
Molecular and Biochemical Pathways of Catalpol in Alleviating Diabetes Mellitus and Its Complications

 ,
,  ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
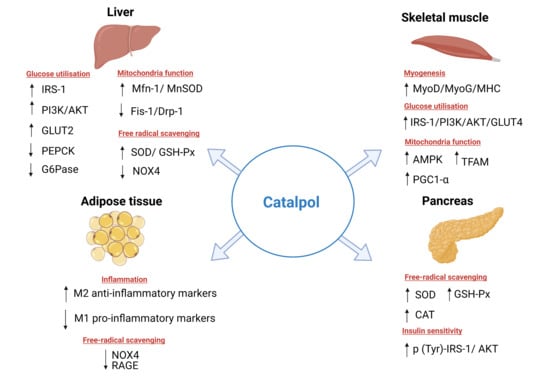
2. Effects of Catalpol in Diabetes Mellitus
2.1. Effects of Catalpol in the Liver
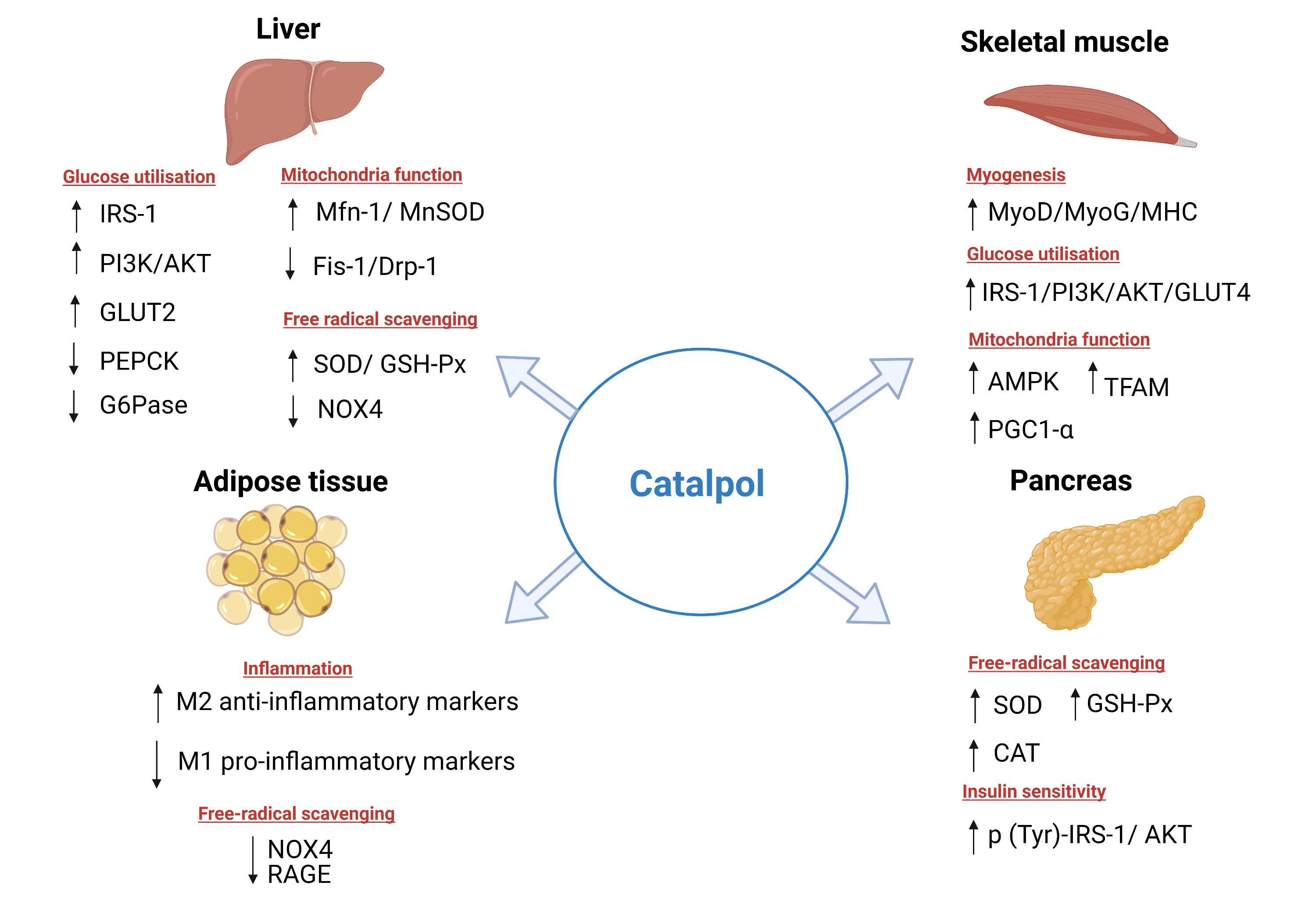
2.1.1. Effect on Glucose Metabolism in the Liver
2.1.2. Effect on Oxidative Stress in the Liver
2.1.3. Effect on Lipid Metabolism in the Liver
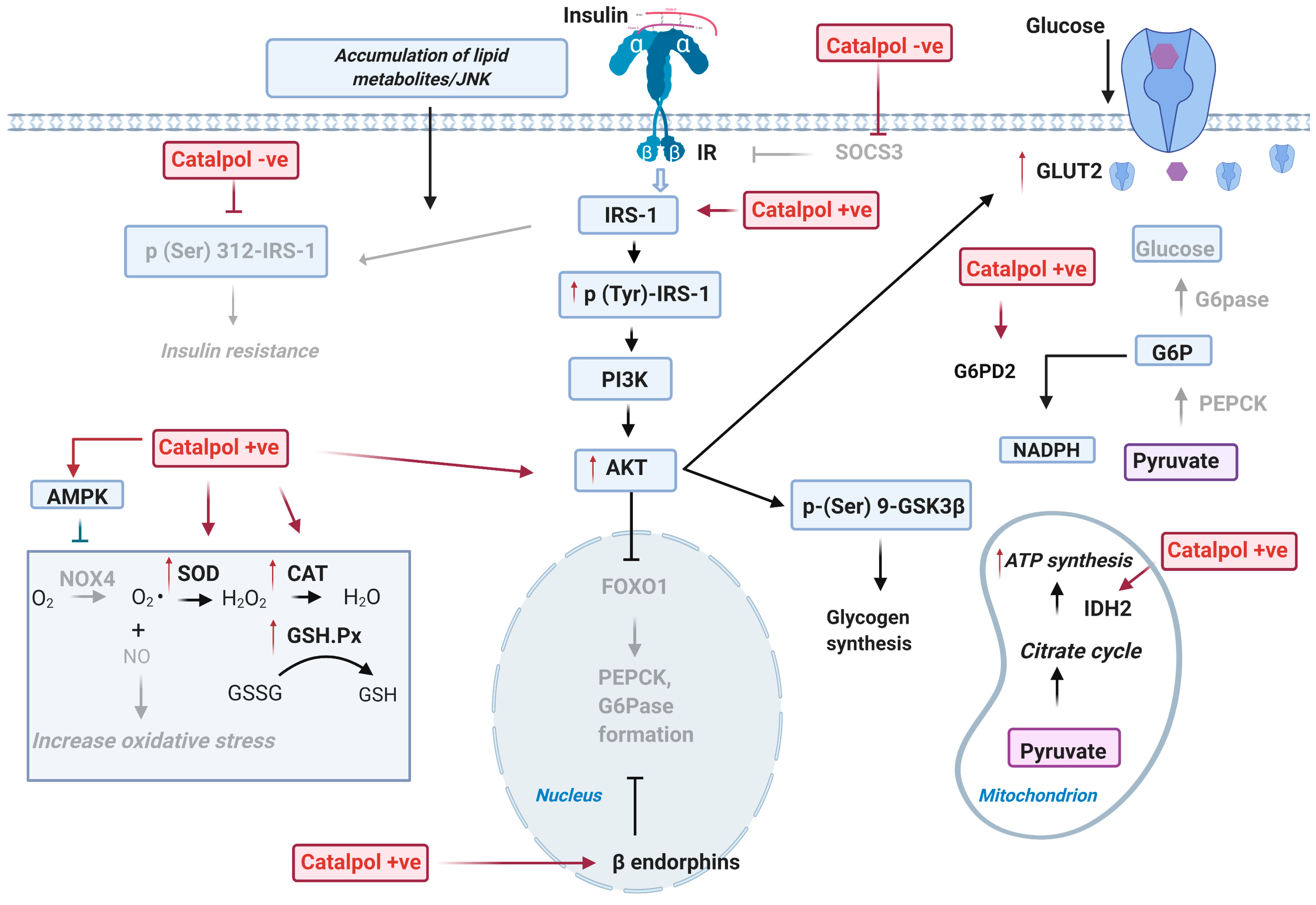
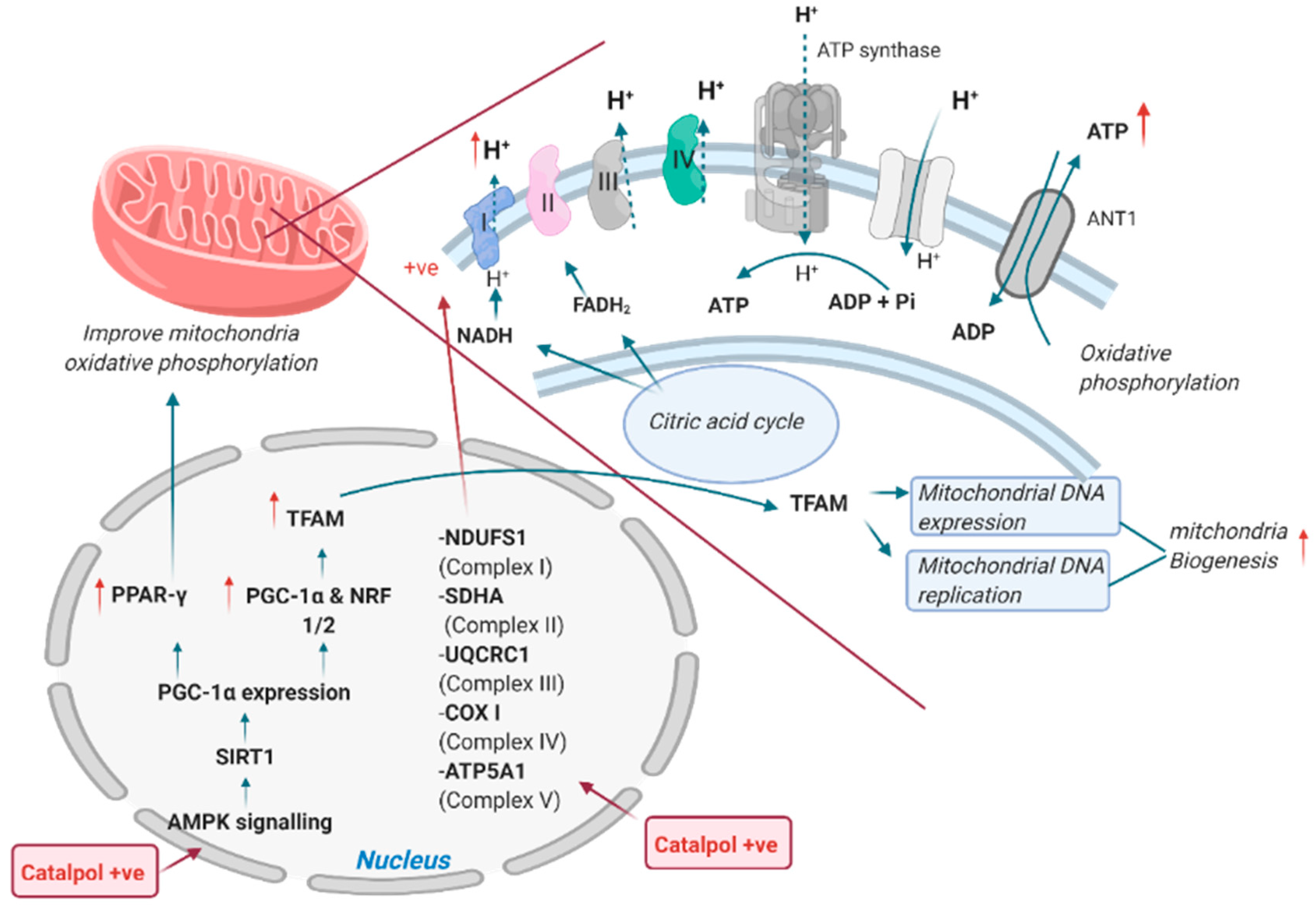
2.2. Effects of Catalpol in the Skeletal Muscle
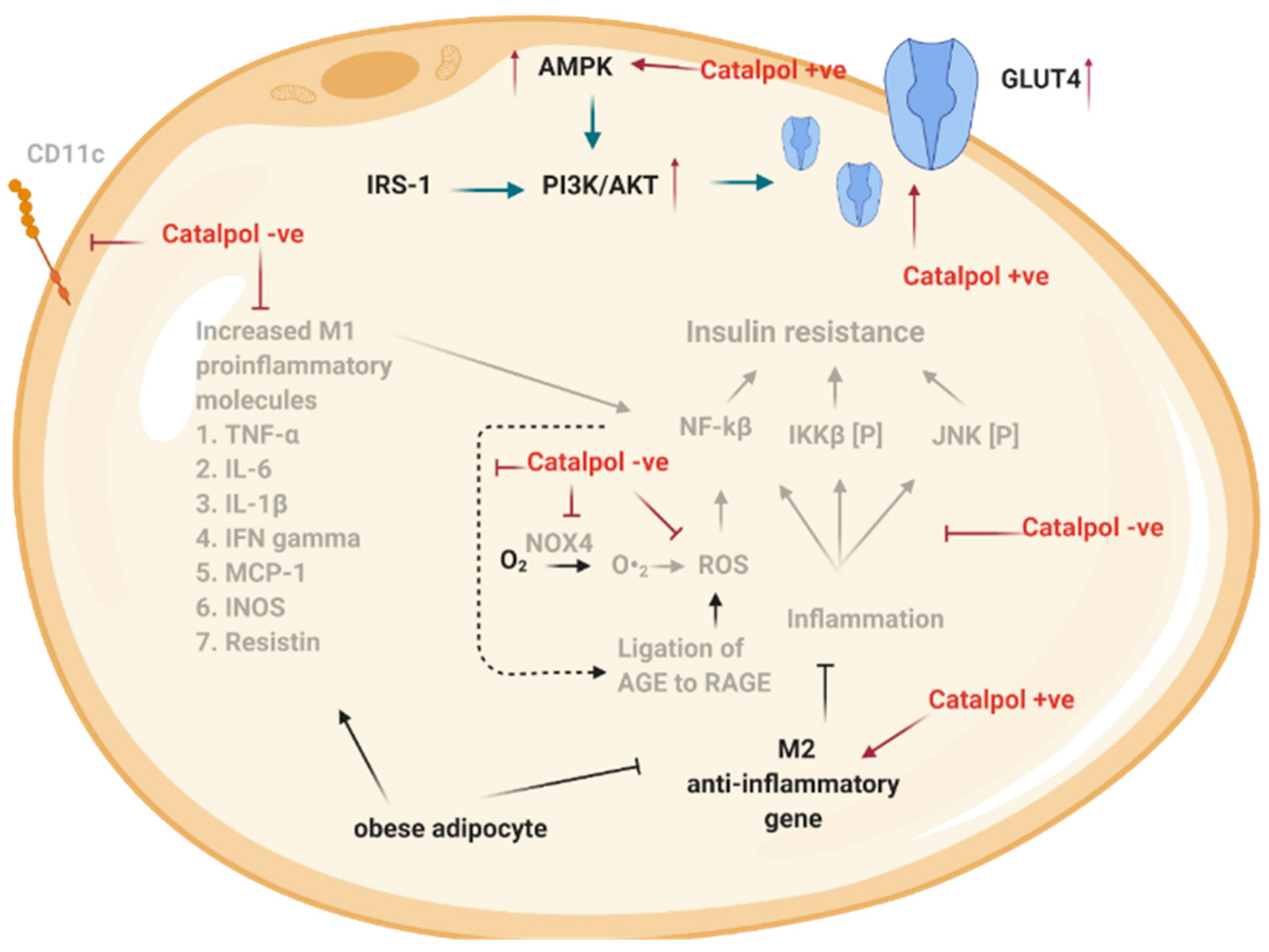
2.3. Effects of Catalpol in the Adipose Tissue
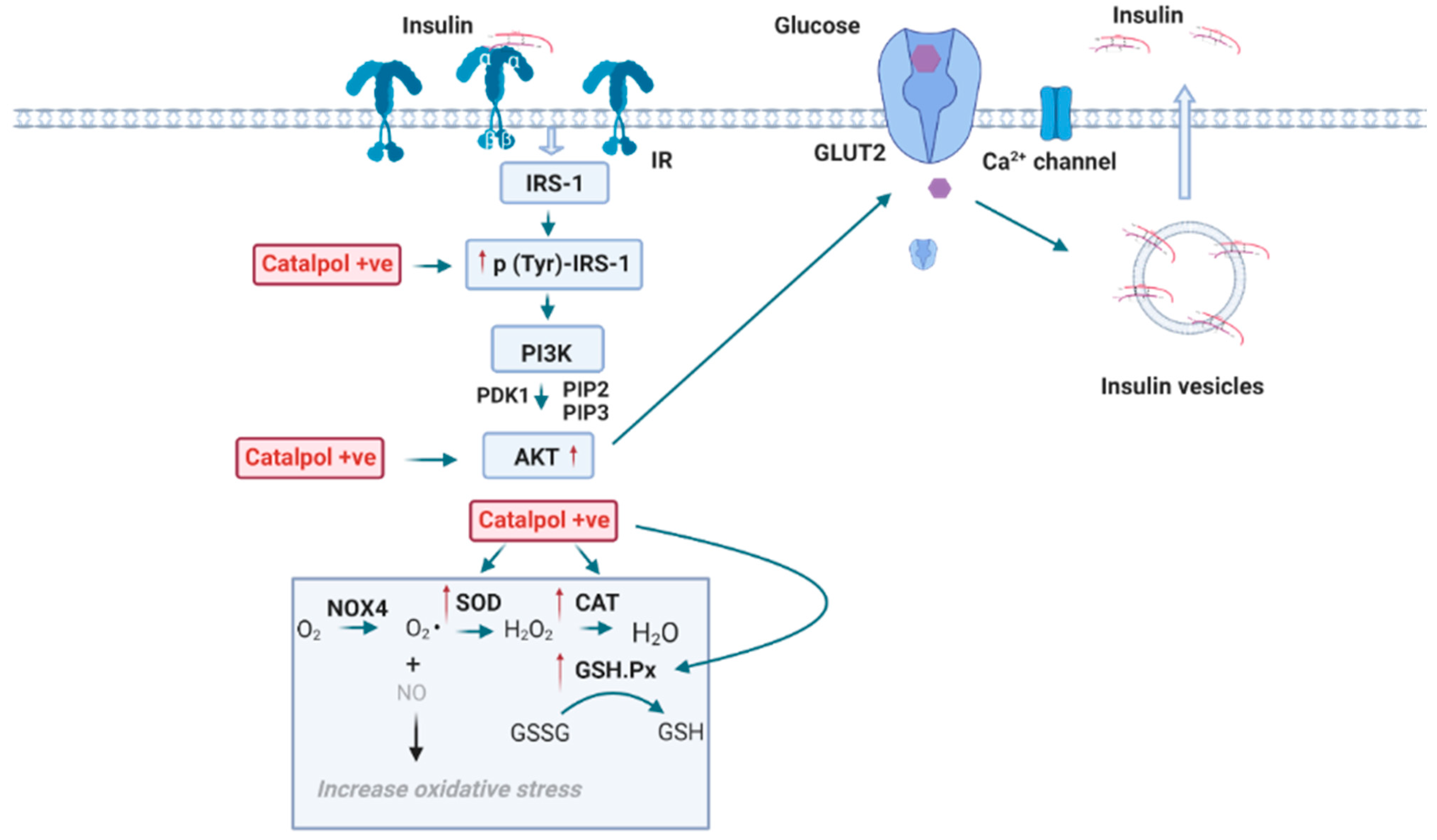
2.4. Effects of Catalpol in the Pancreas
3. Effects of Catalpol in Diabetes Complications
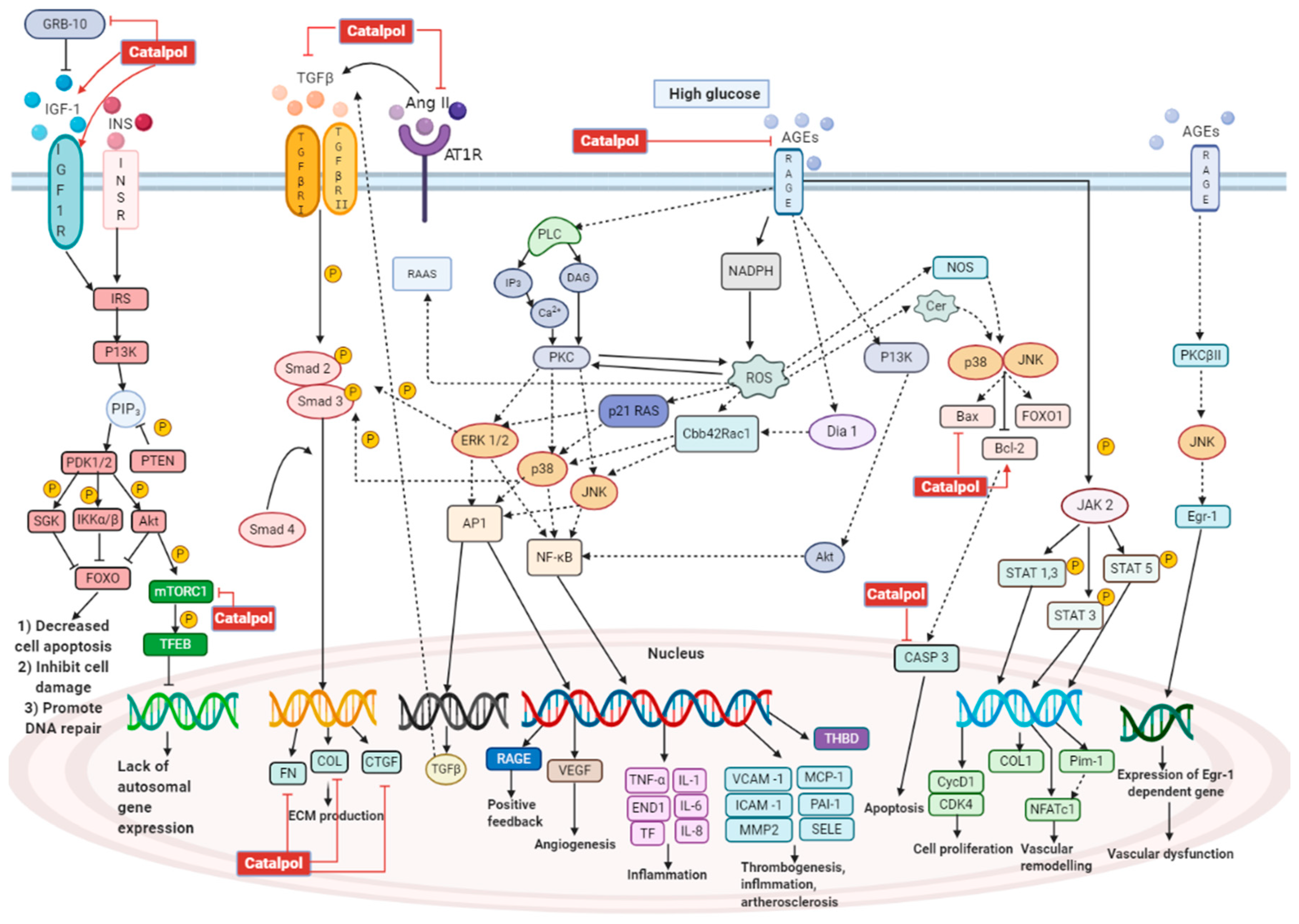
3.1. Effects in Diabetic Nephropathy
3.2. Effects in Diabetic Encephalopathy
3.3. Effects in Diabetic Cardiovascular Conditions
3.3.1. Diabetic Cardiomyopathy
3.3.2. Myocardial Infarction
3.3.3. Diabetic Atherosclerosis and Arrhythmias
3.4. Effects in Other Diabetes Related Complications
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duke, L.; Fereira de Moura, A.; Silvia Gorban de Lapertosa, S.; Lucy Hammond, L.; Jacobs, E.; Abha Kaundal, A. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Cersosimo, E.; Triplitt, C.; Solis-Herrera, C.; Mandarino, L.J.; DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Eds.; MDText.com, Inc.: South Dartmouth (MA), Bristol, 2000. [Google Scholar]
- Nathan, D.M. Long-Term Complications of Diabetes Mellitus. N. Engl. J. Med. 1993, 328, 1676–1685. [Google Scholar] [CrossRef]
- Furman, B.L.; Candasamy, M.; Bhattamisra, S.K.; Veettil, S.K. Reduction of blood glucose by plant extracts and their use in the treatment of diabetes mellitus; discrepancies in effectiveness between animal and human studies. J. Ethnopharmacol. 2020, 247, 112264. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-F.; Yang, Y.-Q.; Xie, D.-F.; Zhu, L.-F.; Zhang, Z.-G.; Lin, W.-X. Identification of Autotoxic Compounds in Fibrous Roots of Rehmannia (Rehmannia glutinosa Libosch.). PLoS ONE 2012, 7, e28806. [Google Scholar] [CrossRef]
- Wang, Z.-J.; Wo, S.-K.; Wang, L.; Lau, C.B.; Lee, V.H.; Chow, M.S.; Zuo, Z. Simultaneous quantification of active components in the herbs and products of Si-Wu-Tang by high performance liquid chromatography–mass spectrometry. J. Pharm. Biomed. Anal. 2009, 50, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.; Liu, K.; Sun, H. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol. Res. 2018, 130, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, L.; Li, X.; Jiang, Z.; Sun, L.; Zhao, G.; Zhou, G.; Zhang, H.; Shang, J.; Wang, T. Mitochondrial fusion/fission process involved in the improvement of ca-talpol on high glucose-induced hepatic mitochondrial dysfunction. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Huang, X.; Hassan, H.M.; Wang, L.; Li, S.; Jiang, Z.; Zhang, L.; Wang, T. Hypoglycaemic effect of catalpol in a mouse model of high-fat di-et-induced pre-diabetes. Appl. Physiol. Nutr. Metab. 2020, 45, 1127–1137. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.; Jiang, Z.; Sun, L.; Ji, J.; Miao, J.; Zhang, X.; Huang, S.; Wang, T.; Zhang, L. Hypoglycemic effect of catalpol on high-fat diet/streptozotocin-induced diabetic mice by increasing skeletal muscle mitochondrial biogenesis. Acta Biochim. et Biophys. Sin. 2014, 46, 738–748. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, G.; Ma, S.; Li, F.; Yuan, M.; Xu, H.; Huang, K. Catalpol ameliorates high-fat diet-induced insulin resistance and adipose tis-sue inflammation by suppressing the JNK and NF-κB pathways. Biochem. Biophys. Res. Commun. 2015, 467, 853–858. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Y.; Liu, Z.; Wang, J.; Wan, D.; Huifeng, Z.; Yang, X.; Wang, T. Antidiabetic and antioxidant effects of catalpol extracted from Rehmannia glutinosa (Di Huang) on rat diabetes induced by streptozotocin and high-fat, high-sugar feed. Chin. Med. 2016, 11, 1–10. [Google Scholar] [CrossRef]
- Bao, Q.; Shen, X.; Qian, L.; Gong, C.; Nie, M.; Dong, Y. Anti-diabetic activities of catalpol in db/db mice. Korean J. Physiol. Pharmacol. 2016, 20, 153–160. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.-R.; Hou, Y.-B.; Jing, X.-L.; Song, X.-Y.; Shen, X.-P. Global gene expression analysis in liver of db/db mice treated with catalpol. Chin. J. Nat. Med. 2018, 16, 590–598. [Google Scholar] [CrossRef]
- Shieh, J.-P.; Cheng, K.-C.; Chung, H.-H.; Kerh, Y.-F.; Yeh, C.-H.; Cheng, J.-T. Plasma Glucose Lowering Mechanisms of Catalpol, an Active Principle from Roots of Rehmannia glutinosa, in Streptozotocin-Induced Diabetic Rats. J. Agric. Food Chem. 2011, 59, 3747–3753. [Google Scholar] [CrossRef]
- Wang, C.-F.; Li, D.-Q.; Xue, H.-Y.; Hu, B. Oral supplementation of catalpol ameliorates diabetic encephalopathy in rats. Brain Res. 2010, 1307, 158–165. [Google Scholar] [CrossRef]
- Melmed, S.; Polonsky, K.S.; Larsen, P.R.; Kronenberg, H.M. Williams Textbook of Endocrinology, 12th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2011. [Google Scholar]
- Trembath, D. Molecular Testing for Glioblastoma. In Diagnostic Molecular Pathology; Coleman, W.B., Tsongalis, G.J., Eds.; Academic Press: London, UK, 2017; pp. 339–347. [Google Scholar]
- Kim, Y.; Kim, E.-Y.; Seo, Y.-M.; Yoon, T.K.; Lee, W.-S.; Lee, K.-A. Function of the pentose phosphate pathway and its key enzyme, transketolase, in the regulation of the meiotic cell cycle in oocytes. Clin. Exp. Reprod. Med. 2012, 39, 58–67. [Google Scholar] [CrossRef]
- Torisu, T.; Sato, N.; Yoshiga, D.; Kobayashi, T.; Yoshioka, T.; Mori, H.; Iida, M.; Yoshimura, A. The dual function of hepatic SOCS3 in insulin re-sistance in vivo. Genes Cells. 2007, 12, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Zhu, J.S.; Weldon, H.; Maianu, L.; Garvey, W.T. Glucosamine induces insulin resistance in vivo by affecting GLUT 4 translocation in skeletal muscle. Implications for glucose toxicity. J. Clin. Investig. 1995, 96, 2792–2801. [Google Scholar] [CrossRef]
- Sprouse-Blum, A.S.; Smith, G.; Sugai, D.; Parsa, F.D. Understanding Endorphins and Their Importance in Pain Management. Hawaii Med. J. 2010, 69, 70–71. [Google Scholar]
- Ramnanan, C.J.; Edgerton, D.S. The role of insulin in the regulation of PEPCK and gluconeogenesis in vivo. US Endocrinol. 2010, 5, 34–39. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.J.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef]
- Smriti, K.; Pai, K.M.; Ravindranath, V.; Pentapati, K.C. Role of salivary malondialdehyde in assessment of oxidative stress among diabetics. J. Oral Biol. Craniofacial Res. 2016, 6, 42–45. [Google Scholar] [CrossRef]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef]
- Candas, D.; Li, J.J. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox Signal. 2014, 20, 1599–1617. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Relationship between mitofusin 2 and cancer. Adv. Protein Chem. Struct. Biol. 2019, 116, 209–236. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2017, 9, 6128–6143. [Google Scholar] [CrossRef]
- Lipogenesis in diabetes and obesity. Nutr. Rev. 2009, 49, 255–257. [CrossRef]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl coA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: A bedside to bench investigation. Cell Metab. 2017, 26, 394–406. [Google Scholar] [CrossRef]
- Erion, D.M.; Park, H.-J.; Lee, H.-Y. The role of lipids in the pathogenesis and treatment of type 2 diabetes and associated co-morbidities. BMB Rep. 2016, 49, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver Peroxisome Proliferator-activated Receptor γ Contributes to Hepatic Steatosis, Triglyceride Clearance, and Regulation of Body Fat Mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef]
- Yap, K.; Gan, S.; Candasamy, M.; Md, S.; Majeed, A.A.; Bhattamisra, S. Effect of catalpol on liver glucose homeostasis in high fat diet/low dose streptozotocin-induced type 2 diabetes mellitus. Br. J. Pharmacol. 2019, 176, 3051–3052. [Google Scholar]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of Muscle Glycogen Synthesis in Normal Subjects and Subjects with Non-Insulin-Dependent Diabetes by13C Nuclear Magnetic Resonance Spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef]
- Xu, D.; Wang, L.; Jiang, Z.; Zhao, G.; Hassan, H.M.; Sun, L.; Fan, S.; Zhou, Z.; Zhang, L.; Wang, T. A new hypoglycemic mechanism of catalpol revealed by enhanc-ing MyoD/MyoG-mediated myogenesis. Life Sci. 2018, 209, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Eftimie, R.; Brenner, H.R.; Buonanno, A. Myogenin and MyoD join a family of skeletal muscle genes regulated by electrical activity. Proc. Natl. Acad. Sci. USA 1991, 88, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Edwards, K.A.; Bernstein, S.I. Myosin heavy chain isoforms regulate muscle function but not myofibril assembly. EMBO J. 1996, 15, 4454–4459. [Google Scholar] [CrossRef]
- Bonnieu, A.; Carnac, G.; Vernus, B. Myostatin in the Pathophysiology of Skeletal Muscle. Curr. Genom. 2007, 8, 415–422. [Google Scholar] [CrossRef]
- Cartee, G.D. Roles of TBC1D1 and TBC1D4 in insulin- and exercise-stimulated glucose transport of skeletal muscle. Diabetol. 2015, 58, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. C2C12 cell model: Its role in understanding of insulin resistance at the molecular level and pharmaceutical development at the preclinical stage. J. Pharm. Pharmacol. 2020, 72, 1667–1693. [Google Scholar] [CrossRef]
- Szendroedi, J.; Schmid, A.I.; Chmelík, M.; Tóth, C.; Brehm, A.; Krššák, M.; Nowotny, P.; Wolzt, M.; Waldhäusl, W.; Roden, M. Muscle Mitochondrial ATP Synthesis and Glucose Transport/Phosphorylation in Type 2 Diabetes. PLoS Med. 2007, 4, e154. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.Q.; Li, C.J.; Jiang, Z.Z.; Wang, L.; Huang, H.F.; Li, Z.J.; Sun, L.X.; Fan, S.S.; Zhang, L.Y.; Wang, T. The hypoglycemic mechanism of catalpol involves increased AMPK-mediated mitochondrial biogenesis. Acta. Pharmacol. Sin. 2020, 41, 791–799. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Section 22.5 Acetyl coenzyme a carboxylase plays a key role in controlling fatty acid metabolism. In Biochemistry, 5th ed.; Berg, J.M., Tymoczko, J.L., Stryer, L., Eds.; W H Freeman: New York, NY, USA, 2002. [Google Scholar]
- Yap, K.H.; Yee, G.S.; Candasamy, M.; Tan, S.C.; Md, S.; Abdul Majeed, A.B.; Bhattamisra, S.K. Catalpol ameliorates insulin sensitivity and mitochondrial respiration in skeletal muscle of type-2 diabetic mice through insulin signaling pathway and AMPK/SIRT1/PGC-1α/PPAR-γ activation. Biomolecules 2020, 10, 1360. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free. Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.H.; Mathews, S.T.; Camp, H.S.; Ding, L.; Leff, T. Selective activation of PPARγ in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am. J. Physiol. Metab. 2010, 298, E28–E37. [Google Scholar] [CrossRef]
- Hajer, G.R.; Van Haeften, T.W.; Visseren, F.L.J. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur. Hear. J. 2008, 29, 2959–2971. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1 (LPS+) vs. classi-cally and M2 (LPS–) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Satoh, N.; Shimatsu, A.; Himeno, A.; Sasaki, Y.; Yamakage, H.; Yamada, K.; Suganami, T.; Ogawa, Y. Unbalanced M1/M2 Phenotype of Peripheral Blood Monocytes in Obese Diabetic Patients: Effect of pioglitazone. Diabetes Care 2009, 33, e7. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central, role for JNK in obesity and insulin re-sistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef]
- Choi, H.-J.; Jang, H.-J.; Chung, T.-W.; Jeong, S.-I.; Cha, J.; Choi, J.-Y.; Han, C.W.; Jang, Y.-S.; Joo, M.; Jeong, H.-S.; et al. Catalpol suppresses advanced glycation end-products-induced inflammatory responses through inhibition of reactive oxygen species in human monocytic THP-1 cells. Fitoter. 2013, 86, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; RGilbert, E.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Sabra, G.; Dubiel, E.A.; Kuehn, C.; Khalfaoui, T.; Beaulieu, J.-F.; Vermette, P. INS-1 cell glucose-stimulated insulin secretion is re-duced by the downregulation of the 67 kDa laminin receptor. J. Tissue Eng. Regen. Med. 2015, 9, 1376–1385. [Google Scholar] [CrossRef]
- Elhassan, S.A.M.; Candasamy, M.; Ching, T.S.; Heng, Y.K.; Bhattamisra, S.K. Effect of madecassoside and catalpol in amelioration of insulin sensitivity in pancreatic (INS-1E) β-cell line. Nat. Prod. Res. 2019, 30, 1–5. [Google Scholar] [CrossRef]
- Haller, H.; Ji, L.; Stahl, K.; Bertram, A.; Menne, J. Molecular Mechanisms and Treatment Strategies in Diabetic Nephropathy: New Avenues for Calcium Dobesilate—Free Radical Scavenger and Growth Factor Inhibition. BioMed Res. Int. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Tan, A.L.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in Diabetic Nephropathy. Semin. Nephrol. 2007, 27, 130–143. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Padilla-Tejeda, D.E.; Carrillo-Ibarra, S.; Miranda-Díaz, A.G. Oxidative Stress, Apoptosis, and Mitochondrial Function in Diabetic Nephropathy. Int. J. Endocrinol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tessari, P. Nitric oxide in the normal kidney and in patients with diabetic nephropathy. J. Nephrol. 2014, 28, 257–268. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Xiang, L.; Chen, Z.; Lu, H.; Zhou, L.; Yang, L.; Ji, Y.; Liu, Y.; Sun, X.; Deng, Y.; et al. Catalpol alleviates renal damage by improving lipid metabolism in diabetic db/db mice. Am. J. Transl. Res. 2018, 10, 1750–1761. [Google Scholar] [PubMed]
- Yang, S.; Deng, H.; Zhang, Q.; Xie, J.; Zeng, H.; Jin, X.; Ling, Z.; Shan, Q.; Liu, M.; Ma, Y.; et al. Amelioration of diabetic mouse nephropathy by catalpol correlates with down-regulation of GRB10 expression and activation of insulin-like growth factor 1/insulin-like growth factor 1 re-ceptor signaling. PLoS ONE 2016, 17, e0151857. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, C.X. Effect of catalpol on diabetic nephropathy in rats. Phytomedicine 2013, 20, 1023–1029. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Jiang, M.; Fu, Y.; Zhu, Y.; Jiao, N.; Liu, L.; Du, Q.; Wu, H.; Xu, H.; et al. Loganin and catalpol exert cooperative ameliorating effects on podocyte apoptosis upon diabetic nephropathy by targeting AGEs-RAGE signaling. Life Sci. 2020, 252, 117653. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Shan, Z.; Mi, W.; Zhao, Y.; Li, M.; Wang, B.; Zheng, X.; Feng, W. Catalpol ameliorates podocyte injury by stabilizing cytoskeleton and en-hancing autophagy in diabetic nephropathy. Front. Pharmacol. 2019, 10, 1477. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, Y.; Lv, Z.; Shu, A.; Du, Q.; Wang, W.; Chen, Y.; Xu, H. Study on the inhibitive effect of Catalpol on diabetic nephropathy. Life Sci. 2020, 257, 118120. [Google Scholar] [CrossRef]
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Rüster, C.; Wolf, G. Angiotensin II as a Morphogenic Cytokine Stimulating Renal Fibrogenesis. J. Am. Soc. Nephrol. 2011, 22, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yoon, S.-O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate That Negatively Regulates Insulin Signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Schertzer, J.D. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: Im-plications for diabetes and cardiovascular disease. Immunol. Cell Biol. 2014, 92, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241. [Google Scholar]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Guo, G.; Morrison, D.J.; Licht, J.D.; Quaggin, S.E. WT1 Activates a Glomerular-Specific Enhancer Identified from the Human Nephrin Gene. J. Am. Soc. Nephrol. 2004, 15, 2851–2856. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- De La Monte, S.M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alz-heimer’s disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Bahniwal, M.; Little, J.P.; Klegeris, A. High Glucose Enhances Neurotoxicity and Inflammatory Cytokine Secretion by Stimulated Human Astrocytes. Curr. Alzheimer Res. 2017, 14, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.; Nunes, S.; Reis, F.; Pereira, F.C. Diabetic encephalopathy: The role of oxidative stress and inflammation in type 2 di-abetes. Int. J. Interferon Cytokine Mediat. Res. 2012, 4, 75–85. [Google Scholar]
- Zhou, H.; Liu, J.; Ren, L.; Liu, W.; Xing, Q.; Men, L.; Song, G.; Du, J. Relationship with spatial memory in diabetic rats and protein kinase Cγ, caveolin-1 in the hippocampus and neuroprotective effect of catalpol. Chin. Med. J. (Engl.) 2014, 127, 916–923. [Google Scholar]
- Wang, S.; Leem, J.; Fannon, M.; Kleschevnikova, N.; Head, B. Neuron-targeted caveolin-1 gene therapy preserves cognitive func-tion and synaptic plasticity in a mouse model of Alzheimer’s disease (AD). Alzheimer’s Dement. 2019, 15, 246–248. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Morato, M.; Reina-Couto, M.; Pinho, D.; Teixeira, A.A.; Sousa, T. Regulation of the Renin-Angiotensin-Aldosterone System by Reactive Oxygen Species. Renin-Angiotensin Syst.-Past Present Future 2017, 119–157. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of Cell Death in Heart Disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef]
- Zou, G.; Zhong, W.; Wu, F.; Wang, X.; Liu, L. Inhibition of lncRNA Neat1 by catalpol via suppressing transcriptional activity of NF-κB attenuates cardiomyocyte apoptosis. Cell Cycle 2019, 18, 3432–3441. [Google Scholar] [CrossRef]
- Zou, G.L.; Zhong, W.L.; Wu, F.; Wang, X.X.; Liu, L. Catalpol attenuates cardiomyocyte apoptosis in diabetic cardiomyopathy via Neat1/miR-140–5p/HDAC4 axis. Biochimie 2019, 165, 90–99. [Google Scholar] [CrossRef]
- Leon, B.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Tabas, I. Insulin Resistance, Hyperglycemia, and Atherosclerosis. Cell Metabol. 2011, 14, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Stein, B.; Ambrose, J.A.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. Atherosclerotic plaque rupture and thrombosis. Evolving concepts. Circulation 1990, 82, II47–II59. [Google Scholar] [PubMed]
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial infarction-from atherosclerosis to thrombosis: Uncovering new diag-nostic and therapeutic approaches. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e176–e185. [Google Scholar] [CrossRef]
- Bi, F.; Xu, Y.; Sun, Q. Catalpol pretreatment attenuates cardiac dysfunction following myocardial infarction in rats. Anatol. J. Cardiol. 2018, 19, 296–302. [Google Scholar] [PubMed]
- Huang, C.; Cui, Y.; Ji, L.; Zhang, W.; Li, R.; Ma, L.; Xing, W.; Zhou, H.; Chen, B.; Yu, J.; et al. Catalpol decreases peroxynitrite formation and consequently exerts cardi-oprotective effects against ischemia/reperfusion insult. Pharm. Biol. 2013, 51, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lu, Y.; Yan, X.; Wu, X.; Kuai, M.; Sun, X.; Chen, Q.; Kong, X.; Liu, Z.; Tang, Y.; et al. Catalpol protects glucose-deprived rat embryonic cardiac cells by inducing mitophagy and modulating estrogen receptor. Biomed. Pharmacother. 2017, 89, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2008, 81, 457–464. [Google Scholar] [CrossRef]
- Bi, F.J.; Zhang, H.; Xu, Y.J.; Hu, J. Protective effect of catalpol on isoproterenol-induced myocardial injury in Wistar rats. African J. Biotechnol. 2012, 11, 9270–9275. [Google Scholar]
- Zeng, J.; Huang, F.; Tu, Y.; Wu, S.; Li, M.; Tong, X. Protective Effect of Catalpol on Myocardium in Rats with Isoprenaline-Induced Myocardial Infarcts via Angiogenesis through Endothelial Progenitor Cells and Notch1 Signaling Pathway. Pharmacol. Pharm. 2013, 4, 619–627. [Google Scholar] [CrossRef][Green Version]
- Badadani, M. Autophagy Mechanism, Regulation, Functions, and Disorders. ISRN Cell Biol. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Saito, T.; Sadoshima, J. Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Mythili, S.; Malathi, N. Diagnostic markers of acute myocardial infarction. Biomed. Rep. 2015, 3, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Lei, M.; Hu, W.; Han, S.; Wang, Q. A brief review: The therapeutic potential of bone marrow mesenchymal stem cells in myocardial infarction. Stem Cell Res. Ther. 2017, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.; Wang, W.E.; Zeng, C. How to Improve the Survival of Transplanted Mesenchymal Stem Cell in Ischemic Heart? Stem Cells Int. 2015, 2016, 1–14. [Google Scholar] [CrossRef]
- Ju, X.; Xue, D.; Wang, T.; Ge, B.; Zhang, Y.; Li, Z. Catalpol Promotes the Survival and VEGF Secretion of Bone Marrow-Derived Stem Cells and Their Role in Myocardial Repair After Myocardial Infarction in Rats. Cardiovasc. Toxicol. 2018, 18, 471–481. [Google Scholar] [CrossRef]
- Cochain, C.; Channon, K.M.; Silvestre, J.-S. Angiogenesis in the Infarcted Myocardium. Antioxidants Redox Signal. 2013, 18, 1100–1113. [Google Scholar] [CrossRef]
- Goldberg, I.J. Why does diabetes increase atherosclerosis? I don’t know! J. Clin. Investig. 2004, 114, 613–615. [Google Scholar] [CrossRef][Green Version]
- Barrett, E.J.; Liu, Z. The endothelial cell: An “early responder” in the development of insulin resistance. Rev. Endocr. Metab. Disord. 2013, 14, 21–27. [Google Scholar] [CrossRef]
- Aronson, D.; Rayfield, E.J. How hyperglycemia promotes atherosclerosis: Molecular mechanisms. Cardiovasc. Diabetol. 2002, 1, 1. [Google Scholar] [CrossRef]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Zheng, C.-Z.; Hao, X.-P.; Zhang, D.-J.; Mao, A.-W.; Yuan, P. Catalpol ameliorates diabetic atherosclerosis in diabetic rabbits. Am. J. Transl. Res. 2016, 8, 4278–4288. [Google Scholar]
- Lan, T.-H.; Huang, X.-Q.; Tan, H.-M. Vascular fibrosis in atherosclerosis. Cardiovasc. Pathol. 2013, 22, 401–407. [Google Scholar] [CrossRef]
- Agarwal, G.; Singh, S.K. Arrhythmias in type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2017, 21, 715–718. [Google Scholar] [CrossRef]
- Yang, L.; Feng, X.; Li, Y.; Zhang, S.; Ying, Y. Therapeutic efficacy of catalpol against apoptosis in cardiomyocytes derived from human induced pluripotent stem cells. AMB Express 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Wongdee, K. Osteoporosis in diabetes mellitus: Possible cellular and molecular mechanisms. World J. Diabetes 2011, 2, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Wongdee, K.; Charoenphandhu, N. Update on type 2 diabetes-related osteoporosis. World J. Diabetes 2015, 6, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Xu, H.Y.; Liu, M.M.; Cai, J.P.; Wang, L.; Hua, Z.; Wu, X.D.; Huo, W.L.; Lv, N.N. Catalpol promotes the proliferation and differentiation of osteo-blasts induced by high glucose by inhibiting kdm7a. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration–What Do We Know So Far. Front. Cell Dev. Biol. 2019, 6, 170. [Google Scholar] [CrossRef]
- Manolagas, S.C. Wnt signaling and osteoporosis. Maturitas 2014, 78, 233–237. [Google Scholar] [CrossRef]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef]
- Safi, S.Z.; Qvist, R.; Kumar, S.; Batumalaie, K.; Ismail, I.S.B. Molecular mechanisms of diabetic retinopathy, general preventive strategies, and novel therapeutic targets. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef]
- Catalani, E.; Cervia, D. Diabetic retinopathy: A matter of retinal ganglion cell homeostasis. Neural Regen. Res. 2020, 15, 1253–1254. [Google Scholar] [CrossRef]
- Shao, Y.; Zhang, Y.; Yu, Y.; Xu, T.T.; Wei, R.; Zhou, Q. Impact of catalpol on retinal ganglion cells in diabetic retinopathy. Int. J. Clin. Exp. Med. 2016, 9, 17274–17280. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattamisra, S.K.; Koh, H.M.; Lim, S.Y.; Choudhury, H.; Pandey, M. Molecular and Biochemical Pathways of Catalpol in Alleviating Diabetes Mellitus and Its Complications. Biomolecules 2021, 11, 323. https://doi.org/10.3390/biom11020323
Bhattamisra SK, Koh HM, Lim SY, Choudhury H, Pandey M. Molecular and Biochemical Pathways of Catalpol in Alleviating Diabetes Mellitus and Its Complications. Biomolecules. 2021; 11(2):323. https://doi.org/10.3390/biom11020323
Chicago/Turabian StyleBhattamisra, Subrat Kumar, Hui Min Koh, Shin Yean Lim, Hira Choudhury, and Manisha Pandey. 2021. "Molecular and Biochemical Pathways of Catalpol in Alleviating Diabetes Mellitus and Its Complications" Biomolecules 11, no. 2: 323. https://doi.org/10.3390/biom11020323
APA StyleBhattamisra, S. K., Koh, H. M., Lim, S. Y., Choudhury, H., & Pandey, M. (2021). Molecular and Biochemical Pathways of Catalpol in Alleviating Diabetes Mellitus and Its Complications. Biomolecules, 11(2), 323. https://doi.org/10.3390/biom11020323