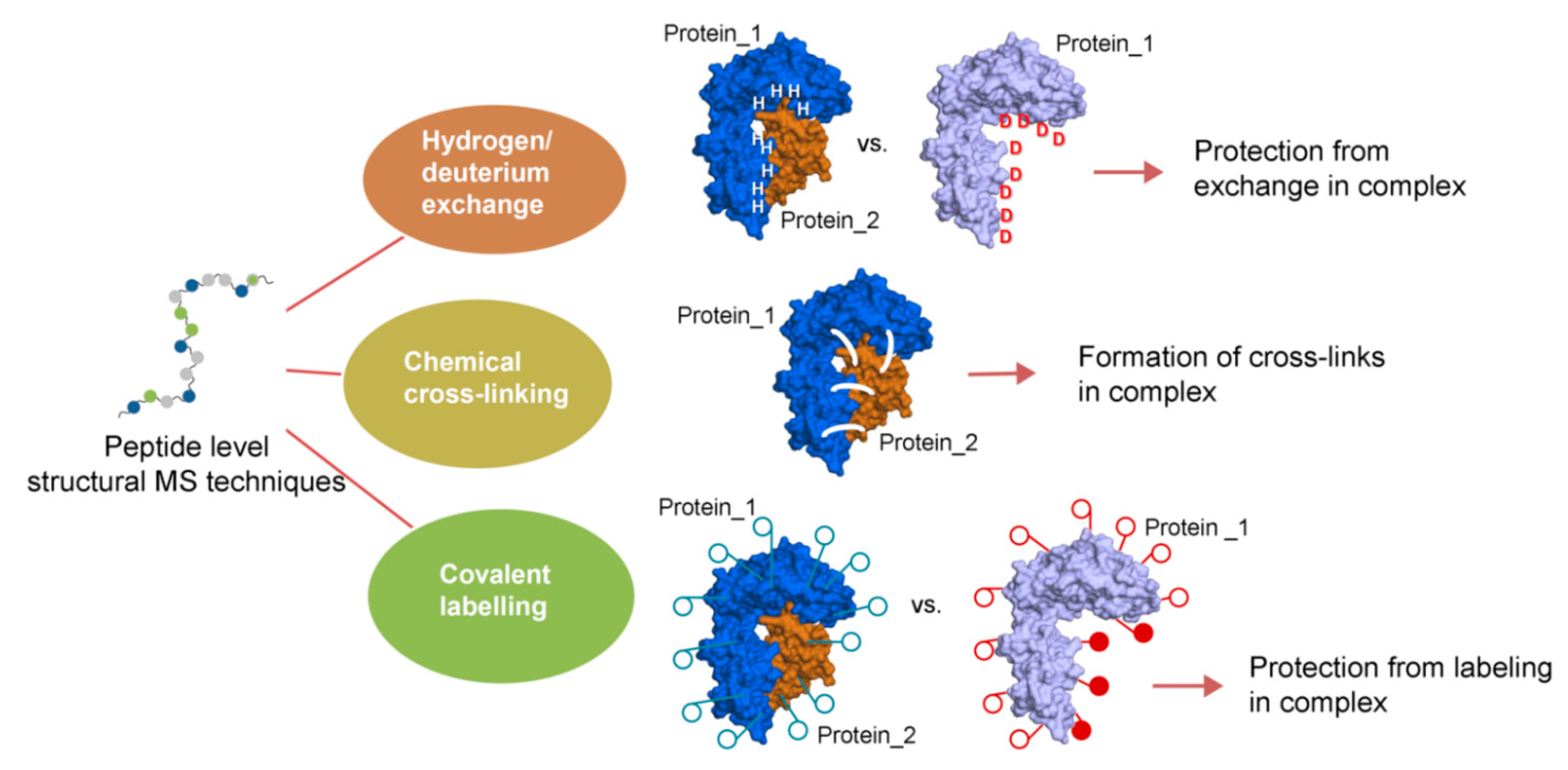
2. Concept and Perspectives of Cross-Linking Mass Spectrometry
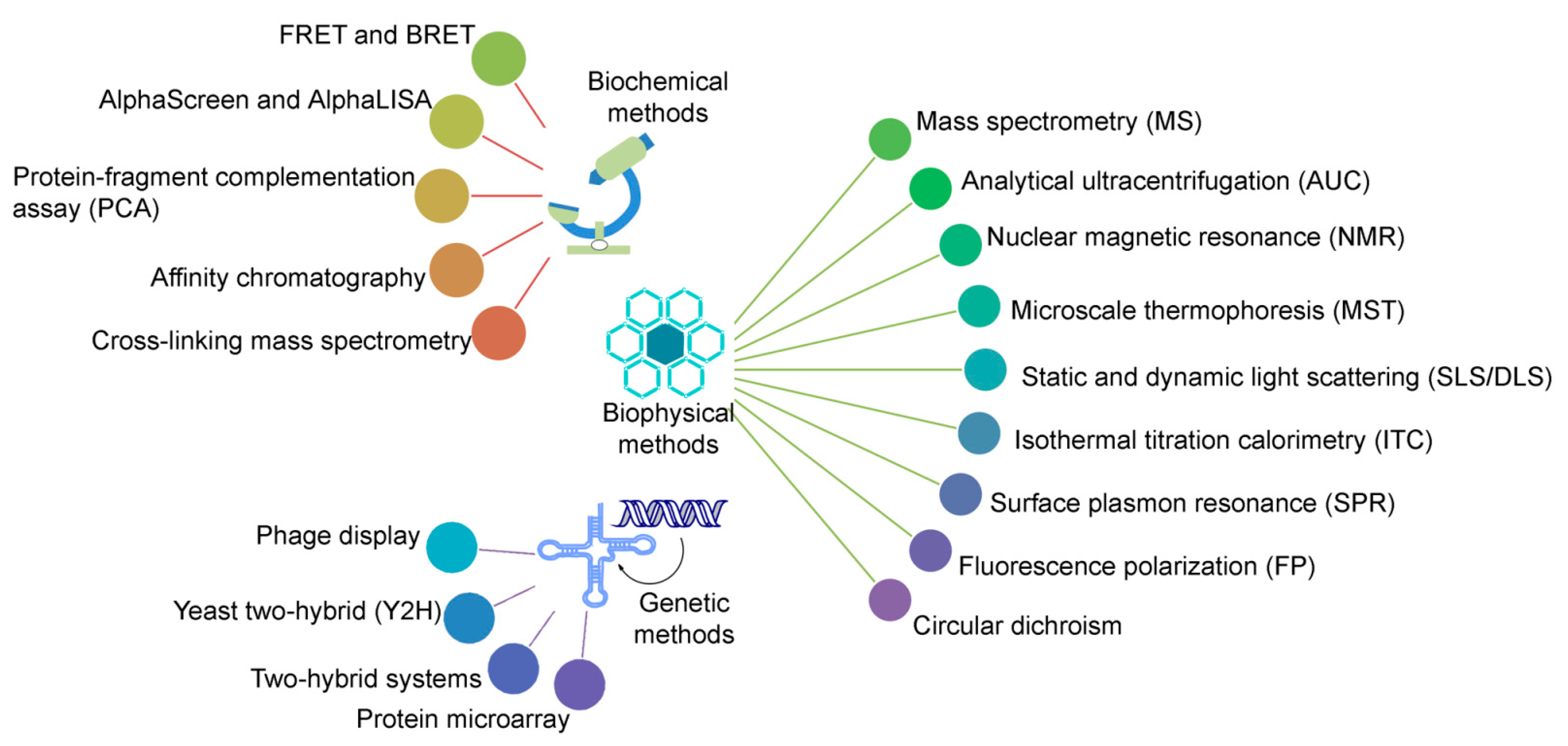
2.1. Associate Methods for CLMS Technique
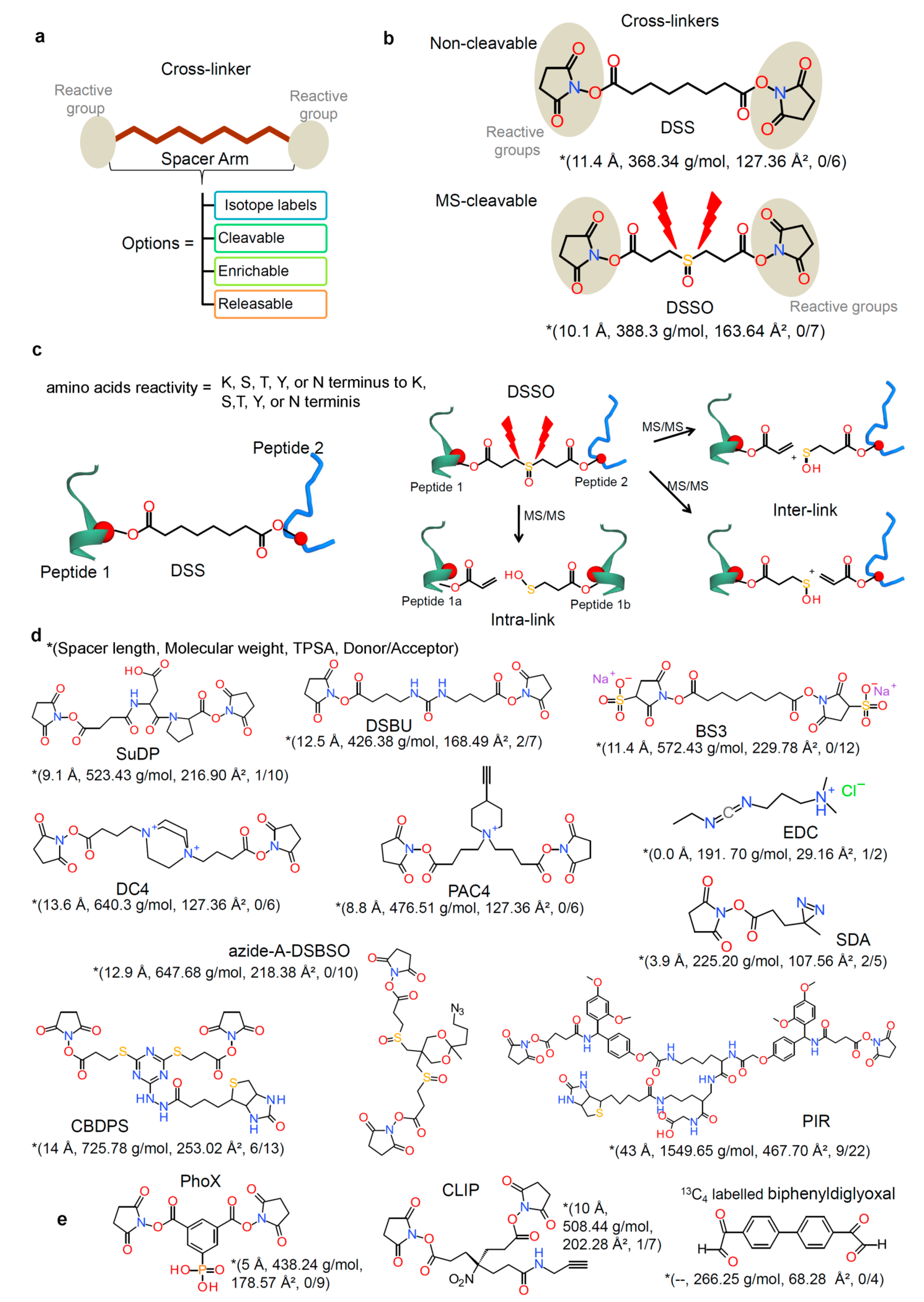
2.2. Chemical Cross-Linkers Structure and Chemistry

2.3. Cross-Linked Sample Preparation for MS (Protein–Protein) Analysis
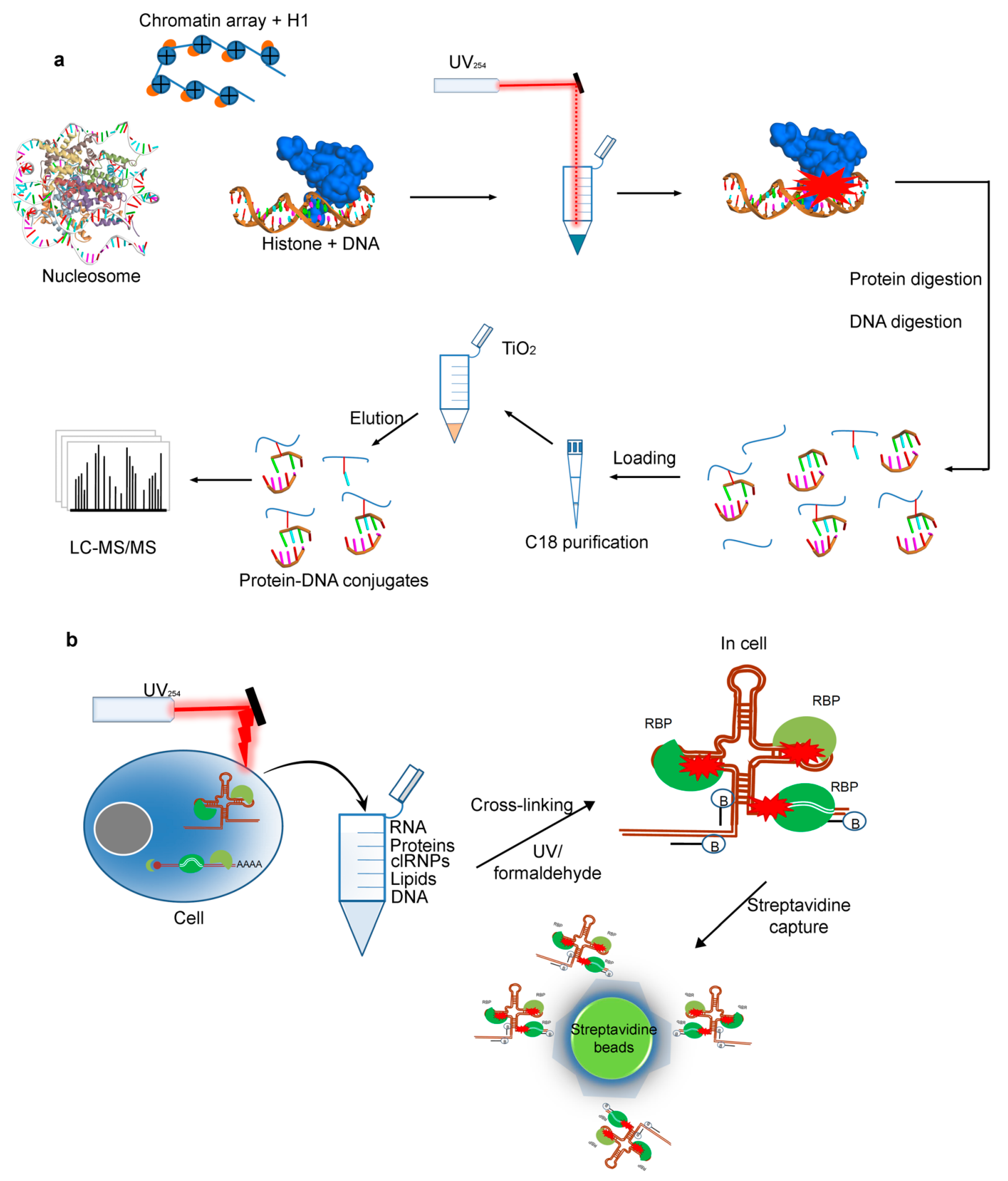
2.4. Cross-Linking by UV for Protein–DNAInteractions
2.5. Protein–RNA Interactions Identified by Cross-Linking MS Technique
2.5.1. The RNA-Centric Cross-Linking

2.5.2. The Protein-Centric Cross-Linking
2.6. Pairing the CLMS Methodologies with Molecular Dynamics Simulations
3. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, M.; Li, Q.; Wang, R. Current experimental methods for characterizing protein-protein interactions. Chem. Med. Chem. 2016, 11, 738–756. [Google Scholar] [CrossRef] [PubMed]
- Berggård, T.; Linse, S.; James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 16, 2833–2842. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 12, 862–873. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef]
- Brooke, M.A.; Nitoiu, D.; Kelsell, D.P. Cell-cell connectivity: Desmosomes and disease. J. Pathol. 2012, 226, 158–171. [Google Scholar] [CrossRef]
- Heck, A.J. Native mass spectrometry: A bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927–933. [Google Scholar] [CrossRef]
- Pirrone, G.F.; Iacob, R.E.; Engen, J.R. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem. 2015, 87, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Tugarinov, V.; Hwang, P.M.; Kay, L.E. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu. Rev. Biochem. 2004, 73, 107–146. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Savino, C. Protein structure determination by x-ray crystallography. Methods Mol. Biol. 2008, 452, 63–87. [Google Scholar] [CrossRef] [PubMed]
- Walian, P.; Cross, T.A.; Jap, B.K. Structural genomics of membrane proteins. Genome Biol. 2004, 5, 215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol. 2020, 63, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hino, N.; Okazaki, Y.; Kobayashi, T.; Hayashi, A.; Sakamoto, K.; Yokoyama, S. Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid. Nat. Methods 2005, 2, 201–206. [Google Scholar] [CrossRef]
- Farrell, I.S.; Toroney, R.; Hazen, J.L.; Mehl, R.A.; Chin, J.W. Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat. Methods 2005, 2, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.L.; Borgnia, M.J.; Bartesaghi, A.; Tran, E.E.; Earl, L.A.; Schauder, D.M.; Lengyel, J.; Pierson, J.; Patwardhan, A.; Subramaniam, S. Cryo-electron microscopy-a primer for the nonmicroscopist. FEBS J. 2013, 280, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Hura, G.L.; Menon, A.L.; Hammel, M.; Rambo, R.P.; Poole, F.L., II; Tsutakawa, S.E.; Jenney, F.E., Jr.; Classen, S.; Frankel, K.A.; Hopkins, R.C.; et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 2009, 6, 606–612. [Google Scholar] [CrossRef]
- Uetrecht, C.; Rose, R.J.; Van Duijn, E.; Lorenzen, K.; Heck, A.J. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc. Rev. 2010, 39, 1633–1655. [Google Scholar] [CrossRef]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef]
- Gau, B.C.; Sharp, J.S.; Rempel, D.L.; Gross, M.L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 2009, 81, 6563–6571. [Google Scholar] [CrossRef]
- Perrin, F. Polarisation of fluorescence and mean life of excited molecules. Polarisation de la lumière de fluorescence. Vie moyenne des molécules dans l’etat excite. J. Phys. Radium. 1926, 7, 390–401. [Google Scholar]
- Du, Y. Protein-Protein Interactions, of the Series Methods in Molecular Biology; Meyerkord, C.L., Fu, H., Walker, J.M., Eds.; Springer: New York, NY, USA, 2015; Volume 1278, pp. 529–544. [Google Scholar]
- Madeira, A.; Ohman, E.; Nilsson, A.; Sjögren, B.; Andrén, P.E.; Svenningsson, P. Coupling surface plasmon resonance to mass spectrometry to discover novel protein-protein interactions. Nat. Protoc. 2009, 4, 1023–1037. [Google Scholar] [CrossRef]
- Boozer, C.; Kim, G.; Cong, S.; Guan, H.; Londergan, T. Looking towards label-free biomolecular interaction analysis in a high-throughput format: A review of new surface plasmon resonance technologies. Curr. Opin. Biotechnol. 2006, 17, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Beck, M.R.; Campbell, S.L. Protein-Protein Interactions, of the Series Methods in Molecular; Springer: New York, NY, USA, 2015; Volume 1278, pp. 267–279. [Google Scholar]
- Ranjbar, B.; Gill, P. Circular dichroism techniques: Biomolecular and nanostructural analyses—A review. Chem. Biol. Drug. Des. 2009, 74, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Some, D. Light-scattering-based analysis of biomolecular interactions. Biophys. Rev. 2013, 5, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Laue, T. Analytical ultracentrifugation: A powerful ‘new’ technology in drug discovery. Drug Discov. Today Technol. 2004, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Characterization of protein-protein interactions by isothermal titration calorimetry. Methods Mol. Biol. 2015, 1278, 183–204. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef]
- Selvin, P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 2000, 7, 730–734. [Google Scholar] [CrossRef]
- Pfleger, K.D.; Eidne, K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174. [Google Scholar] [CrossRef]
- Yasgar, A.; Jadhav, A.; Simeonov, A.; Coussens, N.P. AlphaScreen-based assays: Ultra-high-throughput screening for small-molecule inhibitors of challenging enzymes and protein-protein interactions. Methods Mol. Biol. 2016, 1439, 77–98. [Google Scholar] [CrossRef]
- Remy, I.; Campbell-Valois, F.X.; Michnick, S.W. Detection of protein-protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat. Protoc. 2007, 2, 2120–2125. [Google Scholar] [CrossRef]
- Beeckmans, S. Chromatographic methods to study protein-protein interactions. Methods 1999, 19, 278–305. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Sholukh, M.; Korpela, T. Use of protein-protein interactions in affinity chromatography. J. Biochem. Biophys. Methods 2001, 49, 29–47. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef]
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, A.; Colas, P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol. Sci. 2012, 33, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Ptacek, J.; Snyder, M. Protein microarray technology. Mech. Ageing Dev. 2007, 128, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bilgin, M.; Bangham, R.; Hall, D.; Casamayor, A.; Bertone, P.; Lan, N.; Jansen, R.; Bidlingmaier, S.; Houfek, T.; et al. Global analysis of protein activities using proteome chips. Science 2001, 293, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef]
- Bich, C.; Zenobi, R. Mass spectrometry of large complexes. Curr. Opin. Struct. Biol. 2009, 19, 632–639. [Google Scholar] [CrossRef]
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Protein and polymer analyses up to m/z 100,000 by laser ionization time of flight mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153. [Google Scholar] [CrossRef]
- Glish, G.L.; Vachet, R.W. The basics of mass spectrometry in the twenty-first century. Nat. Rev. Drug Discov. 2003, 2, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Kukacka, Z.; Rosulek, M.; Strohalm, M.; Kavan, D.; Novak, P. Mapping protein structural changes by quantitative cross-linking. Methods 2015, 89, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Chen, Z.A.; Rappsilber, J. Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteom. 2013, 88, 120–128. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Chavez, J.D.; Schweppe, D.K.; Eng, J.K.; Bruce, J.E. In Vivo Conformational Dynamics of Hsp90 and Its Interactors. Cell Chem. Biol. 2016, 23, 716–726. [Google Scholar] [CrossRef]
- Pearson, K.M.; Pannell, L.K.; Fales, H.M. Intramolecular cross-linking experiments on cytochrome c and ribonuclease A using an isotope multiplet method. Rapid Commun. Mass Spectrom. 2002, 16, 149–159. [Google Scholar] [CrossRef]
- Müller, F.; Graziadei, A.; Rappsilber, J. Quantitative Photo-crosslinking Mass Spectrometry Revealing Protein Structure Response to Environmental Changes. Anal. Chem. 2019, 91, 9041–9048. [Google Scholar] [CrossRef]
- Chen, Z.A.; Rappsilber, J. Quantitative cross-linking/mass spectrometry to elucidate structural changes in proteins and their complexes. Nat. Protoc. 2019, 14, 171–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fischer, L.; Tahir, S.; Bukowski-Wills, J.C.; Barlow, P.; Rappsilber, J. Quantitative cross-linking/mass spectrometry reveals subtle protein conformational changes. Wellcome Open Res. 2016, 1, 5. [Google Scholar] [CrossRef]
- Chen, Z.A.; Rappsilber, J. Protein Dynamics in Solution by Quantitative Crosslinking/Mass Spectrometry. Trends Biochem. Sci. 2018, 43, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, R.E.; Hang, L.E.; Molloy, K.R.; Chait, B.T.; Kapoor, T.M. A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells. Cell Chem. Biol. 2018, 25, 110–120.e3. [Google Scholar] [CrossRef]
- Yu, C.; Huszagh, A.; Viner, R.; Novitsky, E.J.; Rychnovsky, S.D.; Huang, L. Developing a Multiplexed Quantitative Cross-Linking Mass Spectrometry Platform for Comparative Structural Analysis of Protein Complexes. Anal. Chem. 2016, 88, 10301–10308. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Rappsilber, J. A protocol for studying structural dynamics of proteins by quantitative crosslinking mass spectrometry and data-independent acquisition. J. Proteom. 2020, 218, 103721. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef]
- Shaw, P.E. Peptidyl-prolyl isomerases: A new twist to transcription. EMBO Rep. 2002, 3, 521–526. [Google Scholar] [CrossRef]
- Lin, W.; Bonin, M.; Boden, A.; Wieduwild, R.; Murawala, P.; Wermke, M.; Andrade, H.; Bornhäuser, M.; Zhang, Y. Peptidyl prolyl cis/trans isomerase activity on the cell surface correlates with extracellular matrix development. Commun. Biol. 2019, 2, 58. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.D.; Bruce, J.E. Chemical cross-linking with mass spectrometry: A tool for systems structural biology. Curr. Opin. Chem. Biol. 2019, 48, 8–18. [Google Scholar] [CrossRef]
- Keller, A.; Chavez, J.D.; Tang, X.; Bruce, J.E. Leveraging the entirety of the protein data bank to enable improved structure prediction based on cross-link data. J. Proteome Res. 2021, 20, 1087–1095. [Google Scholar] [CrossRef]
- Chavez, J.D.; Mohr, J.P.; Mathay, M.; Zhong, X.; Keller, A.; Bruce, J.E. Systems structural biology measurements by in vivo cross-linking with mass spectrometry. Nat. Protoc. 2019, 14, 2318–2343. [Google Scholar] [CrossRef]
- Keller, A.; Chavez, J.D.; Felt, K.C.; Bruce, J.E. Prediction of an upper limit for the fraction of interprotein cross-links in large-scale in vivo cross-linking studies. J. Proteome Res. 2019, 18, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lössl, P.; Scheltema, R.; Viner, R.; Heck, A. Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 2017, 8, 15473. [Google Scholar] [CrossRef]
- Klykov, O.; Steigenberger, B.; Pektaş, S.; Fasci, D.; Heck, A.; Scheltema, R.A. Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 2018, 13, 2964–2990. [Google Scholar] [CrossRef]
- Demartino, G.N. Reconstitution of PA700, the 19S regulatory particle, from purified precursor complexes. Methods Mol. Biol. 2012, 832, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Oeffinger, M. Two steps forward-one step back: Advances in affinity purification mass spectrometry of macromolecular complexes. Proteomics 2012, 12, 1591–1608. [Google Scholar] [CrossRef]
- Mishra, P.K.; Yoo, C.M.; Hong, E.; Rhee, H.W. Photo-crosslinking: An emerging chemical tool for investigating molecular networks in live cells. ChemBioChem 2020, 21, 924–932. [Google Scholar] [CrossRef]
- Tang, X.; Bruce, J.E. Mass Spectrometry of Proteins and Peptides, of the Series Methods in Molecular; Lipton, M.S., Paša-Tolic, L., Walker, J.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 492, pp. 283–293. [Google Scholar]
- Guerrero, C.; Tagwerker, C.; Kaiser, P.; Huang, L. An integrated mass spectrometry-based proteomic approach: Quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell Proteom. 2006, 5, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Vasilescu, J.; Guo, X.; Kast, J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 2004, 4, 3845–3854. [Google Scholar] [CrossRef]
- Van den Heuvel, R.H.; Heck, A.J. Native protein mass spectrometry: From intact oligomers to functional machineries. Curr. Opin. Chem. Biol. 2004, 8, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Benesch, J.L.; Robinson, C.V. Mass spectrometry of macromolecular assemblies: Preservation and dissociation. Curr. Opin. Struct. Biol. 2006, 16, 245–251. [Google Scholar] [CrossRef]
- Sharon, M.; Robinson, C.V. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu. Rev. Biochem. 2007, 76, 167–193. [Google Scholar] [CrossRef]
- Synowsky, S.A.; Van den Heuvel, R.H.; Mohammed, S.; Pijnappel, P.W.; Heck, A.J. Probing genuine strong interactions and post-translational modifications in the heterogeneous yeast exosome protein complex. Mol. Cell Proteom. 2006, 5, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Huang, L. Cross-linking mass spectrometry: An emerging technology for interactomics and structural biology. Anal. Chem. 2018, 90, 144–165. [Google Scholar] [CrossRef]
- Sinz, A. Investigation of protein-protein interactions in living cells by chemical crosslinking and mass spectrometry. Anal. Bioanal. Chem. 2010, 397, 3433–3440. [Google Scholar] [CrossRef]
- Bruce, J.E. In vivo protein complex topologies: Sights through a cross-linking lens. Proteomics 2012, 12, 1565–1575. [Google Scholar] [CrossRef]
- Piersimoni, L.; Sinz, A. Cross-linking/mass spectrometry at the crossroads. Anal. Bioanal. Chem. 2020, 412, 5981–5987. [Google Scholar] [CrossRef]
- Leitner, A. Cross-linking and other structural proteomics techniques: How chemistry is enabling mass spectrometry applications in structural biology. Chem. Sci. 2016, 7, 4792–4803. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Bonvin, A.M.J.J.; Borchers, C.H.; Chalkley, R.J.; Chamot-Rooke, J.; Combe, C.W.; Cox, J.; Dong, M.Q.; Fischer, L.; Götze, M.; et al. Toward increased reliability, transparency, and accessibility in cross-linking mass spectrometry. Structure 2020, 28, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.; Chiu, C.L.; Vellucci, D.; Yang, Y.; Patel, V.R.; Guan, S.; Randall, A.; Baldi, P.; Rychnovsky, S.D.; Huang, L. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell Proteom. 2011, 10, M110.002212. [Google Scholar] [CrossRef]
- Leitner, A.; Joachimiak, L.A.; Unverdorben, P.; Walzthoeni, T.; Frydman, J.; Förster, F.; Aebersold, R. Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9455–9460. [Google Scholar] [CrossRef]
- Pinard, R.; Lambert, D.; Heckman, J.E.; Esteban, J.A.; Gundlach, C.W., 4th; Hampel, K.J.; Glick, G.D.; Walter, N.G.; Major, F.; Burke, J.M. The hairpin ribozyme substrate binding-domain: A highly constrained D-shaped conformation. J. Mol. Biol. 2001, 307, 51–65. [Google Scholar] [CrossRef]
- Harris, M.E.; Christian, E.L. RNA crosslinking methods. Methods Enzymol. 2009, 468, 127–146. [Google Scholar] [CrossRef]
- Buxbaum, E. Cross-linkers. In Biophysical Chemistry of Proteins; Springer: Boston, MA, USA, 2011. [Google Scholar]
- Chen, F.; Nielsen, S.; Zenobi, R. Understanding chemical reactivity for homo- and heterobifunctional protein cross-linking agents. J. Mass Spectrom. 2013, 48, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Belsom, A.; Rappsilber, J. Anatomy of a crosslinker. Curr. Opin. Chem. Biol. 2020, 60, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Steigenberger, B.; Albanese, P.; Heck, A.J.R.; Scheltema, R.A. To cleave or not to cleave in XL-MS? J. Am. Soc. Mass Spectrom. 2020, 31, 196–206. [Google Scholar] [CrossRef]
- Iacobucci, C.; Piotrowski, C.; Aebersold, R.; Amaral, B.C.; Andrews, P.; Bernfur, K.; Borchers, C.; Brodie, N.I.; Bruce, J.E.; Cao, Y.; et al. First community-wide, comparative cross-linking mass spectrometry study. Anal. Chem. 2019, 91, 6953–6961. [Google Scholar] [CrossRef] [PubMed]
- Trester-Zedlitz, M.; Kamada, K.; Burley, S.K.; Fenyö, D.; Chait, B.T.; Muir, T.W. A modular cross-linking approach for exploring protein interactions. J. Am. Chem. Soc. 2003, 125, 2416–2425. [Google Scholar] [CrossRef]
- Tang, X.; Bruce, J.E. A new cross-linking strategy: Protein interaction reporter (PIR) technology for protein-protein interaction studies. Mol. Biosyst. 2010, 6, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Soderblom, E.J.; Goshe, M.B. Collision-induced dissociative chemical cross-linking reagents and methodology: Applications to protein structural characterization using tandem mass spectrometry analysis. Anal. Chem. 2006, 78, 8059–8068. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, C.; Sweedler, J.V.; Goshe, M.B. An enhanced protein crosslink identification strategy using CID-cleavable chemical crosslinkers and LC/MS(n) analysis. Proteomics 2012, 12, 401–405. [Google Scholar] [CrossRef]
- Müller, M.Q.; Dreiocker, F.; Ihling, C.H.; Schäfer, M.; Sinz, A. Cleavable cross-linker for protein structure analysis: Reliable identification of cross-linking products by tandem MS. Anal. Chem. 2010, 82, 6958–6968. [Google Scholar] [CrossRef]
- Petrotchenko, E.V.; Serpa, J.J.; Borchers, C.H. An isotopically coded CID-cleavable biotinylated cross-linker for structural proteomics. Mol. Cell Proteom. 2011, 10, M110.001420. [Google Scholar] [CrossRef]
- Clifford-Nunn, B.; Showalter, H.D.; Andrews, P.C. Quaternary diamines as mass spectrometry cleavable crosslinkers for protein interactions. J. Am. Soc. Mass Spectrom. 2012, 23, 201–212. [Google Scholar] [CrossRef][Green Version]
- Burke, A.M.; Kandur, W.; Novitsky, E.J.; Kaake, R.M.; Yu, C.; Kao, A.; Vellucci, D.; Huang, L.; Rychnovsky, S.D. Synthesis of two new enrichable and MS-cleavable cross-linkers to define protein-protein interactions by mass spectrometry. Org. Biomol. Chem. 2015, 13, 5030–5037. [Google Scholar] [CrossRef]
- Hagen, S.E.; Liu, K.; Jin, Y.; Piersimoni, L.; Andrews, P.C.; Showalter, H.D. Synthesis of CID-cleavable protein crosslinking agents containing quaternary amines for structural mass spectrometry. Org. Biomol. Chem. 2018, 16, 8245–8248. [Google Scholar] [CrossRef] [PubMed]
- Steigenberger, B.; Pieters, R.J.; Heck, A.J.R.; Scheltema, R.A. PhoX: An imac-enrichable cross-linking reagent. ACS Cent. Sci. 2019, 5, 1514–1522. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.M.; Du, X.; Tolić, N.; Wu, S.; Moore, R.J.; Mayer, M.U.; Smith, R.D.; Adkins, J.N. Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal. Chem. 2009, 81, 5524–5532. [Google Scholar] [CrossRef]
- Holding, A.N. XL-MS: Protein cross-linking coupled with mass spectrometry. Methods San Diego Calif. 2015, 89, 54–63. [Google Scholar] [CrossRef]
- Turnbough, C.L., Jr.; Switzer, R.L. Regulation of pyrimidine biosynthetic gene expression in bacteria: Repression without repressors. Microbiol. Mol. Biol. Rev. 2008, 72, 266–300. [Google Scholar] [CrossRef] [PubMed]
- Sinz, A.; Arlt, C.; Chorev, D.; Sharon, M. Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology. Protein Sci. 2015, 24, 1193–1209. [Google Scholar] [CrossRef]
- Götze, M.; Pettelkau, J.; Fritzsche, R.; Ihling, C.H.; Schäfer, M.; Sinz, A. Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom. 2015, 26, 83–97. [Google Scholar] [CrossRef]
- Lima, D.B.; De Lima, T.B.; Balbuena, T.S.; Neves-Ferreira, A.; Barbosa, V.C.; Gozzo, F.C.; Carvalho, P.C. SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. J. Proteom. 2015, 129, 51–55. [Google Scholar] [CrossRef]
- Yılmaz, Ş.; Drepper, F.; Hulstaert, N.; Černič, M.; Gevaert, K.; Economou, A.; Warscheid, B.; Martens, L.; Vandermarliere, E. Xilmass: A New Approach toward the Identification of Cross-Linked Peptides. Anal. Chem. 2016, 88, 9949–9957. [Google Scholar] [CrossRef]
- Rinner, O.; Seebacher, J.; Walzthoeni, T.; Mueller, L.N.; Beck, M.; Schmidt, A.; Mueller, M.; Aebersold, R. Identification of cross-linked peptides from large sequence databases. Nat. Methods 2008, 5, 315–318. [Google Scholar] [CrossRef]
- Mendes, M.L.; Fischer, L.; Chen, Z.A.; Barbon, M.; O’Reilly, F.J.; Giese, S.H.; Bohlke-Schneider, M.; Belsom, A.; Dau, T.; Combe, C.W.; et al. An integrated workflow for crosslinking mass spectrometry. Mol. Syst. Biol. 2019, 15, e8994. [Google Scholar] [CrossRef]
- Hoopmann, M.R.; Zelter, A.; Johnson, R.S.; Riffle, M.; MacCoss, M.J.; Davis, T.N.; Moritz, R.L. Kojak: Efficient analysis of chemically cross-linked protein complexes. J. Proteome Res. 2015, 14, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Meng, J.M.; Cao, Y.; Yin, J.L.; Fang, R.Q.; Fan, S.B.; Liu, C.; Zeng, W.F.; Ding, Y.H.; Tan, D.; et al. A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 2019, 10, 3404. [Google Scholar] [CrossRef]
- Müller, F.; Kolbowski, L.; Bernhardt, O.M.; Reiter, L.; Rappsilber, J. Data-independent acquisition improves quantitative cross-linking mass spectrometry. Mol. Cell Proteom. 2019, 18, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Maiolica, A.; Cittaro, D.; Borsotti, D.; Sennels, L.; Ciferri, C.; Tarricone, C.; Musacchio, A.; Rappsilber, J. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol. Cell Proteom. 2007, 6, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- Algret, R.; Fernandez-Martinez, J.; Shi, Y.; Kim, S.J.; Pellarin, R.; Cimermancic, P.; Cochet, E.; Sali, A.; Chait, B.T.; Rout, M.P.; et al. Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway. Mol. Cell Proteom. 2014, 13, 2855–2870. [Google Scholar] [CrossRef]
- Pettelkau, J.; Thondorf, I.; Theisgen, S.; Lilie, H.; Schröder, T.; Arlt, C.; Ihling, C.H.; Sinz, A. Structural analysis of guanylyl cyclase-activating protein-2 (GCAP-2) homodimer by stable isotope-labeling, chemical cross-linking, and mass spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Arlt, C.; Ihling, C.H.; Sinz, A. Structure of full-length p53 tumor suppressor probed by chemical cross-linking and mass spectrometry. Proteomics 2015, 15, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Kurokawa, M. Separation of fucose and acetylhexosamines by two-dimensional thin-layer chromatography. Anal. Biochem. 1968, 26, 472–474. [Google Scholar] [CrossRef]
- Petrotchenko, E.V.; Serpa, J.J.; Cabecinha, A.N.; Lesperance, M.; Borchers, C.H. “Out-gel” tryptic digestion procedure for chemical cross-linking studies with mass spectrometric detection. J. Proteome Res. 2014, 13, 527–535. [Google Scholar] [CrossRef]
- Parfentev, I.; Schilbach, S.; Cramer, P.; Urlaub, H. An experimentally generated peptide database increases the sensitivity of XL-MS with complex samples. J. Proteom. 2020, 220, 103754. [Google Scholar] [CrossRef]
- Rappsilber, J. The beginning of a beautiful friendship: Cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol. 2011, 173, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Mintseris, J.; Gygi, S.P. High-density chemical cross-linking for modeling protein interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lozano, M.A.; Koopmans, F.; Sullivan, P.F.; Protze, J.; Krause, G.; Verhage, M.; Li, K.W.; Liu, F.; Smit, A.B. Stitching the synapse: Cross-linking mass spectrometry into resolving synaptic protein interactions. Sci. Adv. 2020, 6, eaax5783. [Google Scholar] [CrossRef]
- Slavin, M.; Kalisman, N. Structural analysis of protein complexes by cross-linking and mass spectrometry. Methods Mol. Biol. 2018, 1764, 173–183. [Google Scholar] [CrossRef]
- Jones, A.X.; Cao, Y.; Tang, Y.L.; Wang, J.H.; Ding, Y.H.; Tan, H.; Chen, Z.L.; Fang, R.Q.; Yin, J.; Chen, R.C.; et al. Improving mass spectrometry analysis of protein structures with arginine-selective chemical cross-linkers. Nat. Commun. 2019, 10, 3911. [Google Scholar] [CrossRef]
- Politis, A.; Stengel, F.; Hall, Z.; Hernández, H.; Leitner, A.; Walzthoeni, T.; Robinson, C.V.; Aebersold, R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods 2014, 11, 403–406. [Google Scholar] [CrossRef]
- Rey, M.; Dupré, M.; Lopez-Neira, I.; Duchateau, M.; Chamot-Rooke, J. eXL-MS: An enhanced cross-linking mass spectrometry workflow to study protein complexes. Anal. Chem. 2018, 90, 10707–10714. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, R.; Ihling, C.H.; Götze, M.; Sinz, A. Optimizing the enrichment of cross-linked products for mass spectrometric protein analysis. Rapid Commun. Mass Spectrom. 2012, 26, 653–658. [Google Scholar] [CrossRef]
- Tinnefeld, V.; Venne, A.S.; Sickmann, A.; Zahedi, R.P. Enrichment of cross-linked peptides using charge-based fractional diagonal chromatography (ChaFRADIC). J. Proteome Res. 2017, 16, 459–469. [Google Scholar] [CrossRef]
- Liu, F.; Lössl, P.; Rabbitts, B.M.; Balaban, R.S.; Heck, A.J.R. The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol. Cell Proteom. 2018, 17, 216–232. [Google Scholar] [CrossRef]
- Tan, D.; Li, Q.; Zhang, M.J.; Liu, C.; Ma, C.; Zhang, P.; Ding, Y.H.; Fan, S.B.; Tao, L.; Yang, B.; et al. Trifunctional cross-linker for mapping protein-protein interaction networks and comparing protein conformational states. eLife 2016, 5, e12509. [Google Scholar] [CrossRef]
- Rampler, E.; Stranzl, T.; Orban-Nemeth, Z.; Hollenstein, D.M.; Hudecz, O.; Schlögelhofer, P.; Mechtler, K. Comprehensive cross-linking mass spectrometry reveals parallel orientation and flexible conformations of plant HOP2-MND1. J. Proteome Res. 2015, 14, 5048–5062. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Reischl, R.; Walzthoeni, T.; Herzog, F.; Bohn, S.; Förster, F.; Aebersold, R. Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteom. 2012, 11, M111.014126. [Google Scholar] [CrossRef] [PubMed]
- Herzog, F.; Kahraman, A.; Boehringer, D.; Mak, R.; Bracher, A.; Walzthoeni, T.; Leitner, A.; Beck, M.; Hartl, F.U.; Ban, N.; et al. Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 2012, 337, 1348–1352. [Google Scholar] [CrossRef]
- Banerjee, S.; Mazumdar, S. Electrospray ionization mass spectrometry: A technique to access the information beyond the molecular weight of the analyte. Int. J. Anal. Chem. 2012, 282574. [Google Scholar] [CrossRef] [PubMed]
- Hermans, J.; Ongay, S.; Markov, V.; Bischoff, R. Physicochemical Parameters Affecting the Electrospray Ionization Efficiency of Amino Acids after Acylation. Anal. Chem. 2017, 89, 9159–9166. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Boyes, B.; Fields, T.; Kopkin, R.; Orlando, R. Optimization of data-dependent acquisition parameters for coupling high-speed separations with LC-MS/MS for protein identifications. J. Biomol. Tech. 2013, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-M.; Zhang, J.-Y.; Liu, H.; Sun, H.-C.; Zhu, Y.-P.; Xie, H.-W. Advance of peptide detectability prediction on mass spectrometry platform in proteomics. Chin. J. Anal. Chem. 2010, 38, 286–292. [Google Scholar] [CrossRef]
- Siggers, T.; Gordân, R. Protein-DNA binding: Complexities and multi-protein codes. Nucleic Acids Res. 2014, 42, 2099–2111. [Google Scholar] [CrossRef]
- Stützer, A.; Welp, L.M.; Raabe, M.; Sachsenberg, T.; Kappert, C.; Wulf, A.; Lau, A.M.; David, S.S.; Chernev, A.; Kramer, K.; et al. Analysis of protein-DNA interactions in chromatin by UV induced cross-linking and mass spectrometry. Nat. Commun. 2020, 11, 5250. [Google Scholar] [CrossRef]
- Kramer, K.; Sachsenberg, T.; Beckmann, B.M.; Qamar, S.; Boon, K.L.; Hentze, M.W.; Kohlbacher, O.; Urlaub, H. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 2014, 11, 1064–1070. [Google Scholar] [CrossRef]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The human rna-binding proteome and its dynamics during translational arrest. Cell 2019, 176, 391–403.e19. [Google Scholar] [CrossRef]
- Ide, H.; Shoulkamy, M.I.; Nakano, T.; Miyamoto-Matsubara, M.; Salem, A.M. Repair and biochemical effects of DNA-protein crosslinks. Mutat. Res. 2011, 711, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Angelov, D.; Stefanovsky, V.Y.; Dimitrov, S.I.; Russanova, V.R.; Keskinova, E.; Pashev, I.G. Protein-DNA crosslinking in reconstituted nucleohistone, nuclei and whole cells by picosecond UV laser irradiation. Nucleic Acids Res. 1988, 16, 4525–4538. [Google Scholar] [CrossRef] [PubMed]
- Stützer, A.; Liokatis, S.; Kiesel, A.; Schwarzer, D.; Sprangers, R.; Söding, J.; Selenko, P.; Fischle, W. Modulations of DNA contacts by linker histones and post-translational modifications determine the mobility and modifiability of nucleosomal H3 tails. Mol. Cell 2016, 61, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Hrle, A.; Kramer, K.; Sachsenberg, T.; Staals, R.H.; Randau, L.; Marchfelder, A.; Van der Oost, J.; Kohlbacher, O.; Conti, E.; et al. Analysis of protein-RNA interactions in CRISPR proteins and effector complexes by UV-induced cross-linking and mass spectrometry. Methods 2015, 89, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Veit, J.; Sachsenberg, T.; Chernev, A.; Aicheler, F.; Urlaub, H.; Kohlbacher, O. LFQProfiler and RNP(xl): Open-source tools for label-free quantification and protein-rna cross-linking integrated into proteome discoverer. J. Proteome Res. 2016, 15, 3441–3448. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Wills, K.L.; Furlan, C.; Rappsilber, J. Chromatin enrichment for proteomics. Nat. Protoc. 2014, 9, 2090–2099. [Google Scholar] [CrossRef]
- Ramanathan, M.; Porter, D.F.; Khavari, P.A. Methods to study RNA-protein interactions. Nat. Methods 2019, 16, 225–234. [Google Scholar] [CrossRef]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef]
- Sutherland, B.W.; Toews, J.; Kast, J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass Spectrom. 2008, 43, 699–715. [Google Scholar] [CrossRef]
- Chu, C.; Zhang, Q.C.; Da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, J.; Yi, C. Genome-wide mapping of cellular protein-RNA interactions enabled by chemical crosslinking. Genom. Proteom. Bioinform. 2014, 12, 72–78. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Guttman, M. RAP-MS: A method to identify proteins that interact directly with a specific rna molecule in cells. Methods Mol. Biol. 2018, 1649, 473–488. [Google Scholar] [CrossRef]
- Zeng, F.; Peritz, T.; Kannanayakal, T.J.; Kilk, K.; Eiríksdóttir, E.; Langel, U.; Eberwine, J. A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat. Protoc. 2006, 1, 920–927. [Google Scholar] [CrossRef]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteom. 2011, 10, M110.007385. [Google Scholar] [CrossRef]
- Matia-González, A.M.; Iadevaia, V.; Gerber, A.P. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 2017, 118–119, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Reim, A.; Ackermann, R.; Font-Mateu, J.; Kammel, R.; Beato, M.; Nolte, S.; Mann, M.; Russmann, C.; Wierer, M. Atomic-resolution mapping of transcription factor-DNA interactions by femtosecond laser crosslinking and mass spectrometry. Nat. Commun. 2020, 11, 3019. [Google Scholar] [CrossRef]
- Urdaneta, E.C.; Vieira-Vieira, C.H.; Hick, T.; Wessels, H.H.; Figini, D.; Moschall, R.; Medenbach, J.; Ohler, U.; Granneman, S.; Selbach, M.; et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Butter, F.; Scheibe, M.; Mörl, M.; Mann, M. Unbiased RNA-protein interaction screen by quantitative proteomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10626–10631. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.C.; Aplin, R.T. A mixed photoproduct of uracil and cysteine (5-S-cysteine-6-hydrouracil). A possible model for the in vivo cross-linking of deoxyribonucleic acid and protein by ultraviolet light. Biochemistry 1966, 5, 2125–2130. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Kim, B.; Kim, V.N. fCLIP-seq for transcriptomic footprinting of dsRNA-binding proteins: Lessons from DROSHA. Methods 2019, 152, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.R.; Wilkie, A.M.; Clemens, M.J.; Smith, C.W. Detection of double-stranded RNA-protein interactions by methylene blue-mediated photo-crosslinking. RNA 1996, 2, 611–621. [Google Scholar]
- Zaman, U.; Richter, F.M.; Hofele, R.; Kramer, K.; Sachsenberg, T.; Kohlbacher, O.; Lenz, C.; Urlaub, H. Dithiothreitol (DTT) acts as a specific, UV-inducible cross-linker in elucidation of protein-rna interactions. Mol. Cell. Proteom. 2015, 14, 3196–3210. [Google Scholar] [CrossRef]
- Wower, I.; Wower, J.; Meinke, M.; Brimacombe, R. The use of 2-iminothiolane as an RNA-protein cross-linking agent in Escherichia coli ribosomes, and the localisation on 23S RNA of sites cross-linked to proteins L4, L6, L21, L23, L27 and L29. Nucleic Acids Res. 1981, 9, 4285–4302. [Google Scholar] [CrossRef]
- Bäumert, H.G.; Sköld, S.E.; Kurland, C.G. RNA-protein neighbourhoods of the ribosome obtained by crosslinking. Eur. J. Biochem. 1978, 89, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F.; Chaires, J.B. Functional modification of 16S ribosomal RNA by kethoxal. Proc. Natl. Acad. Sci. USA 1972, 69, 3115–3118. [Google Scholar] [CrossRef]
- Zhang, Q.; Crosland, E.; Fabris, D. Nested Arg-specific bifunctional crosslinkers for MS-based structural analysis of proteins and protein assemblies. Anal. Chim. Acta 2008, 627, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, E.T.; Kellersberger, K.A.; Crosland, E.; Fabris, D. Toward building a database of bifunctional probes for the MS3D investigation of nucleic acids structures. J. Am. Soc. Mass. Spectrom. 2006, 17, 1570–1581. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 19, 1129–1143. [Google Scholar] [CrossRef]
- Padariya, M.; Kalathiya, U.; Houston, D.R.; Alfaro, J.A. Recognition Dynamics of Cancer Mutations on the ERp57-Tapasin Interface. Cancers 2020, 12, 737. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Pawlicka, K.; Verma, C.S.; Houston, D.; Hupp, T.R.; Alfaro, J.A. Insights into the Effects of Cancer Associated Mutations at the UPF2 and ATP-Binding Sites of NMD Master Regulator: UPF1. Int. J. Mol. Sci. 2019, 20, 5644. [Google Scholar] [CrossRef]
- Kapla, J.; Engström, O.; Stevensson, B.; Wohlert, J.; Widmalm, G.; Maliniak, A. Molecular dynamics simulations and NMR spectroscopy studies of trehalose-lipid bilayer systems. Phys. Chem. Chem. Phys. 2015, 17, 22438–22447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bouguet-Bonnet, S.; Buck, M. Combining NMR and molecular dynamics studies for insights into the allostery of small GTPase-protein interactions. Methods Mol. Biol. 2012, 796, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Brodie, N.I.; Popov, K.I.; Petrotchenko, E.V.; Dokholyan, N.V.; Borchers, C.H. Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci. Adv. 2017, 7, e1700479. [Google Scholar] [CrossRef] [PubMed]
- Shirvanyants, D.; Ding, F.; Tsao, D.; Ramachandran, S.; Dokholyan, N.V. Discrete molecular dynamics: An efficient and versatile simulation method for fine protein characterization. J. Phys. Chem. B 2012, 116, 8375–8382. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Belsom, A.; Schneider, M.; Fischer, L.; Brock, O.; Rappsilber, J. Serum albumin domain structures in human blood serum by mass spectrometry and computational biology. Mol. Cell. Proteom. 2016, 15, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, W.; Ferkinghoff-Borg, J.; Lindorff-Larsen, K. Combining experiments and simulations using the maximum entropy principle. PLoS Comput. Biol. 2014, 10, e1003406. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tools | Chemical Cross-Linkers Supported | Website |
|---|---|---|
| MeroX (StavroX included) [106] | BS2G, BS3/DSS, BS3/DSS-D0/D12, CDI, DC4, DSAU, DSBU, DSSO, DST, EDC, Formaldehyde(12), Formaldehyde(24), SDA | http://www.stavrox.com/Download_MeroX_Win.htm (accessed on 23 February 2021) |
| Spectrum Identification Machine for Cross-Linked Peptides (SIM-XL) [107] | DSS, DSG, DSSeb, DSS/DSG/DSSeb (with reporter ions only), XPlex C6N2, XPlex C3N2, XPlex C6Ac2, XPlex C3Ac2, Disulphide, zero-length | http://patternlabforproteomics.org/sim-xl/ (accessed on 23 February 2021) |
| Xilmass [108] | DSS (d0/d12), BS3(d0/d4), EDC and GA | http://compomics.github.io/projects/xilmass.html (accessed on 23 February 2021) |
| xQuest/xProphet [109] | BS3, DSS etc. | http://proteomics.ethz.ch/cgi-bin/xquest2_cgi/download.cgi (accessed on 23 February 2021) |
| XiSEARCH [110] | BS2G, SDA, BS3, DSSO, EDC, NonCovalent, Linear Search | https://www.rappsilberlab.org/software/xisearch/ (accessed on 23 February 2021) |
| Kojak [111] | BS3, DSS etc. | http://www.kojak-ms.org/param/cross_link.html (accessed on 23 February 2021) |
| pLink 2 [112] | BS2G, BS2G_heavy, BS3_heavy, DSS, EDC-DE | http://pfind.ict.ac.cn/software/pLink/ (accessed on 23 February 2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalathiya, U.; Padariya, M.; Faktor, J.; Coyaud, E.; Alfaro, J.A.; Fahraeus, R.; Hupp, T.R.; Goodlett, D.R. Interfaces with Structure Dynamics of the Workhorses from Cells Revealed through Cross-Linking Mass Spectrometry (CLMS). Biomolecules 2021, 11, 382. https://doi.org/10.3390/biom11030382
Kalathiya U, Padariya M, Faktor J, Coyaud E, Alfaro JA, Fahraeus R, Hupp TR, Goodlett DR. Interfaces with Structure Dynamics of the Workhorses from Cells Revealed through Cross-Linking Mass Spectrometry (CLMS). Biomolecules. 2021; 11(3):382. https://doi.org/10.3390/biom11030382
Chicago/Turabian StyleKalathiya, Umesh, Monikaben Padariya, Jakub Faktor, Etienne Coyaud, Javier A. Alfaro, Robin Fahraeus, Ted R. Hupp, and David R. Goodlett. 2021. "Interfaces with Structure Dynamics of the Workhorses from Cells Revealed through Cross-Linking Mass Spectrometry (CLMS)" Biomolecules 11, no. 3: 382. https://doi.org/10.3390/biom11030382
APA StyleKalathiya, U., Padariya, M., Faktor, J., Coyaud, E., Alfaro, J. A., Fahraeus, R., Hupp, T. R., & Goodlett, D. R. (2021). Interfaces with Structure Dynamics of the Workhorses from Cells Revealed through Cross-Linking Mass Spectrometry (CLMS). Biomolecules, 11(3), 382. https://doi.org/10.3390/biom11030382