
Nanobodies Provide Insight into the Molecular Mechanisms of the Complement Cascade and Offer New Therapeutic Strategies


Abstract
1. Introduction
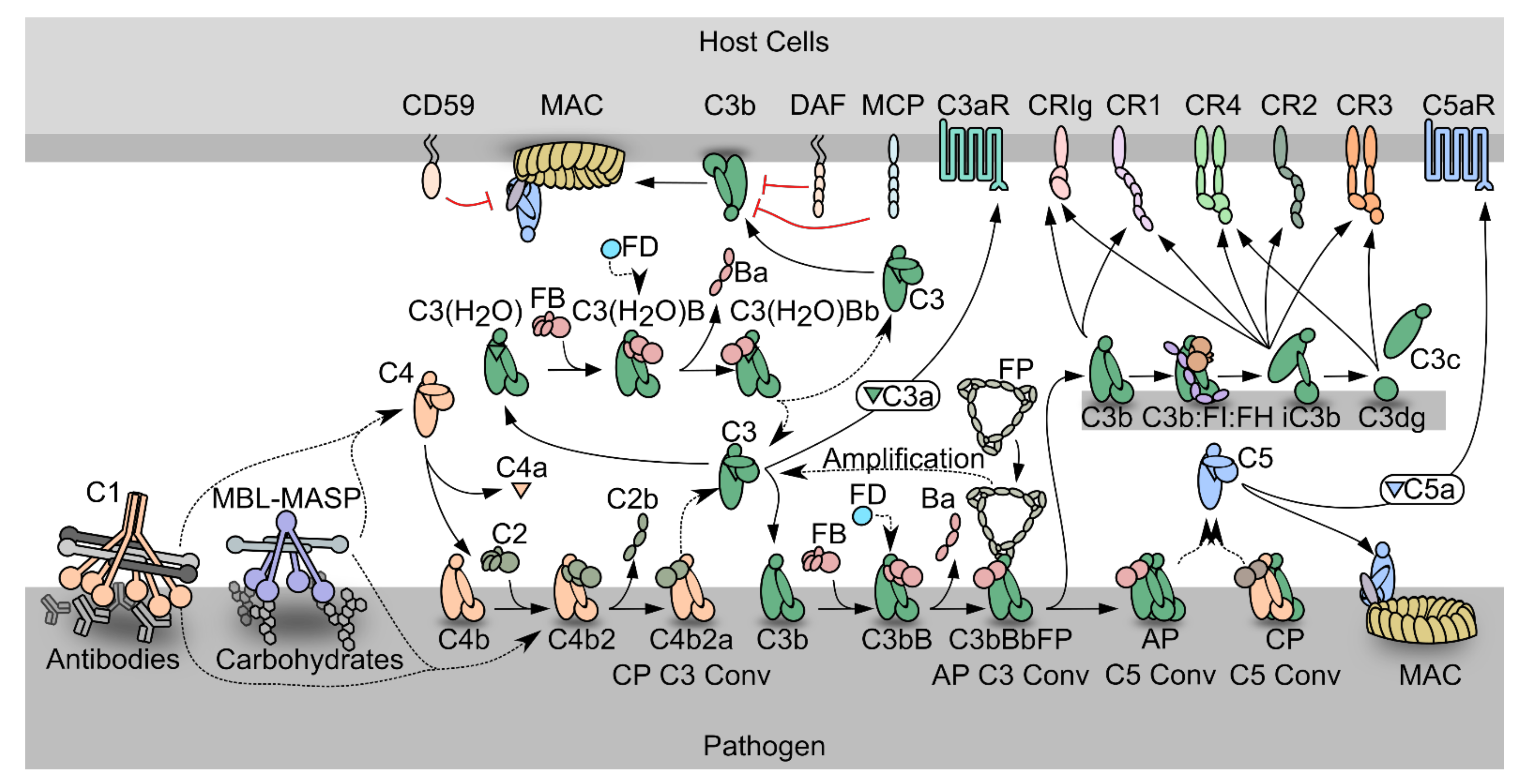
1.1. The Complement System
1.2. The Complement System as a Driver of Pathogenesis
2. The Complement-Targeting Nanobodies
2.1. Complement-Specific Nanobodies for Therapy, Research and Diagnostics
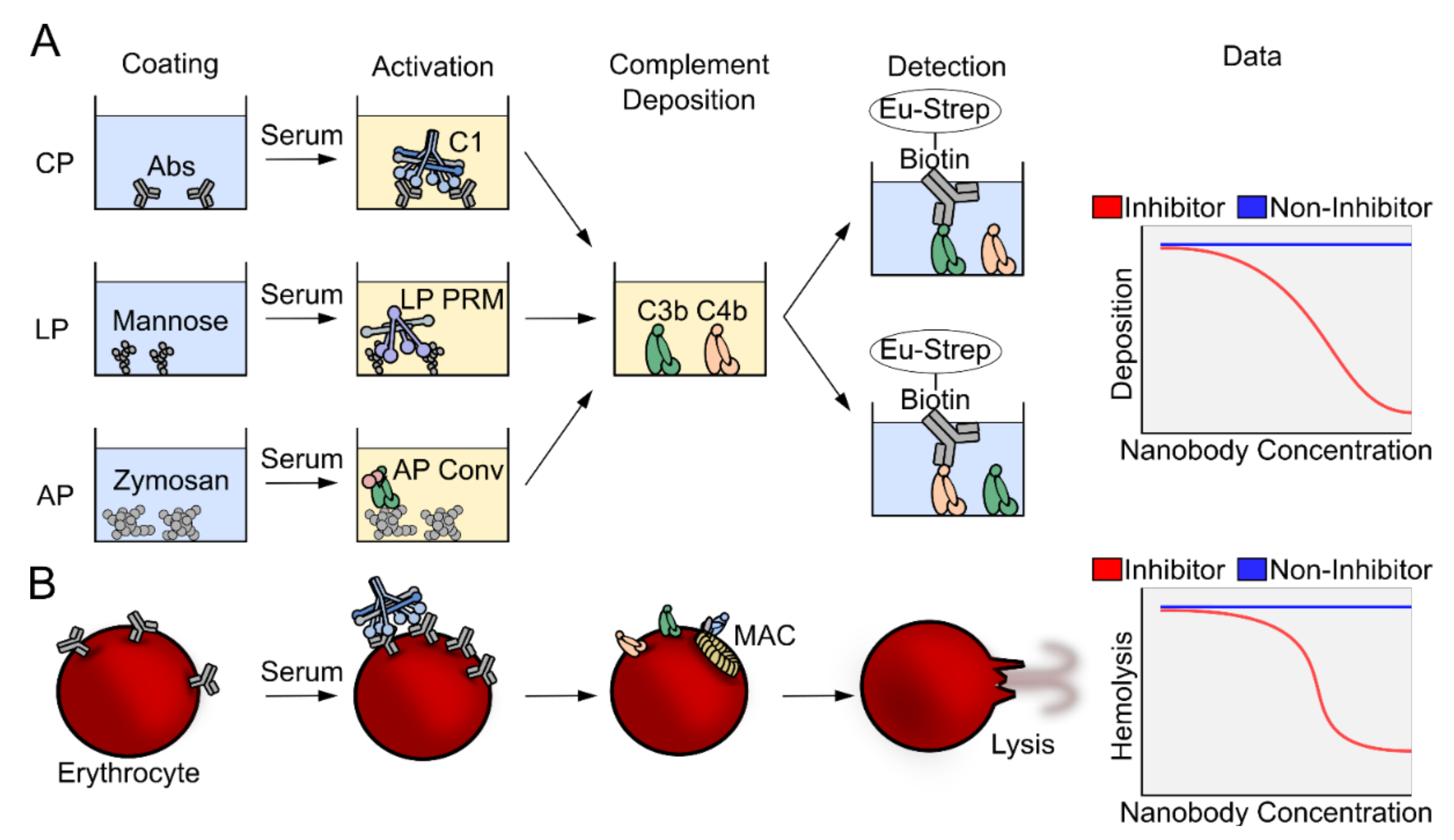
2.2. The Complement Deposition Assays and Hemolytic Assays
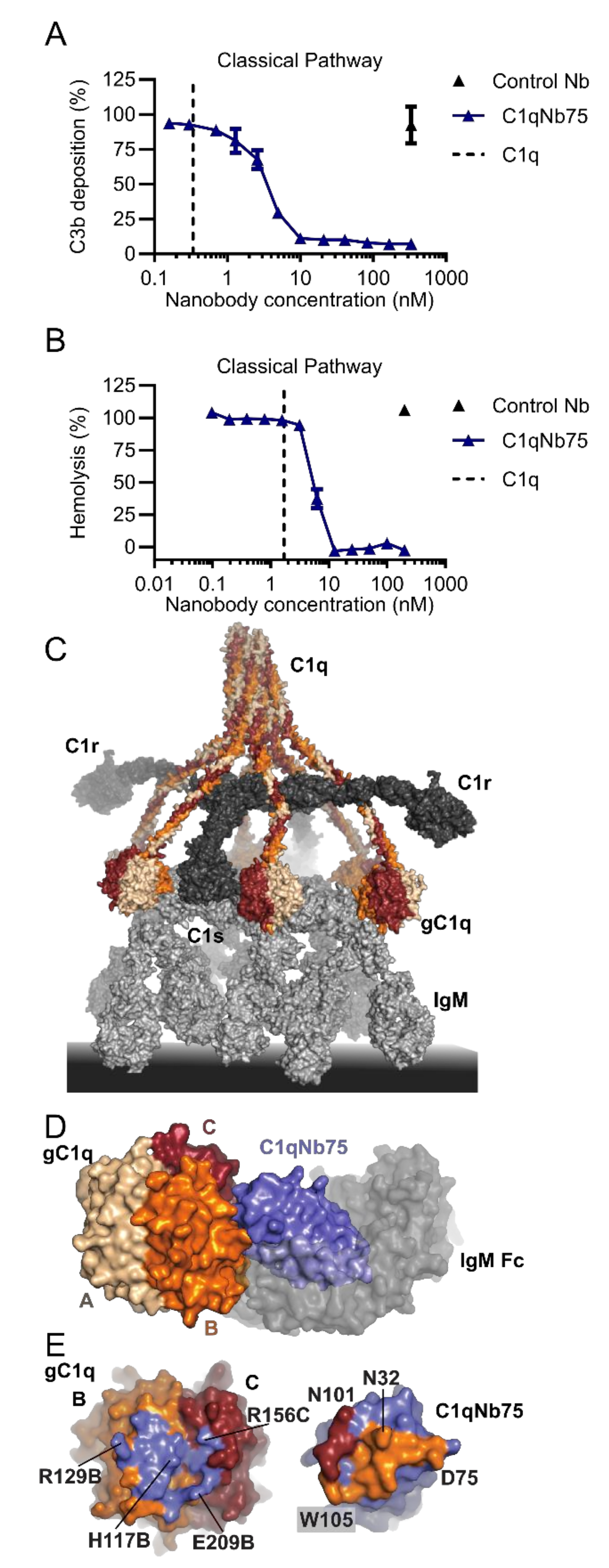
2.3. C1qNb75 Inhibits Initiation of the Classical Pathway
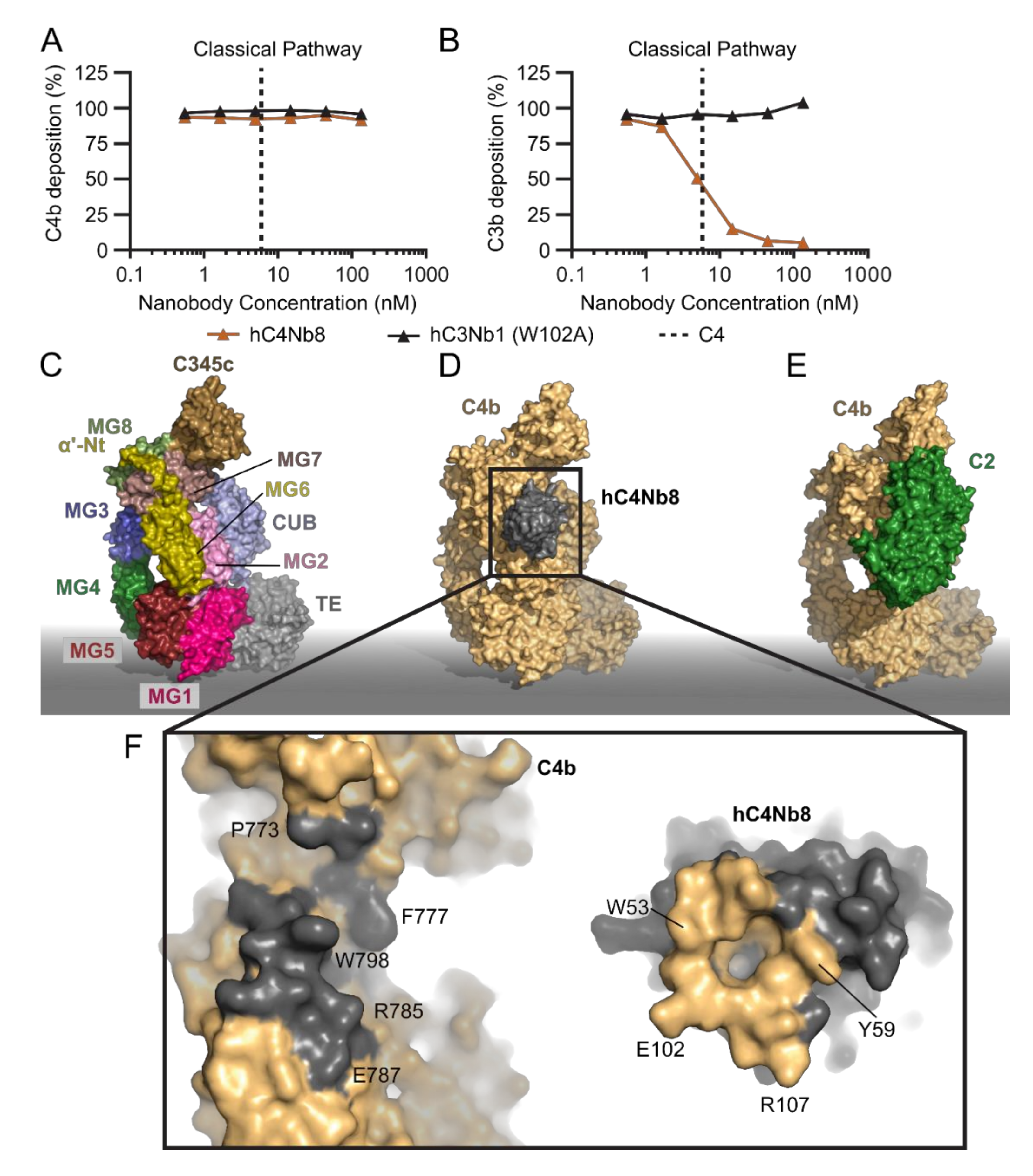
2.4. hC4Nb8 Inhibits In Vivo Assembly of the CP and LP C3 Convertase
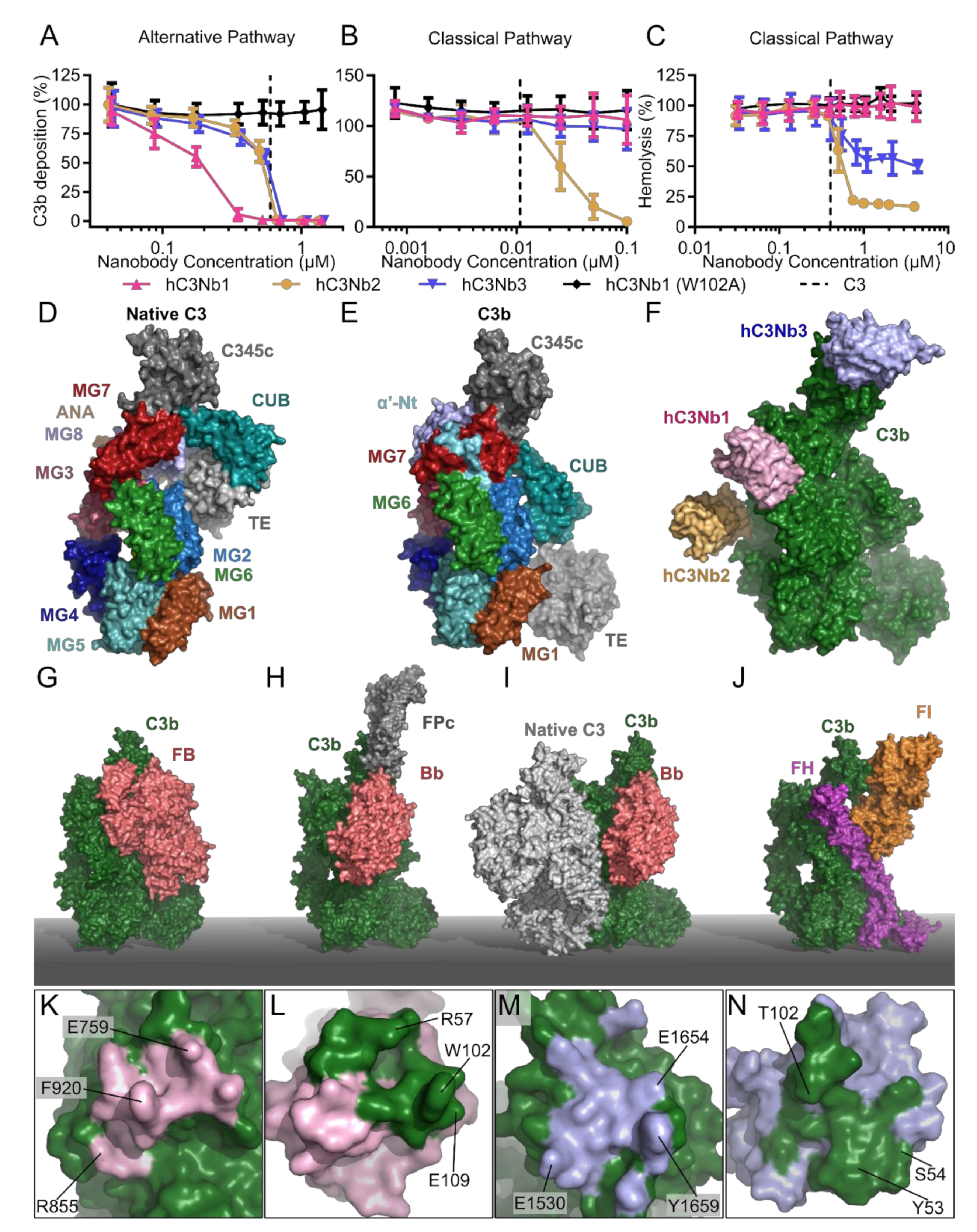
2.5. The hC3Nb1 Is an Inhibitor of AP Proconvertase Assembly
2.6. hC3Nb2 Prevents Substrate Binding to C3 Convertases
2.7. hC3Nb3 Provides Insight into the Role of C3b in C5 Convertases
2.8. The Noninhibitory Properdin-Specific hFPNb1
2.9. Vsig4-Specific Nanobodies
3. Perspectives
3.1. Complement-Specific Nanobodies as Tools in Diagnostics and Research
3.2. Nanobody-Driven Activation of Complement
3.3. Nanobodies as Complement Therapeutics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. Embo. J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef]
- Janeway, C.A. Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Ruseva, M.M.; Zlatarova, A.; Ghai, R.; Kolev, M.; Olova, N.; Gadjeva, M.; Agrawal, A.; Bottazzi, B.; Mantovani, A.; et al. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: Mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry 2006, 45, 4093–4104. [Google Scholar] [CrossRef] [PubMed]
- Païdassi, H.; Tacnet-Delorme, P.; Garlatti, V.; Darnault, C.; Ghebrehiwet, B.; Gaboriaud, C.; Arlaud, G.J.; Frachet, P. C1q Binds Phosphatidylserine and Likely Acts as a Multiligand-Bridging Molecule in Apoptotic Cell Recognition. J. Immunol. 2008, 180, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Gaboriaud, C.; Frachet, P.; Thielens, N.M.; Arlaud, G.J. The human c1q globular domain: Structure and recognition of non-immune self ligands. Front. Immunol. 2011, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Tacnet-Delorme, P.; Chevallier, S.; Arlaud, G.J. β-Amyloid Fibrils Activate the C1 Complex of Complement Under Physiological Conditions: Evidence for a Binding Site for Aβ on the C1q Globular Regions. J. Immunol. 2001, 167, 6374–6381. [Google Scholar] [CrossRef]
- Erlich, P.; Dumestre-Pérard, C.; Ling, W.L.; Lemaire-Vieille, C.; Schoehn, G.; Arlaud, G.J.; Thielens, N.M.; Gagnon, J.; Cesbron, J.-Y. Complement protein C1q forms a complex with cytotoxic prion protein oligomers. J. Biol. Chem. 2010, 285, 19267–19276. [Google Scholar] [CrossRef]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.-J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef]
- Bally, I.; Inforzato, A.; Dalonneau, F.; Stravalaci, M.; Bottazzi, B.; Gaboriaud, C.; Thielens, N.M. Interaction of C1q With Pentraxin 3 and IgM Revisited: Mutational Studies With Recombinant C1q Variants. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905. [Google Scholar] [CrossRef]
- Zlatarova, A.S.; Rouseva, M.; Roumenina, L.T.; Gadjeva, M.; Kolev, M.; Dobrev, I.; Olova, N.; Ghai, R.; Jensenius, J.C.; Reid, K.B.M.; et al. Existence of Different but Overlapping IgG- and IgM-Binding Sites on the Globular Domain of Human C1q. Biochemistry 2006, 45, 9979–9988. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Kidmose, R.T.; Laursen, N.S.; Dobó, J.; Kjaer, T.R.; Sirotkina, S.; Yatime, L.; Sottrup-Jensen, L.; Thiel, S.; Gál, P.; Andersen, G.R. Structural basis for activation of the complement system by component C4 cleavage. Proc. Natl. Acad. Sci. USA 2012, 109, 15425–15430. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilágyi, Á.; Prohászka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496. [Google Scholar] [CrossRef]
- Sitomer, G.; Stroud, R.M.; Mayer, M.M. Reversible adsorption of C′2 by EAC′4: Role of Mg2+, enumeration of competent SAC′4, two-step nature of C′2a fixation and estimation of its efficiency. Immunochemistry 1966, 3, 57–69. [Google Scholar] [CrossRef]
- Müller-Eberhard, H.J.; Polley, M.J.; Calcott, M.A. Formation and functional significance of a molecular complex derived from the second and the fourth component of human complement. J. Exp. Med. 1967, 125, 359–380. [Google Scholar] [CrossRef]
- Wallis, R.; Dodds, A.W.; Mitchell, D.A.; Sim, R.B.; Reid, K.B.M.; Schwaeble, W.J. Molecular Interactions between MASP-2, C4, and C2 and Their Activation Fragments Leading to Complement Activation via the Lectin Pathway. J. Biol. Chem. 2007, 282, 7844–7851. [Google Scholar] [CrossRef]
- Gadjeva, M.; Thiel, S.; Jensenius, J.C. The mannan-binding-lectin pathway of the innate immune response. Curr. Opin. Immunol. 2001, 13, 74–78. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, T. Ficolins and the lectin complement pathway. Immunol. Rev. 2001, 180, 78–85. [Google Scholar] [CrossRef]
- Henriksen, M.L.; Brandt, J.; Andrieu, J.-P.; Nielsen, C.; Jensen, P.H.; Holmskov, U.; Jorgensen, T.J.D.; Palarasah, Y.; Thielens, N.M.; Hansen, S. Heteromeric Complexes of Native Collectin Kidney 1 and Collectin Liver 1 Are Found in the Circulation with MASPs and Activate the Complement System. J. Immunol. 2013, 191, 6117–6127. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Fujita, T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 1992, 176, 1497–1502. [Google Scholar] [CrossRef]
- Thiel, S.; Vorup-Jensen, T.; Stover, C.M.; Schwaeble, W.; Laursen, S.B.; Poulsen, K.; Willis, A.C.; Eggleton, P.; Hansen, S.; Holmskov, U.; et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature 1997, 386, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.R.; Thiel, S.; Willis, A.C.; Vorup-Jensen, T.; Christensen, T.; Petersen, S.V.; Jensenius, J.C. Mannan-binding lectin associated serine protease 3 (MASP-3)—A new component of the lectin pathway of complement activation. Immunopharmacology 2000, 49, 79. [Google Scholar] [CrossRef]
- Janssen, B.J.C.; Huizinga, E.G.; Raaijmakers, H.C.A.; Roos, A.; Daha, M.R.; Nilsson-Ekdahl, K.; Nilsson, B.; Gros, P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 2005, 437, 505–511. [Google Scholar] [CrossRef]
- Janssen, B.J.C.; Christodoulidou, A.; McCarthy, A.; Lambris, J.D.; Gros, P. Structure of C3b reveals conformational changes that underlie complement activity. Nature 2006, 444, 213–216. [Google Scholar] [CrossRef]
- Pangburn, M.K.; Müller-Eberhard, H.J. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem. J. 1986, 235, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.M.; Wu, J.; Ruyken, M.; van Domselaar, R.; Planken, K.L.; Tzekou, A.; Ricklin, D.; Lambris, J.D.; Janssen, B.J.C.; van Strijp, J.A.G.; et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat. Immunol. 2009, 10, 721. [Google Scholar] [CrossRef]
- Forneris, F.; Ricklin, D.; Wu, J.; Tzekou, A.; Wallace, R.S.; Lambris, J.D.; Gros, P. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 2010, 330, 1816–1820. [Google Scholar] [CrossRef]
- Lachmann, P.J. The Amplification Loop of the Complement Pathways. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2009; Chapter 4; Volume 104, pp. 115–149. [Google Scholar]
- Harboe, M.; Garred, P.; Karlstrom, E.; Lindstad, J.K.; Stahl, G.L.; Mollnes, T.E. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol. Immunol. 2009, 47, 373–380. [Google Scholar] [CrossRef]
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446. [Google Scholar] [CrossRef]
- Fishelson, Z.; Pangburn, M.K.; Müller-Eberhard, H.J. Characterization of the initial C3 convertase of the alternative pathway of human complement. J. Immunol. 1984, 132, 1430–1434. [Google Scholar]
- Harrison, R.A. The properdin pathway: An "alternative activation pathway" or a "critical amplification loop" for C3 and C5 activation? Semin. Immunopathol. 2018, 40, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, K.N.; Mohlin, C.; Adler, A.; Åman, A.; Anand Manivel, V.; Sandholm, K.; Huber-Lang, M.; Fromell, K.; Nilsson, B. Is generation of C3(H2O) necessary for activation of the alternative pathway in real life? Mol. Immunol. 2019, 114, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T.; Austen, K.F. Properdin: Binding to C3b and stabilization of the C3b-dependent C3 convertase. J. Exp. Med. 1975, 142, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Hourcade, D.E. The Role of Properdin in the Assembly of the Alternative Pathway C3 Convertases of Complement. J. Biol. Chem. 2006, 281, 2128–2132. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.V.; Gadeberg, T.A.F.; Thomas, C.; Wang, Y.; Joram, N.; Jensen, R.K.; Mazarakis, S.M.M.; Revel, M.; El Sissy, C.; Petersen, S.V.; et al. Structural Basis for Properdin Oligomerization and Convertase Stimulation in the Human Complement System. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Hourcade, D.; Holers, V.M.; Atkinson, J.P. The Regulators of Complement Activation (RCA) Gene Cluster. In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: Cambridge, MA, USA, 1989; Volume 45, pp. 381–416. [Google Scholar]
- Liszewski, M.K.; Leung, M.; Cui, W.; Subramanian, V.B.; Parkinson, J.; Barlow, P.N.; Manchester, M.; Atkinson, J.P. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP.; CD46). J. Biol. Chem. 2000, 275, 37692–37701. [Google Scholar] [CrossRef]
- Krych-Goldberg, M.; Atkinson, J.P. Structure-function relationships of complement receptor type 1. Immunol. Rev. 2001, 180, 112–122. [Google Scholar] [CrossRef]
- Jozsi, M.; Zipfel, P.F. Factor H family proteins and human diseases. Trends Immunol. 2008, 29, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, Y.-Q.; Ricklin, D.; Janssen, B.J.C.; Lambris, J.D.; Gros, P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 2009, 10, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: A defence system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.G.; Woodruff, T.M. Is the Complement Activation Product C3a a Proinflammatory Molecule? Re-evaluating the Evidence and the Myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef] [PubMed]
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006, 124, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Fu, Y.; Yosri, M.; Chen, Y.; Sun, P.; Xu, J.; Zhang, M.; Sun, D.; Strickland, A.B.; Mackey, Z.B.; et al. CRIg plays an essential role in intravascular clearance of bloodborne parasites by interacting with complement. Proc. Natl. Acad. Sci. USA 2019, 116, 24214–24220. [Google Scholar] [CrossRef]
- Vorup-Jensen, T.; Jensen, R.K. Structural Immunology of Complement Receptors 3 and 4. Front. Immunol. 2018, 9, 2716. [Google Scholar] [CrossRef]
- Gonzalez, S.F.; Lukacs-Kornek, V.; Kuligowski, M.P.; Pitcher, L.A.; Degn, S.E.; Turley, S.J.; Carroll, M.C. Complement-Dependent Transport of Antigen into B Cell Follicles. J. Immunol. 2010, 185, 2659–2664. [Google Scholar] [CrossRef] [PubMed]
- Pangburn, M.K.; Rawal, N. Structure and function of complement C5 convertase enzymes. Biochem. Soc. Trans. 2002, 30, 1006–1010. [Google Scholar] [CrossRef]
- Rawal, N.; Pangburn, M.K. Formation of High Affinity C5 Convertase of the Classical Pathway of Complement. J. Biol. Chem. 2003, 278, 38476–38483. [Google Scholar] [CrossRef]
- Fredslund, F.; Laursen, N.S.; Roversi, P.; Jenner, L.; Oliveira, C.L.; Pedersen, J.S.; Nunn, M.A.; Lea, S.M.; Discipio, R.; Sottrup-Jensen, L.; et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat. Immunol. 2008, 9, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Bayly-Jones, C.; Bubeck, D.; Dunstone, M.A. The mystery behind membrane insertion: A review of the complement membrane attack complex. Philos Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 20160221. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, D.A.C.; Angelier, M.L.; Harrison, R.A.; Rooijakkers, S.H.M. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J. Innate Immun. 2018, 10, 455–464. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Hill, A.; DeZern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Primers 2017, 3, 17028. [Google Scholar] [CrossRef] [PubMed]
- Nicholson-Weller, A.; March, J.P.; Rosenfeld, S.I.; Austen, K.F. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factor. Proc. Natl. Acad. Sci. USA 1983, 80, 5066–5070. [Google Scholar] [CrossRef]
- Holguin, M.H.; Fredrick, L.R.; Bernshaw, N.J.; Wilcox, L.A.; Parker, C.J. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J. Clin. Invest. 1989, 84, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lu, Y.; Harley, K.T.; Tran, M.-H. Atypical Hemolytic Uremic Syndrome: A Brief Review. Hematol. Rep. 2017, 9, 7053. [Google Scholar] [CrossRef]
- Bresin, E.; Rurali, E.; Caprioli, J.; Sanchez-Corral, P.; Fremeaux-Bacchi, V.; Rodriguez de Cordoba, S.; Pinto, S.; Goodship, T.H.J.; Alberti, M.; Ribes, D.; et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J. Am. Soc. Nephrol. 2013, 24, 475–486. [Google Scholar] [CrossRef]
- Rodríguez de Córdoba, S.; Hidalgo, M.S.; Pinto, S.; Tortajada, A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin. Thromb. Hemost. 2014, 40, 422–430. [Google Scholar] [CrossRef]
- Fakhouri, F.; Fremeaux-Bacchi, V.; Noel, L.H.; Cook, H.T.; Pickering, M.C. C3 glomerulopathy: A new classification. Nat. Rev. Nephrol. 2010, 6, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.H.; Appel, G.B.; Blom, A.M.; Cook, H.T.; D’Agati, V.D.; Fakhouri, F.; Fremeaux-Bacchi, V.; Józsi, M.; Kavanagh, D.; Lambris, J.D.; et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat. Rev. Nephrol. 2019, 15, 129–143. [Google Scholar] [CrossRef]
- Zhang, Y.; Meyer, N.C.; Wang, K.; Nishimura, C.; Frees, K.; Jones, M.; Katz, L.M.; Sethi, S.; Smith, R.J. Causes of alternative pathway dysregulation in dense deposit disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 265–274. [Google Scholar] [CrossRef]
- Bu, F.; Borsa, N.G.; Jones, M.B.; Takanami, E.; Nishimura, C.; Hauer, J.J.; Azaiez, H.; Black-Ziegelbein, E.A.; Meyer, N.C.; Kolbe, D.L.; et al. High-Throughput Genetic Testing for Thrombotic Microangiopathies and C3 Glomerulopathies. J. Am. Soc. Nephrol. 2016, 27, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Van Lookeren Campagne, M.; Strauss, E.C.; Yaspan, B.L. Age-related macular degeneration: Complement in action. Immunobiology 2016, 221, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439.e2. [Google Scholar] [CrossRef]
- Ogden, C.A.; deCathelineau, A.; Hoffmann, P.R.; Bratton, D.; Ghebrehiwet, B.; Fadok, V.A.; Henson, P.M. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 2001, 194, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Botto, M.; Walport, M.J. C1q, Autoimmunity and Apoptosis. Immunobiology 2002, 205, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.L.; Zwarthoff, S.A.; Rooijakkers, S.H.M.; Geisbrecht, B.V. Novel Evasion Mechanisms of the Classical Complement Pathway. J. Immunol. 2016, 197, 2051–2060. [Google Scholar] [CrossRef]
- Jäger, U.; D’Sa, S.; Schörgenhofer, C.; Bartko, J.; Derhaschnig, U.; Sillaber, C.; Jilma-Stohlawetz, P.; Fillitz, M.; Schenk, T.; Patou, G.; et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: A first-in-human trial. Blood 2019, 133, 893–901. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Complement in Immune and Inflammatory Disorders: Therapeutic Interventions. J. Immunol. 2013, 190, 3839. [Google Scholar] [CrossRef] [PubMed]
- Wouters, D.; Zeerleder, S. Complement inhibitors to treat IgM-mediated autoimmune hemolysis. Haematologica 2015, 100, 1388–1395. [Google Scholar] [CrossRef]
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Johnson, M.B.; Stevens, B. Pruning hypothesis comes of age. Nature 2018, 554, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.L.; Wilton, D.K.; Muthukumar, A.; Fox, R.G.; Carey, A.; Crotty, W.; Scott-Hewitt, N.; Bien, E.; Sabatini, D.A.; Lanser, T.; et al. CUB and Sushi Multiple Domains 1 (CSMD1) opposes the complement cascade in neural tissues. bioRxiv 2020. [Google Scholar] [CrossRef]
- Comer, A.L.; Jinadasa, T.; Sriram, B.; Phadke, R.A.; Kretsge, L.N.; Nguyen, T.P.H.; Antognetti, G.; Gilbert, J.P.; Lee, J.; Newmark, E.R.; et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol. 2020, 18, e3000604. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.-Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Vukojicic, A.; Delestrée, N.; Fletcher, E.V.; Pagiazitis, J.G.; Sankaranarayanan, S.; Yednock, T.A.; Barres, B.A.; Mentis, G.Z. The Classical Complement Pathway Mediates Microglia-Dependent Remodeling of Spinal Motor Circuits during Development and in SMA. Cell Rep. 2019, 29, 3087–3100.e3087. [Google Scholar] [CrossRef] [PubMed]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538. [Google Scholar] [CrossRef]
- Zelek, W.M.; Xie, L.; Morgan, B.P.; Harris, C.L. Compendium of current complement therapeutics. Mol. Immunol. 2019, 114, 341–352. [Google Scholar] [CrossRef]
- Ricklin, D.; Mastellos, D.C.; Reis, E.S.; Lambris, J.D. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 2018, 14, 26–47. [Google Scholar] [CrossRef]
- Mohebnasab, M.; Eriksson, O.; Persson, B.; Sandholm, K.; Mohlin, C.; Huber-Lang, M.; Keating, B.J.; Ekdahl, K.N.; Nilsson, B. Current and Future Approaches for Monitoring Responses to Anti-complement Therapeutics. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.F.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef]
- Moller-Kristensen, M.; Thiel, S.; Hansen, A.G.; Jensenius, J.C. On the site of C4 deposition upon complement activation via the mannan-binding lectin pathway or the classical pathway. Scand. J. Immunol. 2003, 57, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, P.D.; Thiel, S.; Jensen, L.; Hansen, A.G.; Matthiesen, F.; Jensenius, J.C. Quantification of mannan-binding lectin. J. Immunol. Methods 2006, 315, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.V.; Thiel, S.; Jensen, L.; Steffensen, R.; Jensenius, J.C. An assay for the mannan-binding lectin pathway of complement activation. J. Immunol. Methods 2001, 257, 107–116. [Google Scholar] [CrossRef]
- Harboe, M.; Garred, P.; Lindstad, J.K.; Pharo, A.; Müller, F.; Stahl, G.L.; Lambris, J.D.; Mollnes, T.E. The role of properdin in zymosan- and Escherichia coli-induced complement activation. J. Immunol. 2012, 189, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Laursen, N.S.; Pedersen, D.V.; Gytz, H.; Zarantonello, A.; Bernth Jensen, J.M.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Functional and Structural Characterization of a Potent C1q Inhibitor Targeting the Classical Pathway of the Complement System. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Zarantonello, A.; Presumey, J.; Simoni, L.; Yalcin, E.; Fox, R.; Hansen, A.; Olesen, H.G.; Thiel, S.; Johnson, M.B.; Stevens, B.; et al. An Ultrahigh-Affinity Complement C4b-Specific Nanobody Inhibits In Vivo Assembly of the Classical Pathway Proconvertase. J. Immunol. 2020. [Google Scholar] [CrossRef]
- Jensen, R.K.; Pihl, R.; Gadeberg, T.A.F.; Jensen, J.K.; Andersen, K.R.; Thiel, S.; Laursen, N.S.; Andersen, G.R. A potent complement factor C3-specific nanobody inhibiting multiple functions in the alternative pathway of human and murine complement. J. Biol. Chem. 2018, 293, 6269–6281. [Google Scholar] [CrossRef]
- Pedersen, H.; Jensen, R.K.; Hansen, A.G.; Gadeberg, T.A.F.; Thiel, S.; Laursen, N.S.; Andersen, G.R. A C3-specific nanobody that blocks all three activation pathways in the human and murine complement system. J. Biol. Chem. 2020, 295, 8746–8758. [Google Scholar] [CrossRef]
- Pedersen, H.; Jensen, R.K.; Jensen, J.M.B.; Fox, R.; Pedersen, D.V.; Olesen, H.G.; Hansen, A.G.; Christiansen, D.; Mazarakis, S.M.M.; Lojek, N.; et al. A Complement C3-Specific Nanobody for Modulation of the Alternative Cascade Identifies the C-Terminal Domain of C3b as Functional in C5 Convertase Activity. J. Immunol. 2020. [Google Scholar] [CrossRef]
- Zheng, F.; Put, S.; Bouwens, L.; Lahoutte, T.; Matthys, P.; Muyldermans, S.; De Baetselier, P.; Devoogdt, N.; Raes, G.; Schoonooghe, S. Molecular imaging with macrophage CRIg-targeting nanobodies for early and preclinical diagnosis in a mouse model of rheumatoid arthritis. J. Nucl. Med. 2014, 55, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.W.; Damko, E.; Modi, B.; Tu, N.; Meagher, K.; Voronina, V.; Gartner, H.; Ehrlich, G.; Rafique, A.; Babb, R.; et al. A novel bispecific antibody platform to direct complement activity for efficient lysis of target cells. Sci. Rep. 2019, 9, 12031. [Google Scholar] [CrossRef]
- Thielens, N.M.; Tedesco, F.; Bohlson, S.S.; Gaboriaud, C.; Tenner, A.J. C1q: A fresh look upon an old molecule. Mol. Immunol. 2017, 89, 73–83. [Google Scholar] [CrossRef]
- Pan, Q.; Ebanks, R.O.; Isenman, D.E. Two Clusters of Acidic Amino Acids Near the NH Terminus of Complement Component C4 α′-Chain Are Important for C2 Binding. J. Immunol. 2000, 165, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.; Jensen, J.K.; Andersen, G.R. Solution Structures of Complement C2 and Its C4 Complexes Propose Pathway-specific Mechanisms for Control and Activation of the Complement Proconvertases. J. Biol. Chem. 2016, 291, 16494–16507. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Wu, J.; Ricklin, D.; Forneris, F.; Di Crescenzio, P.; Schmidt, C.Q.; Granneman, J.; Sharp, T.H.; Lambris, J.D.; Gros, P. Regulator-dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat. Struct. Mol. Biol. 2017, 24, 643–651. [Google Scholar] [CrossRef]
- Carroll, M.C.; Isenman, D.E. Regulation of humoral immunity by complement. Immunity 2012, 37, 199–207. [Google Scholar] [CrossRef]
- Pedersen, D.V.; Revel, M.; Gadeberg, T.A.F.; Andersen, G.R. Crystallization and X-ray analysis of monodisperse human properdin. Acta Crystallogr. Sect. F 2019, 75. [Google Scholar] [CrossRef]
- Pedersen, D.V.; Roumenina, L.; Jensen, R.K.; Gadeberg, T.A.; Marinozzi, C.; Picard, C.; Rybkine, T.; Thiel, S.; Sorensen, U.B.; Stover, C.; et al. Functional and structural insight into properdin control of complement alternative pathway amplification. Embo. J. 2017, 36, 1084–1099. [Google Scholar] [CrossRef]
- Laursen, N.S.; Andersen, K.R.; Braren, I.; Spillner, E.; Sottrup-Jensen, L.; Andersen, G.R. Substrate recognition by complement convertases revealed in the C5-cobra venom factor complex. Embo. J. 2011, 30, 606–616. [Google Scholar] [CrossRef]
- Schatz-Jakobsen, J.A.; Pedersen, D.V.; Andersen, G.R. Structural insight into proteolytic activation and regulation of the complement system. Immunol. Rev. 2016, 274, 59–73. [Google Scholar] [CrossRef]
- Pangburn, M.K. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J. Immunol. 1989, 142, 202–207. [Google Scholar]
- Wen, Y.; Ouyang, Z.; Schoonooghe, S.; Luo, S.; De Baetselier, P.; Lu, W.; Muyldermans, S.; Raes, G.; Zheng, F. Structural evaluation of a nanobody targeting complement receptor Vsig4 and its cross reactivity. Immunobiology 2017, 222, 807–813. [Google Scholar] [CrossRef]
- Wiesmann, C.; Katschke, K.J.; Yin, J.; Helmy, K.Y.; Steffek, M.; Fairbrother, W.J.; McCallum, S.A.; Embuscado, L.; DeForge, L.; Hass, P.E.; et al. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature 2006, 444, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Van Lookeren Campagne, M.; Katschke, K.J., Jr.; Gullipalli, D.; Miwa, T.; Ueda, Y.; Wang, Y.; Palmer, M.; Xing, G.; Song, W.C. Prevention of Fatal C3 Glomerulopathy by Recombinant Complement Receptor of the Ig Superfamily. J. Am. Soc. Nephrol. 2018, 29, 2053–2059. [Google Scholar] [CrossRef]
- Williams, R.O. Collagen-induced arthritis in mice. Methods Mol. Med. 2007, 136, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Monach, P.A.; Mathis, D.; Benoist, C. The K/BxN arthritis model. Curr. Protoc. Immunol. 2008, 15, Unit 15.22. [Google Scholar] [CrossRef]
- Zheng, F.; Perlman, H.; Matthys, P.; Wen, Y.; Lahoutte, T.; Muyldermans, S.; Lu, S.; De Baetselier, P.; Schoonooghe, S.; Devoogdt, N.; et al. Specificity Evaluation and Disease Monitoring in Arthritis Imaging with Complement Receptor of the Ig superfamily targeting Nanobodies. Sci. Rep. 2016, 6, 35966. [Google Scholar] [CrossRef]
- Bernth Jensen, J.M.; Petersen, M.S.; Ellerman-Eriksen, S.; Møller, B.K.; Jensenius, J.C.; Sørensen, U.B.S.; Thiel, S. Abundant human anti-Galα3Gal antibodies display broad pathogen reactivity. Sci. Rep. 2020, 10, 4611. [Google Scholar] [CrossRef]
- Bernth Jensen, J.M.; Laursen, N.S.; Jensen, R.K.; Andersen, G.R.; Jensenius, J.C.; Sørensen, U.B.S.; Thiel, S. Complement activation by human IgG antibodies to galactose-α-1,3-galactose. Immunology 2020, n/a. [Google Scholar] [CrossRef] [PubMed]
- Lausen, M.; Thomsen, M.E.; Christiansen, G.; Karred, N.; Stensballe, A.; Bennike, T.B.; Birkelund, S. Analysis of complement deposition and processing on Chlamydia trachomatis. Med. Microbiol. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.V.; Rösner, T.; Hansen, A.G.; Andersen, K.R.; Thiel, S.; Andersen, G.R.; Valerius, T.; Laursen, N.S. Recruitment of properdin by bi-specific nanobodies activates the alternative pathway of complement. Mol. Immunol. 2020, 124, 200–210. [Google Scholar] [CrossRef]
- Duggan, S. Caplacizumab: First Global Approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef]
- Hoefman, S.; Ottevaere, I.; Baumeister, J.; Sargentini-Maier, M.L. Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies 2015, 4, 141–156. [Google Scholar] [CrossRef]
- Jovcevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, A.; Langley, E.F.; Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Asavapanumas, N.; Verkman, A.S. Emerging therapeutic targets for neuromyelitis optica spectrum disorder. Expert Opin. Targets 2020, 24, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Werneburg, S.; Jung, J.; Kunjamma, R.B.; Ha, S.K.; Luciano, N.J.; Willis, C.M.; Gao, G.; Biscola, N.P.; Havton, L.A.; Crocker, S.J.; et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2020, 52, 167–182.e7. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Agyemang, A.F.; Alimzhanov, M.B.; Degn, S.; Tsiftsoglou, S.A.; Alicot, E.; Jones, S.A.; Ma, M.; Carroll, M.C. Complement C4 maintains peripheral B-cell tolerance in a myeloid cell dependent manner. Eur. J. Immunol. 2013, 43, 2441–2450. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanobody (Reference) | Mechanism | Epitope | Antigen | Dissociation Constant KD (nM) |
|---|---|---|---|---|
| C1qNb75 [97] | Prevents C1q binding to Fc of IgG and IgM | gC1q chains B and C | hgC1q | 0.3 |
| hC4Nb8 [98] | Blocks CP proconvertase assembly | C4b α’-Nt, MG6 and MG7 domains | hC4 | 2000 |
| hC4b | 0.016 | |||
| mC4b | 0.2 | |||
| hC3Nb1 [99] | Blocks AP proconvertase assembly | C3 and C3b MG6 and MG7 domain | hC3 | 0.9 |
| hC3b | 0.2 | |||
| hC3Nb2 [100] | Prevents substrate binding to C3 convertases | C3 and C3b MG3 and MG4 domains | hC3 | 10 |
| hC3b | 5 | |||
| hC3-MA | 3 | |||
| mC3b | 0.6 | |||
| hC3Nb3 [101] | Blocks AP C3 proconvertase assembly and CP C5 convertase activity | C3 and C3b C345c domain | hC3 | 3 |
| hC3b | 3 | |||
| hC3-MA | 6 | |||
| mC3b | 3 | |||
| hFPNb1 [39] | Non-inhibitory | FP thrombospondin repeat 4 | hFPc | 7.3 |
| Nb119 [102] | Blocks C3b and C3c binding to Vsig4 (CRIg) | Vsig4 (CRIg) ectodomain | hVsig4 | 850 |
| mVsig4 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarantonello, A.; Pedersen, H.; Laursen, N.S.; Andersen, G.R. Nanobodies Provide Insight into the Molecular Mechanisms of the Complement Cascade and Offer New Therapeutic Strategies. Biomolecules 2021, 11, 298. https://doi.org/10.3390/biom11020298
Zarantonello A, Pedersen H, Laursen NS, Andersen GR. Nanobodies Provide Insight into the Molecular Mechanisms of the Complement Cascade and Offer New Therapeutic Strategies. Biomolecules. 2021; 11(2):298. https://doi.org/10.3390/biom11020298
Chicago/Turabian StyleZarantonello, Alessandra, Henrik Pedersen, Nick S. Laursen, and Gregers R. Andersen. 2021. "Nanobodies Provide Insight into the Molecular Mechanisms of the Complement Cascade and Offer New Therapeutic Strategies" Biomolecules 11, no. 2: 298. https://doi.org/10.3390/biom11020298
APA StyleZarantonello, A., Pedersen, H., Laursen, N. S., & Andersen, G. R. (2021). Nanobodies Provide Insight into the Molecular Mechanisms of the Complement Cascade and Offer New Therapeutic Strategies. Biomolecules, 11(2), 298. https://doi.org/10.3390/biom11020298